Обеспечение высоких показателей качества и эффективности лекарственных средств (ЛС) во многом определяется процедурой регистрации, которую обязаны проходить препараты до того, как поступят на рынок. Эта тема актуальна и для Украины, поскольку требования к регистрации ЛС требуют гармонизаци с таковыми, принятыми в развитых странах.
Процесс гармонизации начался в 80-е годы XX-века, что нашло выражение в создании в 90-х годах программы ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use — Международная конференция по гармонизации технических требований к регистрации лекарственных препаратов для человека), которая и в настоящее время является основой для формирования нормативно-регламентирующей базы регистрации фармацевтических препаратов. Таким образом, представители регуляторных органов и фармацевтической промышленности стран ЕС, США и Японии предприняли совместные шаги для дальнейшего развития фармацевтических рынков этих стран — самых крупных в мире.
Это стало возможным благодаря устранению технических баръеров в сфере международного обращения ЛС посредством гармонизации и взаимного признания стандартов и законодательных актов в отношении разработки, проведения клинических исследований, процесса регистрации, формы и структуры регистрационных документов.
Основой регистрационной документации является Common Technical Document (CTD) — Общий технический документ (ОТД), перечень регистрационных документов, созданный в рамках ICH. ОТД на протяжении последних лет органично вписался в пакет важнейших руководств в этой области и по сути является международным стандартом представления данных регистрационного досье препарата.
Главная цель разработки ОТД — гармонизация требований к представлению регистрационных документов, что позволяет направлять их в органы регистрации и лицензирования разных стран в качестве единого досье (для регистрации препаратов).
Представители органов регистрации и лицензирования ЕС, США и Японии согласовали единый формат ОТД для подготовки заявок. Они согласились соответствующим образом изменить действующие в настоящее время национальные законодательные акты и отказаться от существующих форматов в регистрации ЛС.
Для подготовки нормативных документов ОТД ICH были созданы Экспертные рабочие группы (EWGs) по контролю качества, безопасности и эффективности лекарственных препаратов. Последние рабочие встречи состоялись в Сан-Диего (США) — в ноябре 2000 г., в Токио (Япония) — в мае 2001 г. и в составе так называемой ICH Steering Committee (руководящего комитета ICH), в Брюсселе (Бельгия) — в феврале 2002 г.
Компетентные национальные органы получили соответствующие руководства ОТД ICH (полностью все руководства опубликованы на сайте ICH ). В настоящее время ведется подготовка соответствующей документации как для экспертов в нормативно-регламентирующей области, так и для владельцев торговых лицензий (заявителей).
ОБЛАСТЬ ПРИМЕНЕНИЯ ОТД
ОТД, первоначально применимый исключительно к новым химическим соединениям и биотехнологическим препаратам (как указано в разделах Q6A и Q6B ICH), в дальнейшем стали использовать также для других групп препаратов и всех видов торговых лицензий (для оригинальных препаратов, генериков, ОТС-препаратов). В будущем, возможно, ОТД будет применяться при проведении клинических испытаний.
Перспективным является применение ОТД для планирования развития фармацевтической индустрии, также он может быть использован экспертами при подготовке соответствующих рецензий на все типы заявок на регистрацию препаратов.
Имеются некоторые различия в процессе внедрения ОТД в странах, присоединившихся к соглашению ICH.
В странах ЕС ОТД в новой редакции будет применяться ко всем типам торговых лицензий независимо от используемой процедуры — централизованной, децентрализованной (процедуры взаимного признания) или национальной, которые связаны с получением торговой лицензии по полной или сокращенной процедуре. Планируется, что ОТД-формат будет применяться ко всем типам препаратов (новым химическим соединениям, радиофармпрепаратам, вакцинам, фитопрепаратам и т.д.)1.
США
ОТД применим для торговых лицензий на новые лекарственные препараты, сокращенных торговых лицензий на новые лекарственные препараты и торговых лицензий на патентованные биологические препараты (biologic lecenx application — BLA), для новых молекул лекарственных соединений и ранее известных молекул лекарственных соединений, генериков, безрецептурных препаратов, в том числе для изменений, вносимых в способ применения препарата (например, применение препарата по новому показанию, применение новой лекарственной формы, нового пути введения).
Япония
ОТД применим для подготовки к регистрации:
1. Препаратов, в состав которых входят новые ингредиенты.
2. Назначения сопутствующих препаратов.
3. Нового пути введения препарата.
4. Новых показаний к применению препарата.
5. Новых лекарственных форм препарата.
6. Новых доз препарата.
Канада
ОТД применяют при подаче документов на лекарственные препараты (при подаче заявок на выдачу торговой лицензии), в частности на новые ЛС, включая новые химические соединения и препараты, произведенные с использованием биотехнологических методов (биотех-препараты). ОТД необходимо использовать при подготовке к регистрации новых препаратов, представлении документов на новые препараты в сокращенном виде, а также при внесении изменений в регистрационные досье. Причем в двух последних случаях при использовании формата оригинальной заявки. ОТД может быть также использован при представлении документов на комбинированные лекарственные препараты, а также на комбинации препарат/изделие медицинской техники.
СТРУКТУРА ОТД
Модуль 1: Административная и предписывающая информация
A. Оглавление модуля 1
B. Документы, специфичные для каждого региона (например, формы заявок, предписывающая информация) (рисунок)
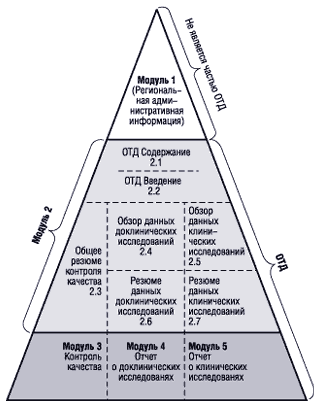 |
Рисунок. Организация и структура ОТД2
Модуль 2: Общий технический документ (резюме)
A. Оглавление общего технического документа
B. Введение
C. Краткий обзор системы обеспечения качества
D. Обзор доклинических данных
E. Обзор клинических данных
F. Резюме данных, полученных в ходе доклинических исследований
1. Фармакология
a. Резюме в текстовом формате
b. Резюме в табличной форме
2. Фармакокинетика
a. Резюме в текстовом формате
b. Резюме в табличной форме
3. Токсикология
a. Резюме в текстовом формате
b. Резюме в табличной форме
G. Резюме данных, полученных в ходе клинических исследований
1. Резюме по биофармацевтическим данным и соответствующим методам аналитических исследований
2. Резюме по данным клинических фармакологических исследований
3. Резюме по клинической эффективности
4. Резюме по клинической безопасности
5. Краткий обзор индивидуальных исследований
Модуль 3: Качество (Система обеспечения качества)
A. Оглавление
B. Основные данные
C. Ключевые ссылки на литературу
Модуль 4: Отчеты по доклиническим исследованиям
A. Оглавление
B. Отчеты по исследованиям
C. Ссылки на литературу
Модуль 5: Отчеты по клиническим исследованиям
A. Содержание отчетов по клиническим исследованиям и относящаяся к ним информация
B. Табличная форма данных по всем клиническим исследованиям
C. Отчеты по клиническим исследованиям
ИНДИВИДУАЛЬНЫЙ МАСТЕР–ФАЙЛ НА ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ
(DRUG MASTER FILE)
В странах ЕС при представлении документации на рассмотрение для получения торговой лицензии файл DMFs (индивидуальный мастер-файл на лекарственный препарат; файл ноу-хау на лекарственный препарат) должен быть оформлен в формате ОТД. В дальнейшем это послужит основанием для уменьшения числа требований, предъявляемых заявителю, так как не будет необходимости измененять представляемую документацию на лекарственную субстанцию, предназначенную для использования в различных целях.
Заявки на получение Сертификата соответствия монографиям Европейской Фармакопеи (Certificate of Suitability — CoS) также могут быть подготовлены с использованием формата ОТД. В настоящее время Европейское управление информации по качеству (European Data Quality Manadgement — EDQM, Страсбург) использует гибкие подходы при приеме документации, подготовленной в том или ином формате.
В странах ЕС
Для того чтобы гарантировать, что вся соответствующая информация при представлении DMF в формате ОТД ЕС будет направлена непосредственно в компетентные органы, заявитель должен консультироваться и осуществлять свою деятельность совместно с лицом, ответственным за производство активного ингредиента. Данная DMF-документация должна быть включена в подробное описание процесса производства, контроля качества препарата на разных этапах производственного процесса, процесса валидации и оценки данных (Модуль 3). Кроме того, Краткий обзор системы обеспечения качества (Модуль 2) должен применяться к каждому из аспектов, отсутствующих в заявке (досье) на получение торговой лицензии на данный препарат. DMF должен полностью соответствовать структуре ОТД3.
ВНЕДРЕНИЕ ОТД — ДОБРОВОЛЬНАЯ И ОБЯЗАТЕЛЬНАЯ ФАЗЫ
Во время рабочей встречи руководящего комитета ICH в Токио было принято решение о том, что с 1 июля 2001 г. нормативно-регламентирующие органы должны принимать заявки во всем регионе ICH (в странах ЕС, включая Норвегию и Исландию, а также в США и Японии), выполненные в формате ОТД. Кроме того, формат ОТД был утвержден к исполнению в Швейцарии и Канаде. В настоящее время, после того, как в ЕС была изменена форма инструкции для заявителей, формату ОТД следует и Австралия.
Таким образом, все основные фармацевтические рынки в настоящее время признают требования формата ОТД и заявителям настоятельно рекомендуется следовать новой структуре представления документации для овладения необходимым опытом в данной области в целях ускорения процесса регистрации при одновременном представлении документации в вышеупомянутых регионах.
Что касается вопроса об окончательных сроках внедрения ОТД как обязательного для исполнения формата оформления регистрационного досье, то представители компетентных уполномоченных органов пришли к единому мнению о том, что начало его действия необходимо отложить до 1 июля 2003 г. Это означает, что представители фармацевтической промышленности будут иметь возможность еще в течение одного года представлять досье на фармацевтические препараты, оформленные в соответствии со старыми «национальными» требованиями.
В противоположность другим нормативно-регламентирующим органам, FDA США дало следующие пояснения: в настоящее время заявителям не обязательно следовать требованиям руководства ОТД относительно формата представляемой заявки. Это означает, что FDA не будет акцентировать внимание на обязательном исполнении требований международного формата ОТД ICH до июля 2003 г.
В течение переходного периода разрешается представление необходимой документации в «смешанном» формате, то есть в виде досье, содержащих блоки как в формате ОТД, так и оформленные в соответствии со старыми требованиями. В любом случае ссылки в досье ОТД на заявку, утвержденную до июля 2001 г., являются правомочными. В Токио придерживаются мнения, что новая документация, касающаяся основных направлений, вариаций и дополнений, может быть представлена в формате ОТД без изменения соответствующих досье, представленных ранее в традиционном формате.
Европейский Союз
Новые полные (стандартные) заявки для получения торговой лицензии на оригинальные препараты и сокращенные заявки (на препараты-генерики, а также заявки, принятые к сведению) должны быть оформлены либо полностью в формате ОТД, либо в «старом» формате ЕС. Однако в течение переходного периода в некоторых случаях могут быть представлены заявки, в которых отдельные модули могут быть оформлены в смешанном формате (в пределах отдельных модулей их содержание должно быть представлено в одном из форматов).
Иначе говоря, в подобных случаях при оформлении регистрационного досье необходимо предоставление полного модуля, включая отчеты экспертов или обзоры (резюме), выполненные в формате соответствующих модулей, заменяющих Модуль 2 (данные контроля качества представлены в формате ОТД, включая резюме контроля качества, данные доклинических и клинических исследований в «старом» формате ЕС).
Для сокращенных вариантов заявок и перекрестных ссылок на оригинальные препараты к предыдущим заявкам принятой формой представления документации является «старый» формат ЕС. Таким образом, не нужно переводить предыдущие заявки в формат ОТД4.
Производители ЛС в регионе ICH должны отдавать себе отчет в том, что все заявки на получение торговой лицензии, представленные после 1 июля 2003 г., должны быть оформлены в формате ОТД.
ИЗМЕНЕНИЕ ЗАКОНОДАТЕЛЬСТВА
Ожидается, что компетентные нормативно-регламентирующие органы стран — участниц ICH должны опубликовать новое руководство, оформленное в соответствии с ОТД ICH с подробным изложением соответствующих требований к Блоку 1, который является административной частью ОТД.
Такая работа уже ведется. Например, Европейская комиссия в мае 2002 г. опубликовала новую версию Инструкции для заявителей4 на своем сайте (http://dg3.eudra.org/F2/eudralex/vol-2/home.htm). Департамент анализа и лицензирования Министерства здравоохранения Японии 21 июня 2001 г. опубликовал извещение «О структурной организации регистрационного досье применительно к новой заявке на фармацевтические препараты, поданное на утверждение».
Более трудной и долговременной задачей является внесение исправлений и изменений в законодательные и нормативные акты. Для этого требуется введение новой терминологии и создание новой административной структуры. В особенности это касается стран ЕС, где необходима детальная разработка формы и содержания регистрационных досье. Изменение директив в странах ЕС является сложным и требует комплексного подхода. Вероятно, этот процесс будет происходить на протяжении всего переходного периода.
Европейский Союз
Инструкция для заявителей была подготовлена Европейской Комиссией в сотрудничестве с компетентными органами государств — членов ЕС, Европейским агентством по оценке лекарственных препаратов (ЕМЕА), другими заинтересованными организациями. Были внесены поправки в Постановление Совета ЕС (ЕЕС) № 2309/93 и в Приложение к Директиве 75/318ЕЕС (в настоящее время вошла в состав Директивы Европейского Парламента и Совета ЕС 2001/83)5.
Опубликованы действующие в настоящее время в странах ЕС требования к оформлению регистрационного досье с внесенными в Приложение поправками к Директиве 75/318/ЕЕС (в настоящее время вошла в Директиву 2001/83). В предисловии к этому документу отмечается, что «подробные отчеты и документы, прилагаемые к заявке на получение торговой лицензии в соответствии со Статьей 4 Директивы Совета ЕС 65/65/ЕЕС (в настоящее время вошла в Директиву 2001/83), должны быть представлены в четырех частях и в соответствии с ныне действующими требованиями помещены в этом Приложении и приняты во внимание при подготовке Инструкции для заявителей»4.
США
В новое руководство FDA были внесены изменения, а именно в инструкции по форматированию, компоновке и представлению Заявок на регистрацию новых лекарственных препаратов и антибиотиков.
Япония
Уведомление Министерства здравоохранения Японии «Нормативно-регламентирующие акты, обязательные для исполнения в области фармацевтического законодательства» составлено с учетом требований ICH СTD.
СТРАНЫ, НЕ ВХОДЯЩИЕ В ICH
Кроме стран региона ICH, законодательные органы других стран, в частности Канады и Швейцарии, объявили о своем участии в этом процессе. Швейцарские власти намереваются с июля 2002 г. сделать формат ОТД обязательным для регистрации NCEs; с 1 января 2003 г. — для регистрации генериков и с 1 июля 2003 г. — для препаратов OTC и ЛС, изготовленных на основе лекарственных растений. Другие страны EFTA (European Free Trade Area — Европейская зона свободной торговли) — Исландия, Норвегия и Лихтенштейн также готовы к внедрению формата ОТД для представления регистрационных досье.
Многие страны Центральной и Восточной Европы, Латинской Америки, Ближнего Востока и Юго-Восточной Азии, ЮАР, Австралия и Новая Зеландия признали или в ближайшее время могут признать формат ОТД.
В.А. Усенко, Д.А. Сухинин
По материалам Regulatory Affairs Journal и сайта ICH
1 Примечание к Торговым лицензиям, Том 2В (май 2002 г.).
2 Инструкция для заявителей, Том 2В, Предисловие и введение (Издание 2002 г.)
3 Инструкция для заявителей, Том 2И (Издание 2002 г.)
4 Инструкция для заявителей, Том 2В (Издание 2002 г.).
5 Полный текст Директивы см.: Фармацевтический сектор: основы современного законодательства в Европейском Союзе. — Киев: Морион, 2002.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим