«Еженедельник АПТЕКА» (№ 28 (299) от 16.07.2001 г.) сообщал о планах Государственного фармакологического центра (ГФЦ) повысить требования к качеству регистрируемых лекарственных препаратов-генериков. В настоящее время разработан проект приказа Министерства здравоохранения Украины о порядке проведения доклинических испытаний лекарственных средств. Согласно положениям проекта приказа, во время процедуры регистрации генерического лекарственного средства более полное представление о его качестве и терапевтической эффективности можно будет получать после проведения фармакологических испытаний, сопоставив данные о фармакокинетике и фармакодинамике регистрируемого препарата с таковыми оригинального препарата.
Планируемые изменения в процедуре регистрации препаратов-генериков комментирует заместитель председателя ГФЦ Виктор Чумак.
— Находящийся в настоящее время на утверждении приказ МЗ о порядке проведения доклинических испытаний лекарственных средств является документом, который станет важным шагом к гармонизации законодательства Украины с законодательством ЕС, в частности, с Директивой Совета ЕС 75/318/ЕЕС «О сближении законов государств ЕС в отношении аналитических, фармакологических и клинических норм и протоколов по испытанию лекарственных препаратов» (далее — Директива). Кроме того, со вступлением в силу этого подзаконного акта будет конкретизирована статья 6 закона «О лекарственных средствах», в которой сказано, что доклиническое изучение лекарственных средств предусматривает химические, физические, биологические, микробиологические, фармакологические, токсикологические и другие научные исследования в целях изучения их специфической активности и безопасности.
До настоящего времени при проведении доклинических исследований внимание было акцентировано на фармакотоксикологических испытаниях лекарственного препарата. Другие виды исследований, обозначенные в Законе, не выполнялись, так как трактовка его положений отсутствовала. Поэтому при создании проекта порядка проведения доклинических испытаний специалисты ГФЦ руководствовались указанной статьей Закона и Директивой.
В Директиве подробно отражены вопросы химического, фармацевтического, биологического, токсикологического и фармакологического испытаний, данные (отчеты) о которых необходимо предоставить заявителю при получении торговой лицензии. В частности, в разделе «Контрольные испытания готовой продукции» указано, что твердые лекарственные формы, предназначенные для приема внутрь, должны быть исследованы in vitro для определения показателя высвобождения и растворения активных ингредиентов. При необходимости такие исследования могут быть проведены и по поводу других лекарственных форм.
Данные по высвобождению активного вещества должны быть приведены в регистрационном досье лекарственного препарата, поскольку они могут влиять на биодоступность, то есть на концентрацию препарата в плазме крови в тот или иной промежуток времени, что в конечном счете может свидетельствовать о его терапевтической эффективности.
Таким образом, с вступлением в действие порядка проведения доклинических испытаний ГФЦ приступает к практической реализации его положений, предусматривающих включение в регистрационное досье данных о биодоступности лекарственного препарата.
Государственная инспекция по контролю качества лекарственных средств неоднократно обращала внимание на то, что формальный перечень показателей АНД отражает только одну сторону проблемы качества лекарственного препарата. К важным фармацевтическим характеристикам качества готового лекарственного средства относятся растворение или скорость высвобождения активного вещества из твердой лекарственной формы, поскольку они могут влиять на особенности фармакокинетики, биодоступность, а также терапевтическую эффективность лекарственного препарата. На практике зачастую два генерических препарата с одним и тем же действующим веществом имеют разную выраженность терапевтического эффекта, что позволяет рассматривать их как разные препараты.
В качестве примера приведу результаты исследований, проведенных лабораторией «Международная лабораторная группа» (рисунок), в ходе которых сравнивалась кинетика высвобождения действующего вещества — диклофенака — пяти лекарственных препаратов в форме таблеток, покрытых кишечно-растворимой оболочкой. Исследование проведено согласно Американской Фармакопее (USP 24) в условиях in vitro при моделировании pH среды желудка и тонкого кишечника, что соответствовало АНД препарата сравнения, которым в данном случае был Вольтарен. Согласно USP 24, серия препарата отвечает требованиям фармакопеи, если в течение 2 ч в кислой среде (1-я среда) растворилось не более 10%, а в последующие 45 мин в фосфатном буфере (2-я среда) — не менее 75% действующего вещества, содержащегося в лекарственной форме. Проводили по шесть исследований каждого препарата через одинаковые промежутки времени. Для построения итогового графика среднее значение шести исследований для каждого временного промежутка вычисляли по методу медианы. Полученные данные свидетельствуют о различии кинетики высвобождения действующего вещества генерических препаратов и препарата сравнения.
?
|
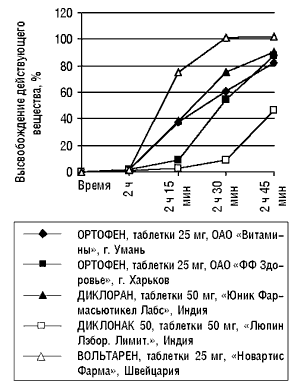
Таким образом, на основании данных о различной кинетике высвобождения действующего вещества нельзя с уверенностью утверждать, что биодоступность, а также терапевтическая эффективность этих препаратов одинаковы.
Показатель растворения/высвобождения действующего вещества из твердой лекарственной формы, предназначенной для перорального применения, является важной фармацевтической характеристикой лекарственного препарата. Определение этого показателя в исследованиях in vitro дает представление о технологии получения препарата и/или субстанции и оно необходимо на стадии производства препарата (для соответствующих твердых лекарственных форм). Показатель растворения/высвобождения обязательно входит в досье лекарственного препарата при подаче заявки на получение торговой лицензии. Кроме того, особенности кинетики растворения/высвобождения действующего вещества могут служить важным источником информации и интерпретации данных исследований по разработке генерических препаратов, поскольку могут влиять на биодоступность, а значит, и на биоэквивалентность или терапевтическую эквивалентность препарата сравнения.
В настоящее время речь не идет о разделении препаратов на «плохие» или «хорошие»; главный критерий для генерика — это его терапевтическая эквивалентность оригинальному препарату или аналогу (препарату сравнения), на которую могут оказывать влияние показатели растворения/высвобождения, фармакодинамики и фармакокинетики. Данные о фармакодинамике и фармакокинетике должны быть отражены в регистрационном досье и в инструкции по медицинскому применению, в противном случае мы дезинформируем врача.
Николай Холоденко
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим