|
Официальное определение протокола КИ можно найти в Руководстве по надлежащей клинической практике (п. 1.44).
|

Протокол КИ — это «документ, описывающий цели, дизайн, методологию, статистические аспекты и организацию КИ. В нем обычно представлены ранее полученные данные об исследуемом препарате и обоснование исследования, однако эта информация может содержаться и в других документах, на которые ссылается протокол» [4, 5].
Успех всего проекта во многом зависит от того, насколько правильно и профессионально был разработан этот документ и насколько точно ему следовали все участники КИ. Планирование КИ и разработка протокола являются обязанностью спонсора исследования (п. 5.4 ICH GCP) [4, 5]. Основная же обязанность исследователя — это соблюдение протокола, утвержденного спонсором и одобренного ЭК и регуляторным органом. Связанным с этим вопросам посвящен раздел 4.5 Руководства по надлежащей клинической практике [4, 5]:
4.5.1 Исследователь/медицинское учреждение должны проводить исследование в соответствии с протоколом, согласованным со спонсором и при необходимости с регуляторными уполномоченными органами и одобренным ЭК. Исследователь/медицинское учреждение и спонсор в подтверждение достигнутой договоренности подписывают протокол или отдельный договор.
|

4.5.2 Исследователь не должен допускать никаких отклонений от протокола или внесения в него изменения без согласия спонсора и без предварительного рассмотрения и документально оформленного одобрения/положительного решения ЭК относительно поправки, кроме случаев, когда необходимо устранить непосредственную опасность, угрожающую субъектам испытания, или когда изменения затрагивают только вопросы материально-технического обеспечения (логистики) или административные аспекты исследования (например замена монитора(-ов), изменение номера(-ов) телефона).
4.5.3 Исследователь или назначенное им лицо должны регистрировать и объяснять любое отклонение от утвержденного протокола.
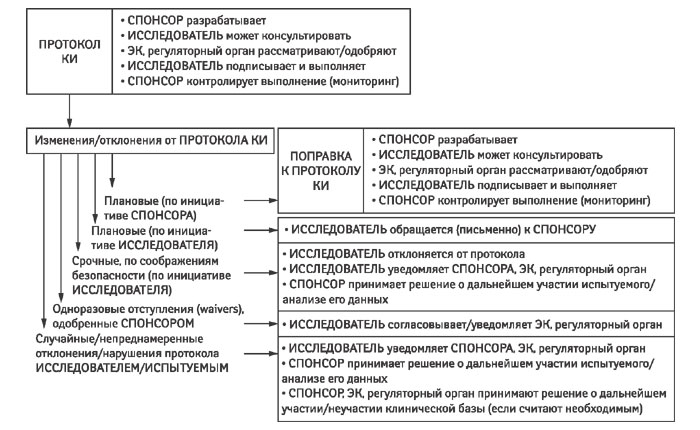
4.5.4 Исследователь может отклониться от протокола или внести в него изменения для устранения непосредственной опасности, угрожающей субъектам испытания (immediate hazard), без предварительного одобрения/положительного решения ЭК. В кратчайшие сроки описание допущенного отклонения или изменения, их причина и при необходимости предлагаемая(-ые) поправка(-и) к протоколу должны быть представлены (рисунок):
a) ЭК для рассмотрения и одобрения/положительного решения;
b) спонсору для согласования;
c) регуляторному(-ым) уполномоченному(-ым) органу(-ам), если требуется.
|
Пункт 4.5.1
До начала каждого КИ представители спонсора просят исследователя подписать протокол рядом с подписью представителей спонсора на так называемой подписной странице. Этим он подтверждает, что ознакомился с представленным протоколом и согласен следовать ему при проведении КИ. Дополнительно обязательство следовать утвержденному протоколу КИ (наравне с требованиями Руководства по надлежащей клинической практике, национальному законодательству и т.д.) содержится также и в контракте, который заключается между спонсором (или его представителем) и ответственным исследователем (и/или медицинским учреждением, в котором он работает). Кроме этих двух документов, исследователя часто просят подписать форму Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) FDA 1572 (для исследований, результаты которых будут подаваться в управление), в которой также прописано обязательство исследователя строго следовать протоколу. Безусловно, это требует от ответственного исследователя, а также его сотрудников и помощников хорошего знания содержания протокола.
Пункт 4.5.2
По причинам, оговоренным выше (этические нормы и обеспечение качества), исследователь и сотрудники исследовательского центра должны строго следовать протоколу, утвержденному спонсором, одобренному регуляторным органом и согласованному с ЭК, и не имеют права самостоятельно изменять процедуры исследования (за исключением случаев необходимости устранения непосредственной опасности для жизни и здоровья испытуемых — см. п. 4.5.4).
Важно!
Испытуемые, в отношении которых не выполняются требования протокола (допускаются отклонения от него), по сути, участвуют в КИ без разрешения регуляторных органов и одобрения ЭК [6].
Однако в ходе проведения КИ в силу объективных обстоятельств может возникнуть необходимость внести какие-либо изменения в уже утвержденный протокол (изменить какой-либо критерий оценки, режим приема препарата или дозу, ввести новый диагностический тест или внести дополнительные вопросы в анкету для испытуемых и др.). Инициатором изменений может выступать спонсор КИ. В таком случае разрабатывается отдельный документ, описывающий и обосновывающий изменения/дополнения к протоколу — поправка к протоколу КИ (Protocol Amendment).
Каждая новая поправка к протоколу должна быть подписана спонсором и ответственными исследователями, а если она содержит существенные изменения (в соответствии с локальными регуляторными определениями), подвергнуться отдельному рассмотрению ЭК и если необходимо — регуляторным органом (с оформлением благоприятного мнения/разрешения в письменном виде). С содержанием поправки к протоколу должны быть ознакомлены все участники исследовательской команды. Если внесенные изменения затрагивают интересы испытуемых, то последние должны быть проинформированы об этих изменениях путем переподписания обновленной формы информированного согласия, также предварительно одобренной ЭК (подробнее о процедуре — см. в главе, посвященной информированным согласиям).
Исследователи могут вносить изменения в одобренный протокол КИ по двум сценариям:
1) в случае экстренной ситуации для устранения непосредственной опасности испытуемому(-ым) — этот вариант рассмотрен в п. 4.5.4 и в главе, посвященной медицинской помощи субъектам исследования;
2) в плановом порядке путем внесения своих предложений спонсору в письменном виде? (в таком случае за спонсором остается решение о внесении/невнесении предложенных изменений) и в случае положительного решения они оформляются в виде официальной поправки к протоколу, требующей отдельного рассмотрения регуляторным органом и ЭК (до получения одобрения/положительного мнения которых реализовать изменения исследователь не имеет права).
Отдельно рассматриваются непреднамеренные «случайные» отклонения и нарушения протокола (см. п. 4.5.3) и варианты отступления от протокола, одобренные спонсором (так называемые разрешенные отступления, waivers) — см. ниже.
Вопрос-ответ
1. Существует ли формальное определение существенной поправки, требующей дополнительного рассмотрения и одобрения ЭК и регуляторным органом?
Пунктом 4.5.2 ICH GCP оговаривается, что любая поправка к протоколу КИ может внедряться только после одобрения ЭК и, если требуется, регуляторного органа. Однако одним из исключений является незначимость поправки для хода исследования («когда изменения затрагивают только вопросы материально-технического обеспечения (логистики) или административные аспекты исследования»). В последнем случае говорят о том, что поправка несущественна. При анализе положений ICH GCP всегда необходимо помнить о том, что они являются только рамочными требованиями, которые внедряются в практику на основании существующей национальной нормативно-правовой базы. Так, например, в ст. 10 (а) европейской директивы по КИ [7] указано, что существенной является поправка, влияющая на безопасность испытуемых или изменяющая интерпретацию научных документов, поддерживающих проведение исследования, или являющаяся существенной по какому-либо другому параметру. В Украине перечень аспектов КИ, относительно которых спонсор может внести существенные поправки, приведен в Порядке проведения клинических исследований лекарственных средств [8].
2. Как оцениваются разрешенные отступления (waivers) с точки зрения необходимости придеживаться протокола? На что должен обращать внимание исследователь?
Так называемые разрешенные отступления (waivers) являются, по сути, выданными в каждом отдельном случае разрешениями спонсора на отклонения от утвержденного протокола. Один из наиболее серьезных случаев — включение в исследование пациентов, не соответствующих одному или нескольким критериям включения/исключения (например, уровень гемоглобина пациента 68,5 г/л, а протоколом разрешено включение пациентов с таковым более 70 г/л и т.п.).
Независимо от обоснования такого решения исследователем и/или спонсором (включая этические или социальные соображения, заботу о благополучии пациента, который иначе не сможет получить дорогостоящую (пусть даже экспериментальную) терапию и т.п.), всегда необходимо помнить, что такое действие является преднамеренным нарушением одобренного регуляторным органом и согласованного с ЭК протокола (если только такая возможность не прописана в самом одобренном протоколе), а следовательно, вмешивается в систему защиты прав и благополучия испытуемых. Исследователь обязан относиться к таким ситуациям со всей серьезностью и ответственностью. Возвращаясь к п. 4.5.2 ICH GCP, видно, что исследователь имеет право отступать от одобренного протокола только после одобрения спонсора (это требование выполнено в случае разрешенного отступления), а также согласования с ЭК (и регуляторным органом, где это необходимо).
Таким образом, представляется оправданным (с точки зрения надлежащей клинической практики и этических принципов) согласовывать каждый случай разрешенного отступления не только со спонсором, но и с ответственным ЭК (и регуляторным органом) до включения испытуемого или в крайнем случае сообщать ответственному ЭК о таких отклонениях от протокола. Ответственным исследователям каждой клинической базы рекомендуется согласовать с ответственным ЭК тактику поведения в случае возникновения разрешенных отступлений до начала КИ (в письменном виде).
Кроме того, одним из важных факторов защиты испытуемых является страховое покрытие. Поэтому исследователю перед включением такого пациента в исследование рекомендуется перечитать страховой договор, чтобы убедиться, что пациент, не строго соответствующий критериям, все же будет покрываться страховкой (об этом нужно помнить и спонсору, оговаривая договор со страховой компанией).
Пункт 4.5.3
При проведении исследования могут возникнуть ситуации, когда протокол все же нарушается. Условно нарушения утвержденного протокола принято делить на отклонения от протокола (protocol deviation) и собственно нарушения протокола (protocol violation), предполагая, что только во втором случае ставится под угрозу целостность полученных данных. Официального определения этим понятиям нет ни в ICH GCP, ни в европейских, американских или украинских нормативно-правовых актах, хотя документы ICH (E6 и E3) [4, 9] оперируют термином «protocol deviation», разделяя отклонения на важные и неважные («все важные отклонения от протокола, связанные с критериями включения/исключения, проведением исследования, ведением испытуемых или оценкой их состояния, должны быть описаны» [в отчете о КИ]). Различными авторами и рабочими группами предлагались варианты трактовки этих двух терминов, приведем только некоторые из них. Так, Norman Goldfarb в «Journal of Clinical Research Best Practices» предложил следующие определения: «Отклонение от протокола (protocol deviation): отклонение от протокола возникает тогда, когда без существенных последствий процедуры исследования отклоняются от протокола, одобренного ЭК, например, пациент пришел на запланированный визит с некоторым опозданием*. Не так серьезно, как нарушение протокола.
Нарушение протокола (protocol violation) — расхождение с протоколом, которое:
а) существенно снижает качество и полноту данных,
б) приводит к неточности формы информированного согласия или
в) влияет на безопасность, права или благополучие испытуемых.
Примерами нарушения протокола могут служить: неадекватное проведение/документирование процедуры информированного согласия, нарушение критериев включения/исключения, несообщение о серьезных побочных явлениях, неправомочное расслепление, использование запрещенных препаратов, неправильное выполнение или пропуск анализов, неправильное обращение с биообразцами, большое количество визитов, пропущенных или проведенных несвоевременно, неадекватное хранение документов, намеренное отступление от протокола, GCP или регуляторных требований персоналом клинической базы, повторяемое нарушение режима лечения/обследования, предусмотренного протоколом».
Рабочая группа по аудиту (Audit Working Party) Европейского форума надлежащей клинической практики (European Forum for Good Clinical Practice) в 2001 г. предложила более короткие определения: «Нарушение протокола (protocol viola-
tion): серьезное отступление — может привести к признанию пациентов непригодными к анализу и/или выбыванию их из исследования. Отклонение от протокола (protocol deviation): менее серьезное отступление — не может поставить под угрозу пригодность данных пациента» [10]. В отсутствие официально принятых определений существенную помощь могут оказать стандартные операционные процедуры спонсора/контрактной исследовательской организации и/или ЭК, а также полученные исследователем письменные инструкции.
Любое отступление от утвержденного протокола (будь то отклонение или нарушение протокола), допущенное исследователем в ходе КИ, должно регистрироваться в первичной документации пациентов, при необходимости — в других материалах исследования с указанием причин. Ни при каких обстоятельствах исследователь или сотрудники исследовательского центра не должны скрывать допущенную ошибку, так как в таком случае даже случайное незначительное отклонение от протокола, выявленное при рутинном мониторинге, аудите или инспекции, может быть расценено как намеренное нарушение (protocol violation) с соответствующими последствиями (в некоторых случаях вплоть до дисквалификации исследователя). Количество и характер отклонений или нарушений протокола, допущенных в ходе исследования на каждой клинической базе, являются одной из ключевых характеристик качества работы исследователя и исследовательского центра в целом. В случае серьезных нарушений протокола исследователем спонсор вправе отстранить его от дальнейшей работы и не компенсировать затраты, так как несоблюдение протокола может привести к получению недостоверных данных и быть причиной отказа в регистрации или перерегистрации препарата.
Вопрос-ответ
3. Следует ли уведомлять об отклонениях от протокола, не вызванных необходимостью устранения непосредственной опасности, угрожающей субъектам испытания (то есть те случаи, причиной которых чаще всего являются ошибки исследователя)?
Хотя в ICH GCP и не оговариваются такие случаи, но представляется логичной и соответствующей принципу главенства заботы о правах, безопасности и здоровье пациента (п. 4.5.2 ICH GCP) необходимость срочного уведомления:
a) ЭК;
б) спонсора;
в) регуляторного(-ых) органа(-ов), если требуется.
В случае, если исследователь задерживает/отказывается сделать такое уведомление, монитор, выявивший данное нарушение (или представители контрактной исследовательской организации/спонсора, уведомленные о нарушении), вправе уведомить ЭК и регуляторные органы самостоятельно.
4. Как часто нарушения протокола являются находкой при инспекционных проверках клинических баз представителями регуляторных органов?
Согласно официальной статистике FDA, нарушения протокола являются наиболее частой находкой при регуляторных инспекциях — так, в 2006 г. на 35% всех клинических баз, проинспектированных сотрудниками Центра по оценке и исследованиям лекарственных средств (Center for Drug Evaluation and Research) FDA, были отмечены существенные нарушения протокола [10].
Пункт 4.5.4
Защита прав и здоровья испытуемых при проведении КИ — основная задача исследователя. Этические нормы проведения КИ являются главенствующими и преобладают над научными целями КИ. В ходе КИ может возникнуть ситуация, когда исследователь обязан отступить от утвержденного протокола. Это может произойти при возникновении угрозы для жизни и здоровья субъекта испытания. При наличии серьезных причин (состояние, угрожающее жизни и безопасности) и определенных условий (необходимость принять решение незамедлительно, отсутствие возможности связаться с представителями спонсора для обсуждения) врач-исследователь может принять самостоятельное решение об отклонении от протокола — например, о назначении препаратов, запрещенных протоколом (исключая случаи запрещения препаратов в связи с известным взаимодействием, которое само по себе может угрожать безопасности испытуемого), об отмене исследуемого лекарственного средства и т.д. Такая тактика исследователя в отношении пациента должна быть обоснована, надлежащим образом задокументирована в первичной документации, информация об отклонении от протокола незамедлительно предоставлена спонсору (монитору и/или медицинскому монитору), ЭК, а также регуляторным органам, если этого требует законодательство страны.
Хотелось бы еше раз отметить, что основными задачами ICH GCP являются обеспечение защиты прав, здоровья и благополучия испытуемых, а также получение достоверных результатов при проведении КИ. Ответственность за реализацию этих задач в значительной степени ложится на исследователя (и других членов исследовательской команды). На каждом этапе проведения КИ протокол со всеми поправками к нему является тем основным документом, к которому постоянно обращаются все участники проекта (спонсор, исследователи, инспекторы, аудиторы, мониторы). На исследователя же ложится основная ответственность по обеспечению соблюдения протокола при проведении исследования на его клинической базе.
Правильное составление протокола КИ спонсором, внимательное его изучение исследователем и членами исследовательского центра до начала КИ, четкое соблюдение протокола во время проведения исследования позволяют получать объективные и достоверные результаты, которые в последующем помогут новому перспективному препарату выйти на фармацевтический рынок и, что еще более важно, защитят потенциальных пациентов от недостаточно эффективных или даже опасных лекарственных средств (ЛС) (или же медицинских приборов, методов лечения и профилактики).
Вопрос-ответ
5. Существуют ли унифицированные требования к содержанию протокола КИ?
В соответствии с требованиями раздела 6 ICH GCP [12, 13] в протоколе должны найти отражение все вопросы, приведенные ниже. В дополнение к перечисленным документам руководство ICH GCP предлагает обращаться еще к одному руководству, разработанному Международной конференцией по гармонизации и посвященному структуре и содержанию отчетов о КИ (Е3) [14].
Общая информация (General Information)
В разделе указываются: название протокола (с обозначением дизайна, фазы, названия и лекарственной формы исследуемого препарата и референтных препаратов, предполагаемого контингента испытуемых), идентификационный номер протокола, дата одобрения спонсором, название/имя и адрес спонсора (фирмы, организации, учреждения), имя, должность и реквизиты лица, уполномоченного спонсором подписывать протокол и поправки к нему, название и реквизиты координирующего центра, медицинского эксперта, контрактной исследовательской организации (в случае привлечения спонсором для проведения испытания). Также в данном разделе могут приводиться имена, должности и реквизиты исследователей, отвечающих за проведение КИ, реквизиты клинических центров (лечебно-профилактических учреждений), клинических лабораторий, других медицинских и технических служб или организаций, вовлеченных в исследование.
Обоснование испытания, исходная информация (Background Information)
В разделе обосновывается целесообразность проведения КИ, приводятся научные сведения по изучаемой проблеме, данные предшествующих исследований, описывается исследуемое ЛС/прибор/метод лечения или профилактики с обоснованием дозы, лекарственной формы и схемы лечения, а также исследуемая популяция. Представляются данные о возможном риске и предполагаемой пользе применения исследуемого ЛС для испытуемых. Приводится указание на то, что испытание будет проводиться в соответствии с протоколом, GCP и нормативными требованиями.
Цели и задачи испытания (Trial Objective and Purpose)
В данном разделе четко описываются основные и второстепенные цели и задачи КИ, которые зависят от его фазы.
Дизайн исследования (Trial Design)
В разделе описывается дизайн КИ (например, двойное слепое плацебо-контролируемое параллельное), как правило, приводится графическая схема, включающая основные процедуры и этапы исследования и их продолжительность, указываются основные параметры, изучаемые в ходе КИ, первичные и вторичные конечные точки/задачи, а также процедура и метод рандомизации, методика ослепления. Указывается планируемая продолжительность участия испытуемых включая период последующего наблюдения (Follow-up Period). Этот раздел включает также характеристику проводимого лечения, указание лекарственной формы, упаковки и маркировки исследуемых ЛС, процедуры учета исследуемых ЛС, хранение рандомизационных кодов с описанием процедуры их раскрытия. Описываются также условия остановки КИ (в целом или его части) и исключения субъектов из исследования. Приводится перечень данных, регистрируемых непосредственно в индивидуальной регистрационной форме и определение первичной документации. Данная информация чрезвычайно важна для планирования разработки препарата, так как научное обоснование испытания и достоверность результатов во многом зависят от дизайна КИ.
Отбор испытуемых и выведение их из исследования (Selection and Withdrawal of Subjects)
В разделе определяются критерии включения и критерии исключения (inclusion criteria and exclusion criteria) субъектов, также описываются условия их досрочного выхода из КИ и возможность/необходимость включения новых субъектов взамен досрочно выбывших, а также процедуры наблюдения субъектов после их выхода из исследования, если такое наблюдение предусмотрено. Обязательным условием включения субъектов исследования в КИ, независимо от его дизайна, является получение от каждого из них (или их законных представителей) согласия на участие в испытании в виде подписанной и датированной формы информированного согласия.
Лечение испытуемых (Treatment of Subjects)
В данном разделе описывается применение исследуемых ЛС/медицинских приборов/методов лечения или профилактических мероприятий, проводимое в рамках испытания. При этом дается характеристика всех продуктов (ЛС/приборов/методов) с указанием дозировки, формы выпуска и способа введения, частоты и длительности приема для каждой группы испытуемых, а также методы контроля соблюдения процедур субъектами исследования (например контроль ведения дневников, анкет, мониторинг эффективности лечения). Представляется перечень ЛС и способов лечения, применение которых разрешено или запрещено до или во время КИ.
Оценка эффективности
(Assessment of Efficacy)
В данном разделе приводится перечень изучаемых параметров эффективности исследуемого ЛС/прибора или мероприятия, методы и сроки их оценки, регистрации и анализа.
Оценка безопасности (Assessment of Safety)
Описываются параметры безопасности, методы и сроки их регистрации, оценки и статистического анализа, порядок сообщения о побочных явлениях/реакциях, интеркуррентных заболеваниях, сроки и условия наблюдения за субъектами исследования, у которых зарегистрированы побочные явления/реакции.
Статистика (Statistics)
Описываются методы, используемые для проведения статистического анализа и отмечены сроки промежуточного анализа. Также указываются планируемое количество испытуемых всего и в каждом центре (при проведении многоцентровых испытаний), обоснование размеров выборки, применяемый уровень значимости, критерии прекращения испытания, принципы и процедуры учета недостающих, не подлежащих анализу и сомнительных данных. Раздел содержит также статистические критерии остановки КИ. Все изменения и нарушения первоначального плана статистической обработки должны быть описаны и обоснованы в поправках к протоколу и/или окончательном отчете об исследовании.
Прямой доступ к первичным данным/документации (Direct Access to Souse Data/Documents)
В данном разделе подчеркивается принцип проведения КИ, согласно которому обязанностью врача-исследователя является предоставление прямого доступа к первичной документации (исходным медицинским данным, медицинским документам пациента) при проведении мониторинга, аудита, инспекции, этической экспертизы.
Контроль качества и обеспечение (гарантия) качества (Quality Control and Quality Assurance)
Описывается процедура обеспечения и контроля качества проведения КИ (мониторинг, аудит, инспекция, этическая экспертиза).
Этика (Ethics)
В данном разделе описываются этические аспекты проведения КИ, в основе которых лежит принцип приоритетности защиты прав субъектов исследования.
Обращение с данными и хранение записей (Data Handling and Record Keeping)
В разделе описываются правила обращения с данными, возможность доступа к ним определенных лиц, порядок и сроки хранения документов КИ после его завершения, соблюдение конфиденциальности информации о субъекте исследования и полученных данных.
Финансирование и страхование (Financing and Insurance)
В данном разделе прописывается процедура финансирования и страхования участников КИ, если это не описано в отдельных договорах.
Публикации (Publication Policy)
В данном разделе описывается возможность публикаций результатов КИ его участниками/исполнителями и права на полученные данные.
Приложения (Supplements or Appendices)
В приложениях, как правило, размещают дополнительную информацию: различные инструкции (например, инструкция для пациента по заполнению дневника), правила обращения с исследуемым препаратом, описание методов КИ, различные документы исследования (информацию для пациента и форму информированного согласия), нормативно-правовые документы.
Приведенная выше структура типичного протокола КИ используется наиболее часто при их составлении. Однако в каждом конкретном случае объем, структура и содержание протокола могут значительно изменяться, что зависит от дизайна, фазы исследования и его особенностей.
Вопрос-ответ
6. Какими средствами пользуется спонсор КИ для проверки соблюдения протокола на каждой клинической базе (в соответствии с п. 5.1.1 ICH GCP)?
В ходе исследования спонсор постоянно контролирует различные аспекты качества проведения КИ, в том числе соблюдение протокола исследователями. В систему контроля и обеспечения качества входит мониторинг и аудит. Монитор является связующим звеном между исследователем и спонсором [15], в ходе исследования он с определенной периодичностью посещает клиническую базу, являясь одновременно и контролирующим звеном, и помощником исследователя. Во время своих визитов монитор должен убедиться в том, что исследователь и все сотрудники центра действуют в соответствии с утвержденным протоколом и поправками к нему. В случае обнаружения нарушений протокола монитор указывает на них и обеспечивает принятие соответствующих мер во избежание повторения подобных нарушений. Проблемы и вопросы, относящиеся к проводимому КИ, исследователь может обсудить с монитором или через монитора переадресовать их спонсору. Аудит является отдельной процедурой обеспечения качества проведения КИ, которая не связана с текущим мониторингом. После проведенного аудита исследователь и монитор получают отчет, в котором описаны проблемы исследования, находки аудитора (findings), рекомендации по их устранению и предупреждению. Исследователь в ответ должен предложить возможные пути решения или устранения проблем и календарный план-график своих действий.
Как показывает практика, перед проведением инспекции или аудита (особенно в случаях, когда эти проверки отсрочены во времени и происходят через несколько месяцев или лет после окончания самого исследования) весьма полезно еще раз внимательно прочитать протокол, чтобы четко ориентироваться в методической части испытания и его особенностях, что значительно сокращает неэффективные затраты времени при проведении инспекции/аудита.
7. Что необходимо учитывать ответственному исследователю для обеспечения соблюдения протокола КИ на своей базе?
С практической точки зрения для исследователя в любом КИ целесообразно выделять периоды планирования, выполнения и оценки полученных результатов. Соответственно такой периодизации планируется и работа исследовательской команды. Ниже приведены рекомендации, которые могут оказаться полезными для работы с протоколом КИ на каждом этапе исследования.
До начала исследования: первое знакомство потенциального исследователя с протоколом предстоящего проекта проводится с целью принятия им решения об участии в КИ и для оценки выполнимости всех пунктов протокола в исследовательском центре. На данном этапе можно объективно оценить возможности проведения КИ в данном центре, задать вопросы спонсору, обсудить неясные/спорные места проекта, выработать общую точку зрения по определенным аспектам медицинского и организационного характера (так как иногда видение и решение одной и той же медицинской проблемы различается между странами). Обсуждение и решение всех спорных вопросов до начала КИ на этапе создания финальной версии протокола очень важны для всех участников проекта. Как правило, спонсор предоставляет потенциальному исследователю протокол на языке его создания (для международных исследований — английском), в некоторых случаях доступен и перевод протокола на русский или украинский языки. Однако руководствоваться всегда лучше оригинальной, английской версией, поскольку в процессе перевода (даже при условии его последующего выверения) возможно возникновение неточностей и ошибок, поэтому так важно знание английского языка для ответственного исследователя и членов его команды.
При ознакомлении с протоколом до начала КИ ответственный исследователь должен уделить особое внимание критериям включения/исключения, оценить распространенность заболевания, его сезонность, доступность целевой группы пациентов, а также постараться спрогнозировать интерес и желание потенциальных испытуемых принять участие в данном исследовании (с учетом соотношения риска/пользы, количества инвазивных процедур, частоты визитов, длительности последующего наблюдения) [16]. Отдельное внимание нужно уделять перечню необходимых диагностических процедур, особенно требующим наличия специальной аппаратуры или выполнения специальных методик (например необходимость проведения компьютерной, магнитно-резонансной томографии, необходимость получения и подготовки для отправки в центральную лабораторию образцов биоматериалов, требующих наличия в исследовательском центре центрифуги и холодильника или морозильной установки, необходимость выполнения лабораторных тестов, которые не выполняются локальной лабораторией рутинно или выполняются с использованием других методик). Кроме того, необходимо изучить условия хранения исследуемого ЛС (например определенный температурный режим, требующий наличия холодильника большого объема или кондиционированного помещения при необходимости соблюдать верхнюю границу температуры 20 С).
Оценка выполнимости, планирование и подготовка к проведению КИ в исследовательском центре являются одним из самых ответственных этапов, от которого во многом зависит конечный результат работы всей команды. Если в силу обстоятельств какой-то из пунктов протокола невыполним в исследовательском центре, следует отказаться от участия в испытании, даже в случае, если исследователь уже дал предварительное согласие. Таким образом демонстрируется профессионализм и ответственность руководителя центра.
В ходе исследования протокол является главным руководством по проведению КИ, что позволяет гармонизировать работу различных людей не только в пределах одной исследовательской команды, но и в разных клиниках, городах, странах (в случае проведения многоцентровых международных КИ), а также функциональных подразделениях (собственно клинических базах, в отделе обработки данных, мониторинга, обеспечения качества и т.д.). Все документы КИ, включая протокол, исследователь должен хранить надлежащим образом в специальном файле исследователя. Если в процессе КИ протокол обновляется (вводятся поправки к нему), в файле исследователя хранятся, как правило, все версии протокола, но на устаревших версиях необходимо сделать соответствующую отметку, чтобы предотвратить ошибочное обращение к недействительному документу. Для каждодневной работы удобно сделать копию фрагмента действующего протокола, например, схему КИ с перечнем процедур, которые должны быть выполнены на каждом визите, или критериями включения/исключения и хранить ее в ближайшей доступности (в кармане врачебного халата, в ящике рабочего стола и т.д.).
После завершения исследования протокол и другие документы КИ должны быть правильно и надежно заархивированы. При архивации документов КИ ответственный исследователь должен убедиться в наличии в исследовательском файле всех версий протокола и поправок. Архивировать документы КИ следует согласно требованиям протокола или другим письменным процедурам, предоставленным спонсором. Целесообразно проверить условия хранения первичной документации в архиве больницы или поликлиники, а также самостоятельно подготовить опись всех документов и выяснить место их хранения в архиве (поскольку к данным документам, возможно, еще предстоит обращаться в случае инспекции или аудита). Залогом качественного архивирования является поддержание исследовательского файла в рабочем порядке со своевременным файлированием всех необходимых документов в соответствующие разделы на протяжении всего времени КИ. n
Ирина Борзенко, Елена Руднева
Авторы выражают благодарность Константину Волковинскому за помощь в подготовке публикации
|
|
|
*В зависимости от дизайна, целей и задач исследования визит, проведенный на несколько дней позже запланированного, может быть расценен не только как отклонение от протокола (например, в исследовании, изучающем гипотензивные препараты с периодом наблюдения пять лет), но и как нарушение протокола (например, в исследовании фармакокинетики препарата, где забор крови должен происходить в четко отведенное (до минут) время).
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим