В течение первых десяти лет работы Плана менеджмента инцидентов в Европейском Союзе (European Union Incident Management plan — EU-IMP) в общей сложности 78 случаев были обработаны через Сеть по изучению инцидентов (Incident Review Network — IRN). Они в основном касались вопросов безопасности (56%), качества (34%) или и того, и другого (5%). При этом несоблюдение надлежащей производственной практики обусловило более трети проблем. Сведения о большинстве из них (70%) поступили от европейских регуляторов и касались продукции, получившей разрешение на маркетинг по централизованной и национальной процедурам. Эти и другие результаты обобщены в статье, опубликованной в журнале «Pharmacoepidemiology»*.
10% инцидентов привело к отзыву регистрации
Инцидент определяется как событие или новая информация об одном или нескольких разрешенных в ЕС лекарствах с потенциальными серьезными последствиями для здоровья населения. Помимо прочего, к ним относят последствия несоблюдения производственных требований или других проблем в цепочке поставок. Хотя сначала может показаться, что некоторые события не имеют серьезных последствий для общественного здравоохранения, они могут вызвать внимание средств массовой информации и обеспокоенность пациентов, а затем негативно повлиять на применение лекарства, отмечено в публикации.
Подавляющее большинство проблем с безопасностью было вызвано сигналами в ходе постмаркетингового наблюдения и исследований, в то время как большинство проблем с качеством и несоответствием стандартам надлежащей производственной практики (Good Manufacturing Practic — GMP) поступило как уведомление о загрязнении и проблемах с данными (data integrity). Выявление серьезных недостатков в исследованиях биоэквивалентности (например проведенных компанией «Semler Research Centre Private Ltd, Индия) или систематических манипуляций с данными в клинических исследованиях (к примеру организованных «GVK Biosciences», Индия) вызвало проблемы несоблюдения требований надлежащей клинической практики (Good Clinical Practice — GCP). Единственная проблема недостаточности поставок, решаемая через IRN, была вызвана ограничениями на рыболовство в Японии после стихийного бедствия в 2011 г., что привело к нехватке сырья для производства сульфата протамина.
Управление инцидентами осуществляется с помощью рутинных мер, таких как:
- инструменты фармаконадзора и контроля качества/производства, например, проведение инспекций;
- регуляторные меры, например, изменение, приостановка или отзыв разрешения на маркетинг;
- выпуск сообщений, предназначенных для пациентов и медицинских работников, например, пресс-релизов, вопросов и ответов (Q&A), прямое общение с медицинскими работниками (Direct Healthcare Professional Communication — DHPC) и т.д.
Зафиксированные за последние 10 лет последствия инцидентов, разрешенных через IRN, включают в 50% случаев изменение разрешений на маркетинг и/или меры по минимизации риска соответствующего лекарства. При этом 10% инцидентов привели к приостановке и 9% — к отзыву разрешения на маркетинг препарата.
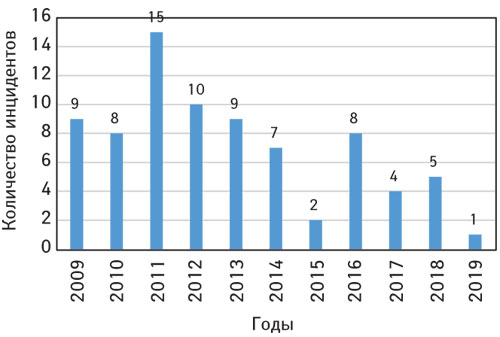
Из-за непредсказуемости инцидентов количество уведомлений значительно варьировало в разные годы наблюдения: максимальное значение составило 15 в 2011 г., а минимальное — 1 в 2019 г. (из которых на отчетный период приходится только 8 мес) (рисунок).
Примеры инцидентов
IRN оказала поддержку при устранении нескольких важных инцидентов. Например, по сигналу Института Пауля Эрлиха (Paul Ehrlich Institut) о случаях возможного иммунного энцефалита в связи с применением даклизумаба для лечения рассеянного склероза. Так, в качестве временной меры разрешение на маркетинг даклизумаба приостановлено на 2 нед. Затем, в результате тесного сотрудничества и обмена информацией между всеми заинтересованными сторонами, а также после тщательной оценки всех имеющихся доказательств Комитет по оценке риска фармаконадзора (Pharmacovigilance Risk Assessment Committee — PRAC) Европейского агентства по лекарственным средствам (European Medicines Agency — EMA) смог окончательно сделать вывод в течение 3 мес, что риск, связанный с продуктом, перевешивает пользу. Между тем, владелец разрешения на маркетинг принял решение добровольно отозвать разрешение на продажу этого продукта.
Второй пример безопасности касается уведомления о намерении владельца разрешения на маркетинг прекратить несколько текущих клинических исследований иделалисиба, применяемого для лечения взрослых пациентов с хроническим лимфолейкозом (ХЛЛ) или фолликулярной лимфомой из-за повышенного риска серьезных респираторных инфекций и связанных с ними случаев смерти, наблюдавшегося в трех исследованиях.
Учитывая серьезность потенциальной проблемы, а также имеющиеся неполные доказательства, PRAC принял решение (в течение 1 нед после первоначального уведомления) принять временную меру (ограничение применения у ранее не леченных пациентов с ХЛЛ и при определенном генотипе) и обратиться с DHPC.
4 мес спустя, после обзора полного набора данных, консультации с соответствующими специалистами и с учетом отсутствия альтернативных подходящих методов лечения PRAC рекомендовал снять временное ограничение. Однако, чтобы лучше защитить пациентов, были усилены меры по минимизации риска. К информации о продукте были добавлены необходимые предупреждения.
Как заключили авторы исследования, в течение первых 10 лет работы EU-IMP постепенно совершенствовался, справляясь со все более сложными и новыми проблемами здравоохранения.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим