 У цій постанові було визначено механізми проведення державної реєстрації медичної техніки та виробів медичного призначення та розмежування понять «медична техніка» та «вироби медичного призначення» в частині вартості оплати за реєстраційну процедуру.
У цій постанові було визначено механізми проведення державної реєстрації медичної техніки та виробів медичного призначення та розмежування понять «медична техніка» та «вироби медичного призначення» в частині вартості оплати за реєстраційну процедуру.
На виконання п. 25–27 Плану заходів щодо виконання у 2013 р. Загальнодержавної програми адаптації законодавства України до законодавства Європейського Союзу, затвердженого розпорядженням КМУ від 25 березня 2013 р. № 157-р, Урядом було прийнято Технічний регламент щодо медичних виробів, затверджений постановою КМУ від 02.10.2013 р. № 753, Технічний регламент щодо медичних виробів для діагностики in vitro, затверджений постановою КМУ від 02.10.2013 р. № 754, та Технічний регламент щодо активних медичних виробів, які імплантують, затверджений постановою КМУ від 02.10.2013 р. № 755 (далі — технічні регламенти), які розроблені на основі директив Ради ЄС від 14 червня 1993 р. № 93/42/ЕЕС, від 27 жовтня 1998 р. № 98/79/ЕЕС та від 20 червня 1990 р. № 90/385/ЕЕС.
Постанова КМУ № 1497 втратила чинність 01.07.2015 р. згідно з постановою КМУ № 753.
З 1 липня 2015 р. вимоги технічних регламентів є обов’язковими.
Відповідно до п. 21 зазначених постанов встановлено, що дія технічних регламентів не поширюється на медичні вироби, активні медичні вироби, які імплантують, та медичні виробів для діагностики in vitro, які пройшли державну реєстрацію, внесені до Державного реєстру медичної техніки та виробів медичного призначення і дозволені до застосування на території України і введення в обіг та/або експлуатацію без проходження процедур оцінки відповідності та маркування національним знаком відповідності:
- до 1 липня 2016 р. — для медичних виробів, строк дії свідоцтва про державну реєстрацію яких необмежений чи закінчується після 1 липня 2016 р.;
- до закінчення строку дії свідоцтва про державну реєстрацію — для медичних виробів, строк дії свідоцтва про державну реєстрацію яких закінчується до 1 липня 2016 р.
Такі медичні вироби дозволяються для реалізації і застосування на території України до закінчення строку їх придатності без проходження процедур оцінки відповідності та маркування національним знаком відповідності.
Наразі проект наказу МОЗ України «Про затвердження Порядку введення в обіг та/або експлуатацію окремих медичних виробів, стосовно яких не виконані вимоги технічних регламентів, але використання яких необхідне в інтересах охорони здоров’я та життя конкретної особи», знаходиться на погодженні в заінтересованих органах виконавчої влади.
Відповідно до технічних регламентів виробники та уповноважені представники мають дотримуватися процедури оцінки відповідності згідно із схемою, що додається (рисунок).
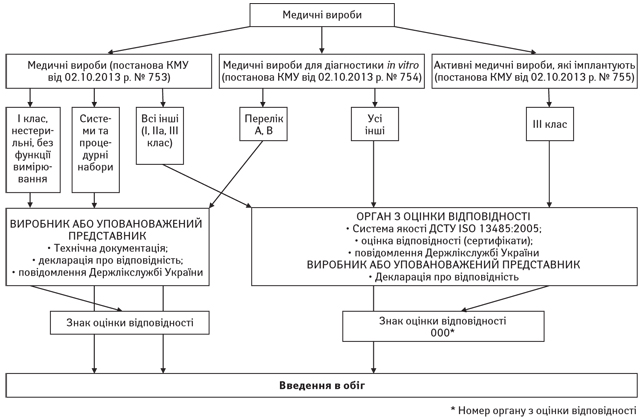
Виробники (уповноважені представники, юридичні особи або фізичні особи – підприємці) згідно з п. 31 Технічного регламенту щодо медичних виробів, п. 23 Технічного регламенту медичних виробів для діагностики in vitro та п. 33–35 Технічного регламенту щодо активних медичних виробів, які імплантують, повідомляють Держлікслужбі України з лікарських засобів (далі — Держлікслужба України) про своє місцезнаходження, перелік і опис відповідних виробів тощо.
У зв’язку з тим, що листом Міністерства юстиції України від 04.06.2015 р. № 15052-0-26-15/10.1 відмовлено в державній реєстрації наказу МОЗ України від 18.05.2015 р. № 282 «Про затвердження Порядку ведення Реєстру осіб, відповідальних за введення медичних виробів, активних медичних виробів, які імплантують, та медичних виробів для діагностики in vitro в обіг» (скасовано наказом МОЗ від 30.06.2015 р. № 399) Держлікслужбою України підготовлено відповідний проект наказу.
Процедури оцінки відповідності вимогам технічних регламентів здійснюють призначені органи з оцінки відповідності (частина 1 ст. 28 Закону України «Про технічні регламенти та процедури оцінки відповідності» (далі — Закон).
Згідно з частинами 8–9 ст. 28 Закону призначені органи заносяться до реєстру призначених органів з оцінки відповідності, який веде Міністерство економічного розвитку і торгівлі України.
Реєстр призначених органів з оцінки відповідності має бути доступним для громадськості в електронній та/або іншій формі та оприлюднюватися на офіційному веб-сайті центрального органу виконавчої влади, що реалізує державну політику у сфері оцінки відповідності.
Перелік призначених органів з оцінки відповідності, зокрема медичних виробів, вимогам технічних регламентів розміщено на сайті Міністерства економічного розвитку і торгівлі України (www.me.gov.ua) у розділі «Діяльність/Технічне регулювання/Оцінка (підтвердження) відповідності/Реєстр призначених органів з оцінки відповідності продукції вимогам технічних регламентів» або на сайті Держлікслужби України (www.diklz.gov.ua) в розділі «Медичні вироби/Технічні регламенти».
Обов’язки призначених органів з оцінки відповідності визначені ст. 29 Закону.
Відповідно до частини 4 ст. 29 Закону призначений орган з оцінки відповідності регулярно звітує про свою діяльність центральному органу виконавчої влади, який подав пропозицію про його призначення (Держлікслужба України), а також Міністерству економічного розвитку і торгівлі України.
Призначені органи з оцінки відповідності ведуть реєстр виданих сертифікатів відповідності та надають копію сертифіката згідно з установленою Міністерством економічного розвитку і торгівлі України процедурою до державного реєстру сертифікатів відповідності. Органи з оцінки відповідності несуть відповідальність за результати оцінки відповідності згідно із законодавством.
Слід зазначити, що з 2011 р. в Україні діє Закон України «Про державний ринковий нагляд і контроль нехарчової продукції», який встановлює правові та організаційні засади здійснення державного ринкового нагляду і контролю нехарчової продукції. Відповідно до постанови КМУ від 01.06.2011 р. № 573 «Про затвердження переліку органів державного ринкового нагляду та сфер їх відповідальності» Держлікслужбу України визначено органом державного ринкового нагляду в сферах медичних виробів, медичних виробів для діагностики in vitro та активних медичних виробів, які імплантують.
Відповідно до частини 1 ст. 4 Закону України «Про державний ринковий нагляд і контроль нехарчової продукції» метою здійснення ринкового нагляду є вжиття обмежувальних (корегувальних) заходів із відповідним інформуванням про це громадськості щодо продукції, яка при її використанні за призначенням або за обґрунтовано передбачуваних умов і при належному встановленні та технічному обслуговуванні становить загрозу суспільним інтересам чи яка в інший спосіб не відповідає встановленим вимогам.
Вжиття заходів ринкового нагляду, порядок та особливості проведення перевірок характеристики продукції у її виробників та розповсюджувачів тощо визначено у розділі IV Закону України «Про державний ринковий нагляд і контроль нехарчової продукції».
Додатково повідомляється, що листом Державної фіскальної служби України від 02.07.2015 р. № 23741/7/99-99-24-03-01-17 митницям надіслано роз’яснення щодо митного оформлення імпорту товарів, які підпадають під дію технічних регламентів.
Коментарі