|
СИСТЕМА ФАРМАКОЛОГИЧЕСКОГО НАДЗОРА В ЕС
Система фармаконадзора и создание Руководства
Согласно Директиве 2001/83/ЕС Европейского парламента и Совета ЕС от 6.11.2001 г. «О своде законов Сообщества в отношении лекарственных препаратов для человека» (раздел IХ «Фармаконадзор», статьи 101–108) (Фармацевтический сектор: основы современного законодательства в Европейском Союзе. — К: МОРИОН, 2002. — 96 с.), система фармакологического надзора — это государственная система сбора данных, которая на основании полученной информации о ПР на лекарственные препараты в условиях их обычного применения обеспечивает принятие соответствующих регуляторных решений в отношении лицензированных в Сообществе лекарственных препаратов. Эта система используется для сбора данных, необходимых для осуществления надзора за ЛС, при особом контроле в отношении ПР у человека, а также для научной оценки этой информации. Такая информация должна быть сопоставлена с данными по изучению использования лекарственных препаратов. Система фармаконадзора также обеспечивает изучение всех имеющихся данных о неправильном применении ЛС и случаях злоупотребления ими, что может повлиять на оценку соотношения польза/риск, связанных с приемом лекарственных препаратов.
 |
Согласно требованиям Директивы Совета ЕС 75/319/ЕЕС с изменениями, внесенными Директивой Комиссии ЕС 2000/38/ЕС от 5 июня 2000 г., Директивы Совета ЕС 81/851/ЕС с изменениями, внесенными Директивой Комиссии ЕС 2000/37/ЕС от 5 июня 2000 г., Комиссия ЕС после консультации с Европейским Агентством по оценке лекарственных препаратов (далее — Агентство), государствами ЕС и заинтересованными сторонами должна была составить руководство по сбору, контролю и представлению сообщений о ПР на лекарственные препараты в целях содействия обмену информацией по фармакологическому надзору в рамках ЕС. Статьи 24 и 46 Постановления Совета ЕС (ЕЕС) № 2309/93 включают такие же требования (Фармацевтический сектор: основы современного законодательства в Европейском Союзе. — К.: МОРИОН, 2002. — 96 с.). В данное руководство необходимо было включить технические требования в отношении электронного обмена информацией по фармаконадзору в соответствии с международно согласованными форматами. Кроме того, Комиссии ЕС было поручено опубликовать справочное руководство по международно согласованной медицинской терминологии.
В связи с этим Комиссия ЕС после подробной консультации с Агентством, государствами ЕС и заинтересованными сторонами подготовила том 9 — «Фармаконадзор. Лекарственные препараты для человека и для применения в ветеринарии» (далее — Руководство). В нем впервые сведены общие руководящие указания в отношении требований, процедур, распределения функций и деятельности в области фармакологического надзора для представителей промышленности и регуляторных органов, а также международные соглашения, достигнутые в рамках Международной конференции по гармонизации технических требований к регистрации лекарственных средств для человека (ICH).
Предпосылки к созданию Руководства
Поскольку новые ЛС поступают на рынки разных стран в разное время, один и тот же препарат может быть в продаже в одной или нескольких странах, в то время как в других странах он все еще находится на стадии клинических испытаний, предоставление информации о клинической безопасности и ее использование следует рассматривать как непрерывный процесс. Необходимо учитывать, что ограниченное число пациентов, включенных в клинические испытания, невключение в испытание (по крайней мере вначале) определенных категорий пациентов, относящихся к группе риска, отсутствие опыта длительного лечения и ограничение применения сопутствующих методов лечения не позволяют сразу всесторонне оценить профиль безопасности ЛС. В таких случаях обнаружить или подтвердить наличие редко наблюдающихся ПР чрезвычайно трудно, а иногда — невозможно.
Регуляторные требования, которые относятся, в частности, к частоте предоставления и содержанию регулярно обновляемых сведений по безопасности ЛС, различны в трех основных экономически развитых регионах (ЕС, Японии, США). Руководство по фармаконадзору, относящееся к формату и содержанию полных и регулярно обновляемых отчетов по безопасности находящихся на рынке ЛС, было разработано для того, чтобы избежать дублирования усилий и гарантировать предоставление регуляторным органам важных данных, которые не являлись бы противоречивыми.
Для того чтобы получить полную картину клинической безопасности, необходимо проводить строгий контроль новых лекарственных препаратов, особенно в течение первых лет их реализации. Надзор за препаратами, выпущенными на рынок, является общей обязанностью регуляторных органов и владельцев торговых лицензий (ТЛ). Они регистрируют информацию по безопасности ЛС, поступающую из различных источников, и разрабатывают процедуры, обеспечивающие своевременное обнаружение и взаимный обмен данными о безопасности ЛС. Поскольку невозможно оценить всю поступающую информацию с одинаковой степенью срочности, регуляторные органы определили объем информации, которую необходимо предоставлять срочно; в большинстве стран экстренная передача информации осуществляется, как правило, в виде экспресс-отчетности о ПР, являющихся непредвиденными и серьезными.
Как правило, невозможно провести повторную оценку соотношения польза/риск при использовании ЛС для каждого случая проявления ПР, даже если она является серьезной. Поэтому в регулярно обновляемых отчетах по безопасности (PSUR) представлен мировой опыт в отношении безопасности применения ЛС через определенные промежутки времени после выдачи ТЛ для того, чтобы:
• сообщить всю релевантную новую информацию по безопасности, полученную из надлежащих источников;
• связать эти данные с воздействием ЛС на пациента;
• подвести итог статуса лицензирования лекарственного препарата в различных странах и любых существенных изменений, относящихся к безопасности;
• периодически обеспечивать возможность повторной оценки общей безопасности;
• указать, необходимо ли внести изменения в информацию о препарате в целях оптимизации использования ЛС.
Однако если предоставление PSUR требуется в разных странах, в которых предъявляются различные требования в отношении формы, содержания, длительности отчетного периода и сроков предоставления отчетов, то владелец ТЛ может быть вынужден очень часто составлять различные отчеты по одному и тому же препарату. Кроме того, в таких условиях различные регуляторные органы могут получать различные виды информации в разном объеме и в разное время. Поэтому необходимо приложить все усилия к тому, чтобы гармонизировать требования к PSUR, которые также позволили бы повысить эффективность составления отчетов.
В трех регионах — участниках ICH к регулярно обновляемым отчетам по безопасности предъявляют различные требования, например:
• Согласно предписаниям, действующим в США, необходимо ежеквартально представлять отчеты в течение первых 3 лет после выдачи лицензии, а затем ежегодно.
• В ЕС, согласно требованиям Директивы Совета ЕС 93/39/ЕЕС и Постановлению Совета ЕС (ЕЕС) 2309/93, необходимо представлять отчеты 1 раз в 6 мес в течение первых 2 лет, 1 раз в год в течение 3 последующих лет, а затем — 1 раз в 5 лет и одновременно подавать заявки на перерегистрацию.
• В Японии регуляторные органы требуют проведения исследования группы, состоящей из нескольких тысяч пациентов, определенным количеством установленных организаций в течение 6 лет после выдачи ТЛ. Систематическую информацию об этой группе, с учетом точного «общего знаменателя», следует предоставлять ежегодно. Что касается других данных, накопленных в процессе реализации препарата, то о ПР, которые являются не серьезными, а умеренными по степени тяжести и не указаны в маркировке препарата, необходимо сообщать 1 раз в 6 мес в течение 3 лет, а затем — ежегодно.
Структура Руководства
Руководство по фармаконадзору изложено в томе 9 «Фармаконадзор. Лекарственные препараты для человека и для применения в ветеринарии». Первоначальная Информация для заявителей (относящаяся к лекарственным препаратам для человека) впервые опубликована в 1986 г. (окончательная версия — в январе 1989 г. с последующим обновлением в 1993, 1994 и 1996 гг.) и включала Часть V, содержащую руководство для владельцев ТЛ по фармаконадзору. Кроме того, Том VB Информации для заявителей относительно лекарственных препаратов для применения в ветеринарии (III/5056/95, опубликован в виде проекта в январе 1995 г.) также включал Часть V, относящуюся к фармаконадзору. Документы, включенные в эти издания, пересмотрены и обновлены в данном Руководстве. Таким образом, назначение тома 9 «Фармаконадзор. Лекарственные препараты для человека и для применения в ветеринарии» — обновление и замена руководств по фармаконадзору, опубликованных в ЕС до 30 сентября 2001 г. В целях прозрачности в данное Руководство включены все документы, важные для промышленности и/или регуляторных уполномоченных органов государств ЕС и Агентства.
Том 9 состоит из четырех частей:
часть I — фармаконадзор за лекарственными препаратами для человека;
часть II — фармаконадзор за лекарственными препаратами для применения в ветеринарии;
часть III — общая информация об электронном обмене данными по фармаконадзору в ЕС;
часть IV — общие ссылки на административную и правовую информацию, относящуюся к ЛС как для человека, так и для применения в ветеринарии.
Необходимо отметить, что данное Руководство, как и все руководящие документы в правовых или технических областях, характеризующихся быстрым развитием, подлежит регулярному обновлению и пересмотру.
Отдельная глава книги посвящена становлению, развитию и современному состоянию системы фармаконадзора в Украине.
СИСТЕМА ФАРМАКОЛОГИЧЕСКОГО НАДЗОРА В УКРАИНЕ
После провозглашения независимости в Украине впервые в истории здравоохранения официально изучать ПР ЛС начали с 1996 г. (подразделение Фармакологического комитета МЗ Украины — Центр побочного действия лекарств). Деятельность Центра была направлена на сбор и анализ в первую очередь собственной информации о ПР, которые регистрировались при проведении официальных клинических испытаний ЛС. Одновременно началось создание базы данных спонтанных сообщений о ПР ЛС. В 1999 г. эта структура была преобразована в Отдел фармакологического надзора в составе Государственного фармакологического центра МЗ Украины (правопреемник Фармакологического комитета; далее — ГФЦ).
Основные задачи и направления деятельности Отдела фармакологического надзора ГФЦ МЗ Украины
Основные задачи Отдела фармакологического надзора:
• внедрение в практику системы здравоохранения фармаконадзора;
• разработка современной методологии по вопросам организации контроля за безопасностью ЛС в клинических испытаниях и при их применении в медицинской практике;
• разработка и внедрение современной научной методологии по вопросам изучения ПР ЛС;
• участие в разработке и внедрении регламентирующих документов по организации и осуществлению контроля за ПР, а также экспертизы материалов относительно ПР в Украине;
• консультативно-методологическая и просветительская деятельность в сфере контроля за ПР ЛС;
• организация совместно с Центром медицинской статистики МЗ Украины ежегодного сбора статистической информации относительно ПР ЛС в соответствии с нормативными документами МЗ Украины;
• проведение постоянного анализа и обобщения информации о ПР ЛС в целях предоставления обоснованных рекомендаций относительно безопасной фармакотерапии и фармакопрофилактики;
• осуществление мероприятий по усовершенствованию пред- и последипломной подготовки врачей в высших медицинских учебных заведениях и учреждениях последипломного образования III–IV уровня аккредитации в части надзора за ПР ЛС совместно с Главным управлением образования, науки и информационно-аналитического обеспечения и Центральным методическим кабинетом высшего медицинского образования МЗ Украины;
• контроль за выполнением врачами и руководителями лечебно-профилактических учреждений Украины Инструкции об осуществлении надзора за ПР ЛС, утвержденной приказом МЗ Украины № 347;
• экспертиза материалов на всех этапах клинических испытаний ЛС в части контроля за ПР ЛС, а также инспекция случаев ПР ЛС при медицинском применении;
• публикация материалов по вопросам ПР ЛС в средствах периодической медицинской информации и других медицинских изданиях.
Контроль безопасности ЛС на этапе клинического применения требует использования унифицированного подхода к терминам, которыми должны оперировать врачи различных специальностей при решении задач фармакологического надзора. В связи с этим в медицинскую практику были внедрены утвержденные соответствующим приказом МЗ Украины термины для каждодневного использования в практической деятельности врача. В официальных документах и в специальной литературе, а также при подготовке соответствующих отчетов, связанных с вопросами фармакологического надзора и контроля за ПР ЛС, при анализе ситуации в стране относительно безопасного использования ЛС можно использовать только следующие термины: безопасность (безвредность) ЛС; возможная ПР; достоверная ПР; непредусмотренное побочное действие; непредусмотренная ПР/явление; несерьезное побочное действие; несерьезная ПР или несерьезное побочное явление; предвиденное побочное действие; предвиденная ПР/явление; побочное действие; побочный эффект; серьезное побочное действие; спонтанные сообщения; сомнительная ПР и др.
Нормативная база фармакологического надзора в Украине
Юридической основой документов в отношении усовершенствования государственной системы фармакологического надзора являются: закон Украины «О лекарственных средствах» (1996); Порядок государственной регистрации (перерегистрации) ЛС, утвержденный постановлением КМ Украины № 1422 от 13.09.2000 г.; Инструкция о проведении клинических испытаний лекарственных средств и экспертизы материалов клинических испытаний, утвержденная приказом МЗ Украины № 281 от 01.11.2000 г. (зарегистрирована Министерством юстиции Украины 17.11.2000 г. под № 830/5051). В целях достижения гармонизации с нормами, которые применяются в международной практике, — руководства ІСН и Директив Совета Европейского Экономического Сообщества по вопросам фармакологического надзора (Директива 75/319/ЕЕС с дополнениями, внесенными Директивами 83/570/ЕЕС и 93/39/ЕЕС), в декабре 2000 г. приказом МЗ Украины (№ 347 от 19.12.2000 г.) была утверждена «Инструкция об осуществлении надзора за побочными реакциями/действиями лекарственных средств» (зарегистрирована Министерством юстиции Украины 26.12.2000 г. под № 947/5168) (далее — Инструкция).
Таким образом, в Украине была регламентирована деятельность системы контроля за ПР ЛС и определены основные требования к сбору данных, необходимых для осуществления особого контроля за ПР ЛС, которые возникают при клинических испытаниях и медицинском применении, а также к проведению научной оценки этой информации. Инструкция применяется в отношении ЛС, которые находятся на этапе клинических испытаний и/или разрешены к медицинскому применению. Контроль за ПР ЛС осуществляется ГФЦ МЗ Украины, который привлекает для этого сотрудников региональных отделений, созданных согласно приказу директора ГФЦ МЗ Украины.
В Украине Инструкцией, утвержденной приказом МЗ № 347, установлен следующий порядок поступления информации о ПР ЛС в МЗ Украины от врачей и лечебно-профилактических учреждений (рис. 1):
1. Вся информация о ПР ЛС поступает в ГФЦ.
2. Информация о ПР ЛС поступает в ГФЦ от:
• врачей, фармацевтов и медицинских работников независимо от ведомственного подчинения и форм собственности;
• производителей/владельцев регистрационного свидетельства или их уполномоченных представителей;
• уполномоченных международных организаций (ВОЗ, ЕС и т.п.);
• медицинских информационных источников и научных изданий;
• общественных организаций, которые представляют интересы потребителей ЛС, а также граждан;
• комиссии по вопросам этики (во время клинических испытаний ЛС);
• региональных отделений.
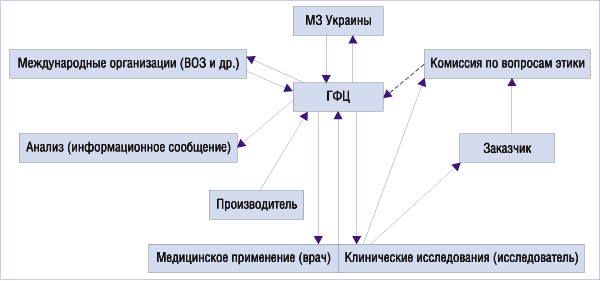 |
Рис. 1. Принципиальная схема поступления информации о ПР ЛС в Украине
3. ГФЦ получает информацию о ПР ЛС, систематизирует и анализирует ее (рис. 2, 3) в соответствии со сроками, представленными в Приложении 1 к данной Инструкции, и готовит информационные сообщения, аналитические обзоры, экспресс-информацию, методические рекомендации, предложения МЗ Украины относительно изменений инструкции к медицинскому применению и обращению ЛС и т.п.
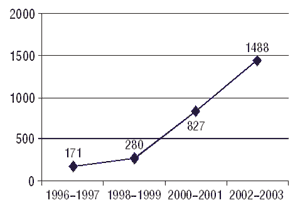 |
Рис. 2. Общее количество спонтанных сообщений о случаях ПР ЛС, поступивших в ГФЦ МЗ Украины в 1996–2003 гг.
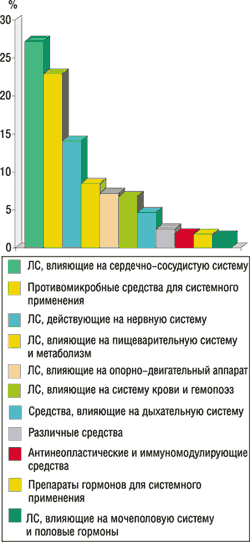 |
Рис. 3. Группы препаратов (согласно АТС), применение которых приводит к развитию ПР
(по данным спонтанных сообщений, 1996–2002 гг.)
4. Отчеты о подозреваемых серьезных ПР ЛС ГФЦ может направлять в ВОЗ через бюро ВОЗ в Украине, а копии — в Агентство ЕС.
В феврале 2001 г. были изданы приказы МЗ Украины № 51 от 08.02.2001 г. «Об организации представления информации о побочном действии лекарственных средств» и № 52 от 08.02.2001 г. «О внесении дополнений в Приложение 3 пункта 5.1 Инструкции об осуществлении надзора за побочными реакциями/действиями лекарственных средств».
Приказом МЗ Украины № 51 от 08.02.2001 г. было дано поручение Министру здравоохранения АР Крым, начальникам управлений здравоохранения областных, Киевской и Севастопольской городских государственных администраций: возложить ответственность за обеспечение предоставления учреждениями здравоохранения информации о ПР ЛС в ГФЦ МЗ Украины на главных штатных (внештатных) специалистов по специальности «Терапия»; обеспечить дооснащение лечебно-профилактических учреждений необходимой медицинской аппаратурой, оборудованием, лекарственными препаратами для оказания неотложной и специализированной медицинской помощи при возникновении ПР ЛС.
Центру медицинской статистики МЗ Украины поручено: предусмотреть в программе отчетности сбор статистической информации относительно ПР ЛС; обобщить предложения относительно внесения изменений в учетно-отчетную документацию о ПР ЛС. Главному управлению образования, науки и информационно-аналитического обеспечения и Центральному методическому кабинету по высшему медицинскому образованию МЗ Украины поручено подать предложения относительно внесения дополнений к учебным программам высших медицинских учебных заведений и учреждений последипломного образования III–IV уровня аккредитации по вопросам надзора за ПР ЛС.
ГФЦ МЗ Украины поручено обеспечить предоставление организационной и методической помощи МЗ АР Крым, управлениям здравоохранения областных, Киевской и Севастопольской городских государственных администраций, учреждениям здравоохранения относительно предоставления информации о ПР ЛС; разработать и подать до 1 сентября 2001 г. на утверждение в МЗ Украины методические рекомендации о сборе и предоставлении информации о ПР ЛС; обеспечить лечебно-профилактические учреждения бланками «Сообщение о побочных реакциях/действии лекарственных средств» для заполнения их на предоставление информации в ГФЦ МЗ Украины на протяжении 2001–2005 гг.
В приказе МЗ Украины № 292 от 16.07.2001 г. «Об усовершенствовании организации представления информации о побочной реакции лекарственных средств» утверждена форма № 69-здоров отраслевой статистической отчетности «Отчет о случаях побочной реакции/действии лекарственных средств в лечебно-профилактических учреждениях», а также в инструкции по составлению отраслевого отчета по форме 69-здоров утвержден годовой «Отчет о случаях побочной реакции/действии лекарственных средств в лечебно-профилактических учреждениях». Утверждены методические рекомендации по заполнению формы 137/о, а также рекомендации по заполнению и предоставлению карты-сообщения «Сообщение о подозреваемых серьезных побочных реакциях/действиях лекарственных средств при проведении клинического испытания», методические рекомендации относительно заполнения и предоставления карты сообщения о любых подозреваемых серьезных и/или непредусмотренных ПР ЛС (заполняет производитель/владелец регистрационного свидетельства или его уполномоченный представитель). Приказом предусмотрен график проведения информационно-учебных совещаний по вопросам организации фармакологического надзора с руководителями органов здравоохранения, госадминистраций регионов и главными штатными (внештатными) специалистами по специальности «Терапия» с участием руководителей лечебно-профилактических учреждений. Проведение этих мероприятий поручено специалистам ГФЦ МЗ Украины. Центром медицинской статистики МЗ Украины внесены дополнения в инструкцию к отчетным формам 003/в, 025/в, 025-2/в и 005-6/в относительно ПР ЛС.
Настоящим приказом врачам и руководителям органов здравоохранения всех звеньев вменяется в обязанность постоянно фиксировать и организовывать сбор информации о ПР ЛС при сопутствующих заболеваниях и осложнениях в медицинской карте стационарного больного (форма 003/о), медицинской карте амбулаторного больного (форма 025/о) и в отчетных формах 066/о «Карта больного, выбывшего из стационара», формах 025-6/о, 025-2/о «Талон амбулаторного пациента», «Статистический талон заключительных (отчетных) диагнозов».
Значимым для системы фармаконадзора в Украине стал Приказ МЗ Украины № 497 от 12.12.01 «Об утверждении Порядка запрета (остановки) и изъятия из обращения лекарственных средств на территории Украины». Целью настоящего приказа является создание комплексного контроля за безопасностью ЛС в Украине, основанного на критериях их качественной (фармацевтической) и «количественной» (медицинской) оценки. Подобный подход позволил оперативно решать вопросы оборота ЛС на фармацевтическом рынке Украины исходя из критериев польза/риск/качество, что особенно актуально в связи с угрозой возрастания в стране случаев фальсификации и недобросовестного производства ЛС. Практическое внедрение механизмов контроля качества и безопасности ЛС, изложенных в этом документе, сделало возможным эффективно влиять на безопасность медицинского применения ЛС, гармонизируя нормативно-правовую базу с требованиями мирового сообщества к системе фармаконадзора.
Сравнительный анализ систем фармаконадзора в ЕС и в Украине
Согласно определению, принятому в Украине, система фармакологического надзора — это государственная система сбора, научной оценки и контроля информации о побочных реакциях/побочном действии лекарственного средства в целях принятия соответствующих решений на этапе клинических испытаний и его медицинского применения в соответствии с действующим законодательством. Сравнивая с определением, принятым в странах ЕС, можно выявить некоторые различия в самих задачах фармаконадзора (см. начало статьи), положенных в основу контроля за безопасностью лекарственных препаратов. Закон Украины «О лекарственных средствах» не содержит положений о фармаконадзоре. Не завершено создание централизованной системы фармаконадзора (для иммунных ЛС существует система, которую курирует Центр иммунобиологических препаратов); в процессе разработки находятся механизмы сопоставления полученной информации о ПР ЛС с данными, отражающими потребление лекарственных препаратов, а также система, которая бы полностью соответствовала принципам предоставления информации по фармаконадзору на международном уровне. Однако уже установлены информационные связи с Главным управлением ВОЗ в Женеве и Центром по сотрудничеству в области международного мониторинга ЛС в Уппсале. Фармаконадзор является неотъемлемой частью системы сертификации ЛС для международной торговли. Поэтому в ЕС ответственность за предоставление данных по фармаконадзору лежит в первую очередь на производителе/владельце ТЛ, а в Украине регулярное поступление информации о ПР обеспечивается в основном за счет сбора врачебных сообщений. Следовательно, потенциал владельцев регистрационных свидетельств еще предстоит задействовать в полной мере.
Гармонизация положений о фармаконадзоре, а самое главное — усовершенствование реальных механизмов, способных гарантировать эффективное функционирование этой системы в Украине, будет способствовать развитию этого направления для обеспечения безопасности и качества ЛС в соответствии с международными стандартами.
Исходя из вышеизложенного, основными задачами формирования гармонизированной системы фармаконадзора в Украине, являются:
• внесение соответствующего раздела в закон Украины «О лекарственных средствах»;
• разработка гармонизированного с ЕС руководства по фармаконадзору;
• создание в рамках национального фармацевтического производства внутренней системы фармаконадзора с обязательным введением должности уполномоченного лица, призванного отвечать за систему фармаконадзора на предприятии, в соответствии с международными стандартами
• усовершенствование государственной системы фармаконадзора, гармонизированной с таковой в ЕС.
Внедрение и скорейшее решение этих задач гарантирует рядовому потребителю контроль безопасности ЛС на фармацевтическом рынке страны. Гармонизация национальных нормативно-законодательных регулирующих положений системы фармаконадзора со стандартами, принятыми в странах ЕС, позволит украинским предприятиям создать условия для производства высококачественных ЛС, повысить заинтересованность производителей в экспорте своей продукции.
Александр Стефанов, директор ГФЦ МЗ Украины, академик АМН Украины
Владимир Мальцев, заведующий сектором клинических испытаний ЛС ГФЦ МЗ Украины, профессор
Алексей Викторов, заместитель заведующего сектором клинических испытаний ЛС ГФЦ МЗ Украины, профессор
Николай Ляпунов, доктор фармацевтических наук, профессор, главный научный сотрудник ГНЦЛС
Людмила Ковтун, кандидат медицинских наук, ведущий научный сотрудник сектора координации и контроля клинических испытаний ЛС ГФЦ
Елена Безуглая, кандидат фармацевтических наук, ведущий научный сотрудник ГНЦЛС
Максим Плошенко, корреспондент «Еженедельника АПТЕКА»
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим