|
Татьяна Бухтиарова, доктор медицинских наук, первый заместитель директора ГФЦ МЗ Украины, осветила основные требования к регистрационному досье. Пакет документов, обеспечивающий работу системы регистрации, включает:
-
Закон Украины «О лекарственных средствах» (Раздел ІІ, ст. 9);
-
Постановление Кабинета Министров Украины от 26.05.2005 г. № 376;
-
Приказ Минздрава Украины от 26.08.2005 г. № 426 (с изменениями, внесенными Приказом МЗ Украины от 01.03.2006 г. № 95), в котором учтены требования:
-
Директив Европейского Парламента и Совета ЕС — 2001/83/ЕЕС с изменениями и дополнениями и 2004/24/ЕС;
-
Постановления Комиссии ЕС № 1085/2003 и другие европейские документы.
Правила регистрации лекарственных средств практически не изменились по сравнению с теми, которые были утверждены Приказом МЗ Украины от 19 сентября 2000 г. № 220, отметила Т. Бухтиарова. Регистрация производится на основании экспертизы материалов относительно лекарственного средства, которая осуществляется в нескольких организациях, причем базовым является ГФЦ МЗ Украины. Сведения о технологии производства проходят экспертизу в Государственной службе лекарственных средств и изделий медицинского назначения, хотя, как отметила Т. Бухтиарова, такое вычленение отдельных фрагментов досье нерационально. Регистрационные материалы (регистрационное досье) — комплект документов на лекарственное средство, экспертиза которых по объему и сути дает возможность сделать вывод об эффективности, безопасности и качестве лекарственного средства с целью рекомендации его к государственной регистрации (перерегистрации) или необходимости проведения дополнительной экспертизы лекарственного средства. Их следует оценивать комплексно, с учетом всех остальных частей. Продолжительность экспертизы составляет не более 210 календарных дней относительно препарата, который подается на государственную регистрацию с полным заявлением и не более 90 дней — другими типами заявлений. Относительно регистрационных материалов, которые сопровождают заявление, Т. Бухтиарова отметила, что следует указывать название, месторасположение и выполняемые функции всех производственных участков; внимательно относиться к выбору названия лекарственного препарата с целью исключить путаницу и потенциальные ошибки и прислушиваться к мнению номенклатурной комиссии ГФЦ МЗ. Основания для отказа присвоения того или иного торгового названия изложены в одном из выпусков журнала «Вестник фармакологии и фармации».
Особенно интересная ситуация возникает, подчеркнула Т. Бухтиарова, если был зарегистрирован препарат в форме in bulk, а отечественный или зарубежный производитель намерен наладить стадию расфасовывания. Заявитель обязательно должен обращать внимание на изменения фармакопейных статей, которые произошли с момента регистрации препарата in bulk, и вносить их в регистрационные документы препарата в дозированных лекарственных формах, иначе заявление будет отклонено. В соответствии с положениями Приказа МЗ Украины от 26.08.2005 № 426 (с изменениями), заявление о проведении экспертизы материалов относительно государственной регистрации лекарственного средства, а также изменений в регистрационные документы, согласно пожеланию заказчика, могут подаваться в так называемом старом европейском формате (согласно приложению 17 Приказа или приложению к Директиве 75/318/ЕЭС с изменениями и дополнениями). Наряду с ним, разрешается и приветствуется подача заявления в формате «Общего технического документа» (Common Technical Document — CTD), созданного в рамках Международной конференции по гармонизации технических требований к регистрации ЛС для человека, производства и продажи ЛС (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use — ICH). Этот формат, представленный в приложении 1 Приказа, практически не отличается от «старого европейского» набором необходимых документов, только структурирование регистрационных материалов другое.
Самый сложный вопрос, отметила Т. Бухтиарова, — соотношение типов заявлений с объемом регистрационных материалов. Приказом выделены следующие типы заявлений.
-
Полное и независимое/автономное заявление (то есть полное досье, содержащее административные, фармацевтические, доклинические и клинические данные) подается, если в состав препарата входит новая активная субстанция (компонент препарата не зарегистрирован в Украине); такое заявление может подаваться и на препарат, содержащий известную активную субстанцию — компонент препарата, зарегистрированного в Украине.
-
Заявление на фиксированную комбинацию — новый препарат, содержащий известные активные субстанции, которые раньше в таком сочетании не использовались с терапевтической целью; предоставляются полные административные, фармацевтические, доклинические и клинические данные только относительно комбинации. Может ли комбинация рассматриваться как по существу аналогичный лекарственный препарат? Да, может, отметила Т. Бухтиарова, если в Украине уже зарегистрирована аналогичная комбинация.
-
Библиографическое заявление — медицинское применение вещества или веществ, входящих в состав лекарственного препарата, хорошо изучено, признаны их эффективность и удовлетворительная степень безопасности; подаются полные административные и фармацевтические данные и подробная библиография относительно фармакологических и клинических характеристик. Если есть необходимость доказательства биоэквивалентности, должны быть приведены соответствующие доклинические или клинические данные.
-
Заявление информированного согласия — лекарственный препарат является по сути аналогичным уже зарегистрированному в Украине референтному лекарственному препарату, и заявитель последнего согласен с тем, чтобы при изучении заявления относительно аналогичного лекарственного препарата использовались данные фармакологических, токсикологических и/или клинических испытаний, которые содержатся в регистрационных материалах на референтный препарат. В ряде случаев необходимы подтверждения биоэквивалентности.
-
Так называемое заявление на генерик (по существу аналогичный лекарственный препарат); подаются полные административные и фармацевтические данные, соответствующие доклинические и клинические данные.
-
Заявление на препарат относительно так называемого оригинального препарата (например, разные лекарственные формы, разные терапевтические показания и др.); предоставляются полные административные и фармацевтические данные, соответствующие доклинические и клинические данные.
Таблица
Документы по проблемам регистрации лекарственных препаратов
|
По качеству |
По доклиническим |
По клиническим |
|
|
Отечественные |
«Лекарственные средства. Надлежащая производственная практика» (Руководство 42-01-2001) (Разработчики: Н.А. Ляпунов и др.— К., 2001) «Лекарственные средства. Технологический процесс. Документация» (Руководство 42-01-2003) (Разработчики: Н.А. Ляпунов и др.— К., 2003) и другие издания |
Доклинические исследования лекарственных средств (методические рекомендации, разработанные ГФЦ МЗ Украины) и другие издания |
«Руководство по клиническим испытаниям лекарственных средств» (Под ред. А. В. Стефанова и др.— К., 2000). «Лекарственные средства. Исследование биодоступности и биоэквивалентности» (Руководство 42-7.1:2005) (Разработчики: В.И. Мальцев и др.— К., 2005) и другие издания |
|
Европейские |
Приложение к Модулю 3 CTD 67 руководств по качеству: • 8 общих; • 11 — по активным субстанциям; • 26 — по лекарственным препаратам 22 руководства по биотехнологическим препаратам |
24 справочных руководства по доклиническим исследованиям: 2 — по фармакологии; 3 — по фармакокинетике; 19 — по токсикологии |
87 Руководств по клиническим исследованиям: 14 — общая эффективность; 8 — клиническая безопасность; 6 — клиническая фармакология; 2 — особые популяции; 52 — другие (по системам организма); 5 — информация, относящаяся к лекарствам. |
В последнее время, в связи с планируемым вступлением Украины во Всемирную торговую организацию (ВТО) ужесточаются требования к регуляторным органам относительно сохранения конфиденциальности информации, а также вводятся дополнительные меры по защите интеллектуальной собственности. В частности, предполагается предоставлять производителю оригинального препарата период маркетинговой эксклюзивности сроком на 5 лет. Эта норма принята практически во всех странах-членах ВТО. Не должна быть исключением и наша страна, поэтому соответствующий пакет документов уже готовится. В завершение доклада были перечислены наиболее важные отечественные и европейские руководства, методические рекомендации и другие документы, к которым следует обращаться при подготовке регистрационного досье.
О современных требованиях к доклиническим (фармокологическим и токсикологическим) исследованиям при регистрации лекарственных средств рассказала Валентина Коваленко, доктор биологических наук, заведующая отделом токсикологии Института фармакологии и токсикологии АМН Украины. Доклинические исследования лекарственного средства — программа, результатом выполнения которой является обеспечение максимальной безопасности использования препарата при проведении клинических испытаний и дальнейшего применения в медицинской практике. К ним относятся химические, физические, биологические, микробиологические, фармакологические, токсикологические и другие экспериментальные исследования, проводимые с целью определения эффективности и безопасности лекарственного средства. На сегодняшний день, отметила В. Коваленко, в Украине насчитывается свыше 30 баз доклинических исследований, расположенных практически во всех крупных городах Украины. Особенно В. Коваленко отметила следующее: «Исследования in vivo ни на одном из видов животных не позволяют предсказать клиническую эффективность или безопасность для людей. Наиболее успешным подходом к оценке безопасности и эффективности биологических агентов является рациональность, которая базируется на данных современной науки».
Татьяна Ефимцева, кандидат медицинских наук, заместитель начальника отдела координации и контроля клинических испытаний ГФЦ МЗ Украины подробно остановилась на этапах становления подходов к проведению клинических испытаний лекарственных средств, которые, оказывается, организовывали еще до нашей эры, а начиная с XVIII века проводят сравнительные испытания. Украинская законодательная база по организации и проведению клинических испытаний сегодня включает «Порядок проведения клинических испытаний лекарственных средств и экспертизы материалов клинических испытаний и типовое положение о комиссии по вопросам этики» (утверждены Приказом МЗ от 13.02.2006 г. № 66); «Руководства по клиническим исследованиям. Лекарственные средства. Надлежащая клиническая практика» (Руководство 42-7. 0:2005, утверждено приказом МЗ Украины от 22.07.2005 г. № 373); «Лекарственные средства. Исследование биодоступности и биоэквивалентности» (Руководство 42-7.1:2005). Порядок проведения клинических испытаний лекарственных средств разработан на основе целого ряда руководств, принятых в ЕС, поэтому с европейскими требованиями максимально гармонизированы заявления на государственную регистрацию, процедура информирования о начале и окончании клинического испытания, процедура предоставления информации о побочных реакциях, досье на исследуемый лекарственный препарат, а также требования по проведению инспекций клинических исследований. За последние 10 лет, отметила Т. Ефимцева, резко возросло количество проводимых в Украине международных многоцентровых исследований, увеличилась доля среди них испытаний II фазы, а также увеличилось количество клинических баз. Это доказывает доверие к украинским клиникам и исследователям, подчеркнула докладчик.
Одним из видов клинических испытаний, которые, естественно, должны соответствовать нормам надлежащей клинической практики (GCP), являются исследования биодоступности и биоэквивалентности, о действующих нормах по проведению которых рассказала Людмила Ковтун, кандидат медицинских наук, руководитель группы аттестации и инспекции клинических баз ГФЦ МЗ Украины. Биологическая неэквивалентность генерического препарата может стать причиной целого ряда неблагоприятных явлений:
-
неэффективность терапии и дальнейшее прогрессирование заболевания;
-
развитие побочных реакций;
-
увеличение расходов на лечение (как основного заболевания, так и осложнений);
-
психологический ущерб
-
для пациента (теряет надежду на выздоровление, недоверие к врачу и методу лечения)
-
для врача (неверно выбрана тактика, метод лечения, неуверенность в своих силах).
Факторы, влияющие на биодоступность препарата и повышающие риск бионеэквивалентности, следующие:
-
физико-химические свойства действующих веществ (степень дисперсности, полиморфизм);
-
вспомогательные вещества (состав покрытия таблеток и капсул);
-
технологический процесс (грануляция порошков);
-
условия хранения;
-
тип упаковки (стекло, пластмасса, бумага и др.).
Как подчеркнула Л. Ковтун, поэтапная гармонизация национальной законодательной и нормативной базы с европейскими требованиями обеспечивает качество, эффективность и безопасность лекарственных средств, а значит, открывает им путь на мировой рынок.
Исследование биодоступности и биоэквивалентности включает две составные части: клиническую, которая выполняется на клинических базах ГФЦ МЗ Украины, и фармакокинетическую (биоаналитическую), которую проводят в фармакокинетических лабораториях. В Украине в достаточной мере разработаны правила функционирования клинической базы изучения биоэквивалентности препаратов, отметила Виктория Либина, кандидат биологических наук, заведующая лабораторией экспериментальной фармакокинетики, биоэквивалентности и токсикогенетики ГНЦЛС. Но работе фармакокинетической лаборатории, в которой должен осуществляться химический анализ (биоаналитическое определение) испытуемого и референтного препаратов в биоматериале добровольцев, в документах уделено недостаточно внимания. Внедрение требований надлежащей лабораторной практики (Good Laboratory Practice — GLP) в их деятельность позволит значительно повысить уровень разработки и контроля качества генерических препаратов. Требования к валидации, которая является «критической точкой» каждого исследования, в отечественном руководстве по исследованиям биодоступности и биоэквивалентности, полностью гармонизированным с европейскими нормами, представлены в самом общем виде, тогда как в правилах, принятых FDA (21 Code of Federal Regulations — CFR, Part 58) — детально. Правила проведения валидации биоаналитических методик, принятые FDA, регламентируют полную (full), частичную (partial) и перекрестную (cross) валидацию.
Основными валидационными критериями биоаналитической методики являются:
-
стабильность (stability) — устойчивость параметров исходных растворов и тестируемого вещества в биоматериале;
-
специфичность (selectivity) — способность метода дифференцированно однозначно определять тестируемое вещество в присутствии других компонентов пробы;
-
чувствительность (sensitivity) — наименьшее количество тестируемого вещества, которое может быть обнаружено и количественно определено данным биоаналитическим методом;
-
правильность (accuracy) — характеризует степень соответствия между известным истинным значением концентрации тестируемого вещества в биопробе и значением, полученным с использованием этой биоаналитической методики;
-
точность (precision) — характеризует степень близости (плотности) результатов индивидуальных повторных измерений, выполненных данным биоаналитическим методом.
Важно отметить, что фармакопейные требования к валидации не могут быть в полной мере отнесены к биоаналитическим методикам, особенно в части критериев приемлемости, сказала В. Либина. Национальные требования по валидации биоаналитических методик должны быть строго гармонизированы с правилами Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) и принятыми в ЕС. В противном случае, материалы и отчеты по биоэквивалентности препаратов-генериков отечественных производителей не будут приниматься в других странах. Соблюдение всех требований GLP позволит обеспечить качество проведения биоаналитических исследований, а проведение внутренних и внешних инспекций — проконтролировать его. В целом, высокое качество исследований по оценке биоэквивалентности генерических препаратов может быть гарантировано только в том случае, если они выполнены с соблюдением требований GLP и с использованием валидированных биоаналитических методов, — подчеркнула В. Либина. В настоящее время усилиями ГФЦ МЗ Украины практически завершено создание двух лабораторий (в Киеве и Харькове) по определению биоэквивалентности, отвечающих требованиям GLP.
«Кто определяет необходимость исследования биоэквивалентности конкретного препарата и выбирает наиболее подходящий метод » — участников семинара очень волновал этот вопрос. Компания должна сама обеспечивать весь процесс разработки лекарственного препарата, подчеркнул Виктор Чумак, кандидат химических наук, директор ГФЦ МЗ Украины. Для этого она может использовать работу собственных сотрудников или привлекать специалистов извне, но ответственность так или иначе лежит на компании-разработчике. Она должна доказать, что достигла поставленной цели, а цель экспертизы, которую проводит регуляторный орган — проверить это.
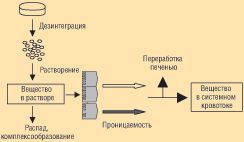
О принципах и критериях отнесения генерических препаратов к биовэйверам (регистрация на основании непрямых доказательств биоэквивалентности) рассказал Николай Головенко, доктор биологических наук, профессор, академик АМН Украины, заведующий отделом физико-химической фармакологии Физико-химического института НАН Украины. Использование альтернативных методов подтверждения биоэквивалентности, которые базируются на биофармацевтической классификационной системе (БКС), является важным шагом на пути внедрения современных методов определения безопасности и эффективности лекарственных генерических препаратов в нашей стране. БКС основана на двух показателях: растворимости лекарственных средств в воде и степени их проникновения через стенку кишечника (всасывание) (рис.). Этот процесс определяется следующими физико-химическими свойствами соединений:
|
-
липофильность;
-
степень ионизации;
-
растворимость;
-
молекулярная масса и объем;
-
химическая стабильность.
Считается, что после того как фармацевтически активный ингредиент всосался, его связь с препаратом (лекарственной формой) закончилась и дальнейшая судьба зависит от организма. Исходя из этого постулата, согласно рекомендациям ВОЗ [Рroposal to waive in vivo bioequivalence requirements for WHO model list of essential medicines immediate release, solid oral dosage forms working document qas/04.109. 11 Оctober 2004], исследование эквивалентности не является необходимым для ряда лекарственных форм. В некоторых случаях лекарственные средства (таблетки, капсулы) с немедленным высвобождением и системным действием могут иметь высокий уровень растворимости и вести себя аналогично водным растворам. Поэтому основным показателем для получения вэйвера является показатель растворимости. На основании показателя растворимости и степени проникновения лекарственную субстанцию относят к тому или другому классу БКС, что позволяет решить вопрос о необходимости проведения исследований биоэквивалентности и биодоступности in vivo:
Класс 1 — высокая растворимость, высокая степень проникновения;
Класс 2 — низкая растворимость, высокая степень проникновения;
Класс 3 — высокая растворимость, низкая степень проникновения;
Класс 4 — низкая растворимость, низкая степень проникновения.
Было доказано, что для лекарственных средств с немедленным высвобождением для перорального приема, которые относятся к классу 1, нет необходимости определять биоэквивалентность in vivo (на людях), так как оценка in vitro полностью ее заменяет. Для лекарственных средств класса 2 и 3 также можно отказаться от изучения биоэквивалентности при определенных показателях растворимости. Для лекарственных средств, относящихся к классу 4, изучение биоэквивалентности является обязательным. Таким образом, если профиль растворимости изучаемого генерика подобен референтному препарату, а в опытах in vitro показано отсутствие бионеэквивалентности, то согласно положениям ВОЗ и порядка регистрации генерических препаратов (приказ МЗ Украины от 26.08.05 г. № 426), изучение биоэквивалентности на людях не является необходимым, а заявка может получить одобрение на основе свидетельства исследования на растворимость и биодоступность в опытах in vitro. В случае возникновения сомнений, касающихся соответствующих предостережений, а не базовых показателей (растворимость, степень проникновения) целесообразно провести экспериментальные исследования специфической фармакологической активности и острой токсичности препарата согласно требованиям ГФЦ МЗ Украины. Изменения, которые предлагаются, будут иметь надлежащее воплощение только в том случае, когда будут выполнены все требования к фармацевтической разработке лечебного средства.
|
РЕЗЮМЕ ИЗ ПЕРВЫХ УСТ |
|||||
|

Дарья Полякова, фото автора
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим