|

Постановлением Кабинета Министров Украины от 28 октября 2004 года № 1419 (далее Постановление) было предусмотрено, что с 1 января 2009 г. оборот лекарственных средств должен осуществляться в соответствии с требованиями надлежащей производственной, дистрибьюторской, лабораторной и клинической практик, которые гармонизированы с соответствующими директивами и руководствами Европейского Союза.
На уровне правительства была запланирована масштабная реформа, однако для ее реализации не были определены и полностью осуществлены необходимые мероприятия. Постановление предусматривало:
- до 1 декабря 2004 г. подать на рассмотрение Кабинета Министров Украины проект Порядка государственного контроля лекарственных средств в случае их ввоза в Украину, предусмотрев проведение в полном объеме посерийного контроля их качества или наличие у производителя сертификата надлежащей производственной практики, выданного или признанного Государственной службой лекарственных средств и изделий медицинского назначения;
- обеспечить вступление указанной службы в международную систему сотрудничества по фармацевтическим инспекциям (PIC/S);
- разработать мероприятия относительно стимулирования отечественных производителей лекарственных средств с целью внедрения надлежащей производственной, дистрибьюторской, лабораторной и клинической практики.
С переходом на работу по правилам GMP, GDP, GCP, GLP мы должны подняться на принципиально новый уровень обеспечения качества, требующий введения европейской системы сертификации лекарственных средств, новых технических регламентов и стандартов и, соответственно, создания и функционирования новой системы стандартизации в рамках Министерства здравоохранения Украины. Однако работы по стандартизации упомянутым выше постановлением запланированы не были.
Лишь в сентябре 2006 г. по результатам совещания у первого вице-премьер-министра Украины Н.Я. Азарова было принято решение обсудить возможность делегирования Министерству здравоохранения функции государственной регистрации стандартов в сфере оборота как лекарственных средств, так и изделий медицинского назначения, включая процедуры сертификации. Было также предложено создать программу поэтапного внедрения стандартов GMP, GLP, GCP, GDP в Украине, что не было осуществлено.
|
Необходимо сразу указать на две принципиальные ошибки в пункте 15 протокольного решения относительно обсуждаемой проблемы:
во-первых, лекарственные средства и изделия медицинского назначения требуют двух совершенно разных систем сертификации и соответственно двух разных систем стандартизации и двух органов по стандартизации. Изделия медицинского назначения стандартизуют в рамках Национальной стандартизации, которой управляет Государственный комитет по вопросам технического регулирования и потребительской политики (Госпотребстандарт). Лекарственные средства требуют построения другой системы стандартизации, которой должно управлять Министерство здравоохранения Украины и представлять ее на международном уровне.
Во-вторых, развитие фармацевтического сектора стали почему-то связывать с внедрением только четырех нормативных документов. При этом не учитывался европейский опыт, в соответствии с которым руководства по GMP, GLP, GCP, GDP находятся в большой и целостной системе нормативных документов, без которой внедрение только надлежащих практик не приведет к ожидаемым результатам.
Появилась идея, что стандартизацией и сертификацией фармацевтической продукции, которая включает лекарственные средства, должен управлять Госпотребстандарт. Несостоятельность этой идеи очевидна, если оценить систему сертификации лекарственных средств для международной торговли, принятую в ЕС. Система гарантии качества и сертификации лекарственных средств в ЕС (далее — Система) включает такие элементы:
- лицензирование (то есть регистрацию) лекарственных средств;
- лицензирование производства и импорта на основании результатов инспектирования на соответствие GMP и регистрационной документации;
- лицензирование дистрибуции на основании результатов инспектирования на соответствие правилам надлежащей дистрибьюторской практики (GDP);
- независимый контроль качества, который связывают с независимостью контроля качества от производства, институтом уполномоченных лиц, а также государственным контролем качества на этапах регистрации и реализации;
- фармакологический надзор (фармаконадзор);
- сертификацию субстанций Европейской Фармакопеей.
Таким образом, система гарантирования качества и сертификации является комплексной, а регистрация, лицензирование, инспектирование, фармаконадзор и другие процедуры — ее составные части. Управлять этой системой должен один орган. Вычленение из Системы элементов и передача их другим органам (например, инспектирования на соответствие требованиям надлежащей производственной практики) приведет к путанице, несоответствиям и разрушению Системы. Кроме того, инспектирование на соответствие GMP в мире проводится в обязательном порядке Инспекторатом по GMP/GDP, а не частными аудиторскими фирмами в рамках добровольной сертификации. Госпотребстандарт не сможет представлять Украину в PIC/S, Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH), Европейской Фармакопее, Европейском агентстве по оценке лекарственных средств (ЕМЕА), Всемирной организации здравоохранения (ВОЗ) и др. Это прерогатива Министерства здравоохранения Украины.
Обеспечение качества лекарственных средств и подтверждение соответствия (сертификация) должны проводиться на соответствие определенным техническим регламентам и стандартам. Поэтому в соответствии с элементами системы гарантирования качества и сертификации должны быть соответствующие блоки нормативно-правовых актов и нормативных документов. Такие блоки нормативных документов приняты в Европейском Союзе, а также в других регионах и странах. В рамках ICH идет постоянная работа по стандартизации, связанная с гармонизацией и/или взаимным признанием нормативных документов в мире.
В ЕС действует стройная система нормативно-правовых актов, технических регламентов и нормативных документов, изложенных в различных томах «Правил, регулирующих лекарственные средства в Европейском Союзе».
Цель настоящей работы — рассмотреть системный подход к стандартизации, который необходим для внедрения GMP, GLP, GCP, GDP.
Определим задачи, стоящие перед стандартизацией (рис. 1).
|
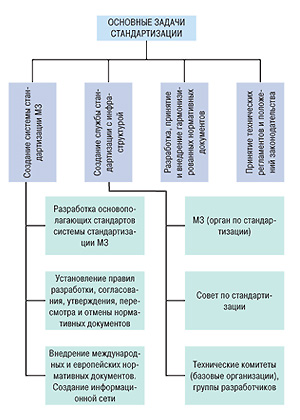
Основная задача стандартизации связана с принятием комплекса гармонизированных руководств и технических регламентов, всесторонне регламентирующих фармацевтическую продукцию. По этому вопросу ведутся дискуссии относительно статуса руководств; предлагается не разрабатывать руководства, а делать приложения к приказам Министерства здравоохранения Украины и регистрировать их в Министерстве юстиции. Как решается этот вопрос в ЕС, четко определено в документе ЕМЕА «Procedure for European Union guidelines and related documents within the pharmaceuticallegislative framework» («Процедура относительно руководств и сопутствующих документов Европейского Союза в рамках фармацевтического законодательства»), перевод которого был опубликован в «Еженедельнике АПТЕКА» 5 февраля 2007 г. № 5 (576).
Если в ICH и ЕС приняты руководства, то аналогичные гармонизированные документы должны быть приняты в Украине. Если в ЕС эти руководства стали обязательными и вошли в законодательство, то необходимо определенными нормативно-правовыми актами вводить их в законодательство Украины; при этом руководство становится частью технического регламента. Если руководства в ЕС имеют рекомендательный характер, то их следует вводить только приказами МЗ Украины. Так, например, в ЕС сначала было издано Руководство по GMP, параллельно были изданы две директивы, в которых указаны только принципы GMP. Поэтому вначале нам тоже необходимо принять Руководство по GMP и решить, каким нормативно-правовым актом сделать положения этого руководства обязательными. Вероятно, вначале ими могут стать Лицензионные условия, в которых надо указать основные принципы GMP и сделать ссылку на обязательность выполнения требований, изложенных в Руководстве по GMP. Так же, видимо, следует поступить в случае Руководства по GDP. Что касается руководств по GLP и GCP приказ об их введении в действие в качестве обязательных необходимо будет, видимо, только зарегистрировать в Министерстве юстиции Украины. Однако очевидно, что это будут только первые шаги для пересмотра Закона Украины «О лекарственных средствах», который необходимо приводить в соответствие с Директивой 2001/83/ЕС Европейского Парламента и Совета ЕС от 6 ноября 2001 г. «О своде законов Сообщества в отношении лекарственных препаратов для человека» с дополняющими ее директивами.
Чтобы эффективно решить основную задачу, необходимо разработать систему стандартизации фармацевтической продукции Министерства здравоохранения Украины и принять основополагающие стандарты, регламентирующие эту систему.
Приказом Министерства здравоохранения Украины от 14 сентября 2005 г. № 471 введен в действие и опубликован один из основополагающих стандартов СТ МОЗУ 42-1.0:2005 «Фармацевтическая продукция. Система стандартизации. Основные положения». Дополнительно необходимо издать приказ, который должен быть зарегистрирован в Министерстве юстиции Украины о том, что Минздрав является органом по стандартизации фармацевтической продукции, формирует систему и службу стандартизации, устанавливает в своих стандартах правила разработки, принятия и актуализации нормативных документов, ведет их учет и поддерживает информационную сеть. Кроме того, в 2008 г. необходимо разработать и принять СТ МОЗУ 42-1.2:2005 «Фармацевтическая продукция. Система стандартизации. Правила разработки и принятия нормативных документов МЗ Украины», а также запланировать разработку других основополагающих стандартов.
И, наконец, третья важная задача. Чтобы эффективно решать проблемы стандартизации фармацевтической продукции, в Украине должна быть создана соответствующая служба стандартизации с инфраструктурой, позволяющей на постоянной основе планировать, разрабатывать и вводить в действие нормативные документы, а также регулярно актуализировать их в соответствии с уровнем развития науки и техники и изменением международных норм. Эта инфраструктура должна включать Министерство здравоохранения Украины как орган по стандартизации фармацевтической продукции, Совет по стандартизации, технические комитеты и базовые организации, а также соответствующую информационную сеть. То есть, Минздрав на постоянной основе должен планировать, финансировать и поручать разработку нормативных документов техническим комитетам (базовым организациям) или назначать для этого группы разработчиков с руководителем (докладчиком) и соруководителем (содокладчиком), как это принято ЕМЕА и ICH.
Рассмотрим структуру комплекса нормативных документов, которые необходимы для перехода на работу по правилам GMP, GLP, GCP, GDP (рис. 2).
|
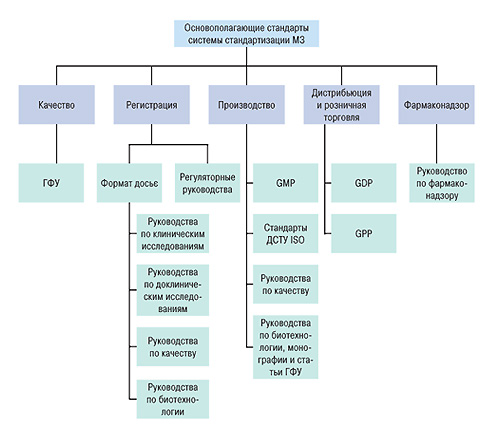
Первый блок составляют основополагающие стандарты, регламентирующие систему стандартизации Министерства здравоохранения Украины.
Следующие блоки нормативных документов фактически соответствуют элементам Системы сертификации лекарственных средств для международной торговли; это нормативные документы, регламентирующие качество, регистрацию, производство, дистрибуцию, розничную торговлю и фармаконадзор.
Чтобы приступить к обеспечению качества на этапе производства, необходимо установить критерии качества. Минимальные показатели качества для лекарственных форм, субстанций, сырья и материалов устанавливают на государственном уровне в Фармакопее. В связи с этим, прежде всего необходимо на постоянной основе разрабатывать и актуализировать фармакопейные статьи и монографии.
Что касается качества конкретных лекарственных средств, то оно устанавливается в регистрационном досье. Чтобы правильно разработать и исследовать препарат, а затем составить регистрационные документы, необходимы 5 блоков специализированных руководств: регуляторные руководства, руководства по качеству, по биотехнологии и биологическим препаратам, по доклиническим исследованиям и по клиническим испытаниям.
Качество лекарственных средств закладывается на этапе фармацевтической разработки, для которой должен быть установлен общий методологический подход и специальные подходы в отношении различных лекарственных форм, препаратов-генериков и оригинальных лекарственных средств. Качество, заложенное при фармацевтической разработке, проходит экспертизу на этапе регистрации и обеспечивается: при производстве лекарственных средств путем соблюдения правил GMP, при дистрибуции — путем соблюдения правил GDP, а при розничной реализации — правил GPP. Эффективность и безопасность разработанных лекарственных средств подтверждается на этапах доклинических и клинических исследований, а безопасность выведенных на рынок препаратов — опытом клинического применения. Объективность данных при этом обеспечивается посредством соблюдения правил GLP и GCP, а также руководства по фармаконадзору. Другими словами, если критерии качества не определены, а качество не заложено, то обеспечение качества и его исследование — это нонсенс.
Таким образом, прослеживается четкая связь и логическое взаимодействие между нормативными документами, регламентирующими критерии качества и контроль качества, требования к фармацевтической разработке и регистрационному досье, обеспечение качества при производстве, дистрибуции, розничной реализации, порядок проведения доклинических и клинических исследований, а также фармакологического надзора. Такая связь требует не просто введения 4 основополагающих руководств по GMP, GDP, GCP, GLP, а принятия и постоянной актуализации системы нормативных документов.

Система отраслевых нормативных документов необходима не только для того, чтобы выставить уровень качества и его обеспечения; она является основой для понимания между разработчиками и экспертами, между производителями и инспекторами, между исследователями и аудиторами.
Без системного подхода успеха быть не может. На этом пути в Украине есть определенные достижения:
- введена Государственная Фармакопея Украины и 2 приложения к ней;
- создана современная структура регистрационного досье (приказы Министерства здравоохранения Украины от 26.08.2005 г. № 426; от 1.03.2006 г. № 95);
- приняты 6 руководств по качеству и 2 клинических руководства (фармацевтическая разработка, спецификации, испытания стабильности, производство ГЛС, валидация процессов, вспомогательные вещества, биодоступность и биоэквивалентность, надлежащая клиническая практика (GCP));
- введены гармонизированные руководства по GMP и GDP.
В настоящее время необходимо актуализировать руководство по GMP и ввести ряд рекомендаций PIC/S; разработать и ввести в действие руководства по GLP и инспектированию на соответствие GLP, руководства по качеству на основании 30 руководств ЕС и ICH; 71 руководство и 140 статей и монографий Государственной Фармакопеи Украины по биологическим/биотехнологическим препаратам; 54 руководства по доклиническим испытаниям; 176 руководств по клиническим испытаниям; гармонизированное руководство по фармаконадзору; руководство по надлежащей аптечной практике (GPP), а также следующие приложения к национальной Фармакопее. Кроме того, Держспоживстандарт должен ввести 34 стандарта ДСТУ ISO по вопросам стерилизации и чистых помещений, на которые ссылается Руководство по GMP; в настоящее время в Украине введено только 11 таких стандартов.
До 1 января 2009 г. осталось около 8 мес. Хотелось бы пожелать руководству Министерства здравоохранения Украины и всей фармацевтической общественности справиться за этот срок с проведением указанных работ по стандартизации, однако весь комплекс этих работ вряд ли удастся сделать за этот период. Поэтому необходимо установить приоритеты и постараться разработать и ввести в действие:
- актуализированные руководства по GMP, GDP, GCP;
- руководства по GLP и инспектированию на соответствие GLP,
- максимально возможное количество руководств по качеству, необходимых для работы по правилам GMP и составлению раздела регистрационного досье, посвященного качеству;
- руководства по биотехнологии и фармакопейные монографии/статьи, на которые ссылается руководство по GMP;
- руководства по клиническим испытаниям, на которые ссылается руководство по GCP.
Эта работа должна быть поручена профессионалам как в научных вопросах, так и в вопросах стандартизации.
Хочется подчеркнуть, что внедрение надлежащих практик GMP, GDP, GCP, GLP имеет две стороны медали:
во-первых, государство должно установить нормы, регламенты и законы, постоянно их актуализируя. Кроме того, государство должно создать соответствующие инспекционные службы и подготовить компетентных инспекторов, экспертов, аудиторов. Министерству здравоохранения Украины необходимо также обеспечить вступление в соответствующие международные организации, например, Инспектората по GMP/GDP в PIC/S для развития международной торговли.
Во-вторых, производители, дистрибьюторы, исследовательские центры и клинические базы должны вложить финансовые средства, приложить усилия и выйти на уровень работы по правилам GMP, GDP, GCP, GLP. Для этого, как указано в постановлении, им должны быть созданы определенные преференции — меры, стимулирующие внедрение надлежащих практик. Такой мерой может быть, например, освобождение от налогов, в частности, от НДС по договорным работам на разработку новых препаратов или закупку оборудования.
В заключение хочется отметить связь науки с новыми стандартами. С введением гармонизированных нормативных документов возрастает наукоемкость фармацевтической продукции. При этом производство в соответствии с этими стандартами будет требовать от науки все более высокого уровня фармацевтической разработки и экспериментальных исследований. Это, в свою очередь, потребует инвестиций в науку. Надеемся, что научные учреждения проведут техническое переоснащение, а научные работники окажутся востребованными на рынке, уровень которого будет определяться гармонизированными стандартами. n
Н.А. Ляпунов, А.И. Гризодуб, Е.П. Безуглая (ГП «ГНЦЛС», ГП «НЭФЦ»)
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим