|
Европейские требования к организации инспекций клинических испытаний
|

Согласно определению Директивы 2001/20/ЕС (1), инспекция – это действия уполномоченного органа по проведению официальной проверки документов, помещений, мер по обеспечению качества, а также всех прочих материалов, которые, по мнению уполномоченного органа, связаны с проведением клинического испытания и которые могут находиться на клинической базе, в офисах спонсора и/или контрактной исследовательской организации или в других учреждениях, которые уполномоченный орган считает необходимыми для проведения инспекции.
В соответствии со статьей 15 Директивы 2001/20/ЕС для проверки соблюдения положений Надлежащей клинической (GCP) и производственной практики (GMP) государства – члены Европейского Союза (ЕС) назначают инспекторов для инспекции мест, имеющих отношение к проводимому клиническому испытанию, особенно клинической базы или баз, производственного участка, где выпускается исследуемое лекарственное средство, лабораторий, используемых для проведения исследований в ходе клинического испытания и/или помещений спонсора [1].
Инспекции должны проводиться официальной инстанцией соответствующего государства ЕС, то есть локально – в каждой стране – члене ЕС и централизованно – в рамках ЕС. Европейское агентство по оценке лекарственных препаратов (ЕМЕА) предпринимает меры по совершенствованию этой процедуры.
После завершения проверки составляется отчет, который должен быть доступен спонсору при условии соблюдения конфиденциальности. Кроме того, отчет может быть предоставлен другим государствам – членам ЕС, Комитету по вопросам этики и ЕМЕА по обоснованному запросу. Для признания результатов инспекций странами – членами ЕС должны выполняться требования к процедурам ее проведения. Этим вопросам посвящены руководства, разработанные во исполнение указанной Директивы, проекты которых в настоящее время подготовлены для одобрения [2, 3]. Основные положения этих документов будут изложены далее.
Подробное руководство по процедурам инспекции для проверки соответствия нормам Надлежащей клинической практики – GCP (Брюссель, ENTR/F/2D(2002))
Положения, которые представлены в этом документе, должны расцениваться как основные требования для взаимного признания результатов инспекций в пределах ЕС. Проверки могут проводиться во время или после клинического испытания и/или как часть проверки заявок на получение торговой лицензии. Результаты их должны признаваться всеми странами – членами ЕС (при условии если все инспекции проводятся по единому принципу). Инспекционные проверки клинического испытания должны проводиться согласно процедурам, установленным в соответствии с требованиями ЕС, чтобы соблюсти признанные стандарты для обеспечения взаимного признания их результатов. Усовершенствование и гармонизация стандартов проведения проверок должны быть достигнуты странами – членами ЕС в сотрудничестве с Европейской Комиссией и ЕМЕА посредством проведения совместных инспекций, согласования процессов и руководств, обмена опытом и обучения.
В руководстве приводится ряд новых терминов, которые приведены ниже:
Национальная программа по соответствию нормам GCP означает особый комплекс мер, предпринимаемых страной – членом ЕС для проверки мест (помещений, учреждений и организаций), имеющих отношение к проводимому клиническому испытанию.
Программа по соответствию нормам GCP ЕМЕА определяется как такой же комплекс мер, предпринимаемый ЕМЕА в рамках его полномочий в соответствии с Постановлением ЕС № 2303/93 и странами – членами ЕС по отношению к исследованию, являющемуся предметом рассмотрения Комитета по патентованным лекарственным препаратам – СРМР.
Замечание/факт нарушения – невыполнение предписанных требований, зарегистрированное во время инспекции и подтвержденное соответствующими доказательствами.
В основном разделе данного Руководства – «Компоненты системы» определено, какие меры должны предпринимать страны – члены ЕС, ЕМЕА и Комиссия для проведения проверок испытаний и их координации в соответствии с принципами GCP. А именно страны – члены ЕС должны:
• публиковать документы относительно принятия принципов GCP на своих территориях;
• определить правовые или административные основы, в рамках которых функционирует инспекционная программа GCP, с установлением полномочий инспекторов, возможности посещения клинических баз и доступа к документации и данным, относящимся к клиническому испытанию;
• определить объем и распространение программы проверки соответствия нормам GCP. Как минимум это включает клинические испытания, которые относятся к сфере действия Директивы 2001/20/ЕС;
• гарантировать, что уполномоченный орган несет ответственность за группу инспекторов, а также за установление соответствия GCP проводимому клиническому испытанию и меры, которые необходимо будет принять на основании результатов инспекции;
• установить перечень процедур для определения соответствия нормам GCP. Этот перечень должен включать процедуры, которые будут применяться для проверки организации клинического испытания, а также состояния их планирования и выполнения;
• опубликовать документы, содержащие критерии, которые формируют правовые или административные основы и в рамках которых функционирует инспекционная программа GCP (законы, нормативные документы, инструкции, правила);
• сохранять отчеты о проведенных национальных и международных инспекциях и их результатах;
• утвердить процедуру подачи запроса на проведение инспекции/оказание помощи другими странами – членами ЕС и возможность сотрудничества в выполнении инспекций мест в разных странах – членах ЕС;
• установить процесс проведения инспекций в третьих странах;
• вводить в базу данных EudraCT ссылки на инспекции, проведенные для проверки соблюдения норм GCP в соответствии с требованиями статьи 11 Директивы 2001/20/ЕС [1].
В свою очередь Европейская Комиссия и ЕМЕА должны установить программу проверки соответствия клинических испытаний нормам GCP, проводить встречи для координации этих инспекций, публиковать документы, формирующие основу программы соблюдения норм GCP, сохранять отчеты и запросы инспекций, установить процедуру организации их проведения в третьих странах.
Относительно соблюдения конфиденциальности указано, что поскольку уполномоченный орган может иметь доступ к персональным медицинским данным испытуемых и информации, имеющей коммерческую ценность, а в некоторых случаях может возникнуть необходимость в снятии копий с конфиденциальных документов клинических баз или подробном цитировании их в отчетах, то страны – члены ЕС должны:
• принимать меры для сохранения конфиденциальности не только инспекторами, но и другими субъектами, получившими доступ к такой информации в результате проведения проверки клинических испытаний;
• гарантировать, что отчеты об инспекциях могут предоставляться только перечисленным в статье 15.2. Директивы 2001/20/ЕС субъектам, в соответствии с их национальным законодательством и в зависимости от соглашений, заключенных между Сообществом и третьими странами. Национальные законодательства могут предусматривать получение отчетов об инспекциях другими субъектами.
Эти же требования в отношении конфиденциальности информации должны выполнять Комиссия, ЕМЕА и этические комитеты.
Так как вопросам квалификации, подготовки инспекторов, их личным качествам посвящено соответствующее подробное руководство [3], в подразделе «Персонал и подготовка» содержатся основные положения, касающиеся этой темы. Так, каждый уполномоченный орган должен гарантировать привлечение необходимого количества квалифицированных и подготовленных инспекторов. При этом инспекторы должны подписывать Декларацию об интересах, относящуюся к неразглашению финансовых и других аспектов инспектируемых объектов, и обеспечиваться соответствующими средствами идентификации (например, идентификационной картой или удостоверением личности).
Уполномоченные органы стран – членов ЕС должны принимать необходимые меры, если обнаружено отклонение от принципов GCP во время или после инспекции. Соответствующие действия должны быть описаны в документах уполномоченных органов.
Европейские требования к квалификации инспекторов, определяющих соответствие клинических исследований требованиям Надлежащей клинической практики для исследуемых продуктов (ENTR/F/2D(2002))
Указанное руководство предназначено в качестве дополнения к любым национальным требованиям и определяет требования к GCP инспекторам [3], а требования к GMP инспекторам изложены в отдельном документе.
В этом руководстве представлены требования к квалификации инспектора, который проводит инспекцию клинического испытания от уполномоченного органа государства – члена ЕС, действующего от имени Сообщества для определения соблюдения норм GCP при проведении клинического испытания.
Указывается, что инспекторы должны быть должностными лицами или назначаться для этого уполномоченным органом государств – членов ЕС в соответствии с национальным законодательством и следовать положениям национальных уполномоченных органов. Все инспекторы должны быть компетентны для выполнения своих обязанностей, иметь соответствующую подготовку, регулярно участвовать в тренингах, а информация о квалификации, навыке и опыте работы каждого инспектора должна сохраняться уполномоченными органами и постоянно обновляться.
Инспекторы должны осознавать и сохранять конфиденциальность в том случае, если они получают доступ к такой информации в результате проведения инспекции в соответствии с действующим национальным законодательством, европейскими требованиями и международными соглашениями. Работа инспекторов должна проводиться в соответствии со Стандартными операционными процедурами – СОП, которые подлежат регулярному обновлению.
Требования к персональным качествам инспекторов в документе рассмотрены достаточно подробно. Так, при проведении инспекции инспектор должен способствовать созданию открытой атмосферы. Инспекторам необходимо оставаться объективными во время инспекции, отвечать на вопросы, но избегать принятия на себя роли консультанта. Инспектор должен обладать честностью, зрелостью, быть объективным, уверенным в себе, иметь аналитические способности, понимать решающее значение инспекции и реалистично воспринимать ситуацию.
Указывается, что инспектор предпочтительно должен иметь законченное высшее образование в области медицины, фармации, фармакологии, токсикологии или других. Уровень профессиональной подготовки инспектора должен способствовать хорошей коммуникации между всеми лицами, занятыми в клиническом испытании со сторон спонсора, исследователя или этических комитетов.
Инспектор должен понимать принципы разработки лекарств, знать процедуры клинических испытаний и требования к ним. К этому относится знание роли фармацевтической, доклинической и клинической документации.
Инспекторы должны хорошо разбираться в терминологии, медицинских процедурах и системах записи клинических данных, а также в законодательном регулировании как национальной системы, так и внутри ЕС в отношении проведения клинических испытаний и функциональных обязанностей работников сферы здравоохранения, предоставляющих медицинскую помощь пациентам.
Инспекторы должны поддерживать свою квалификацию регулярным участием в семинарах, научных встречах, конференциях, приобретением нового практического опыта и участием в подготовке печатных работ. Обмен информацией между инспекторами из разных стран должен осуществляться посредством проведения совместных инспекций, участия в обучающих программах, тренингах, консультациях сотрудников разных инспекторатов на национальном и международном уровнях.
Проведение инспекций клинических испытаний лекарственных средств в Украине
В Украине инспекции клинических испытаний проводятся Государственным фармакологическим центром МЗ Украины, а именно отделом аттестации и инспекции клинических баз (далее – отдел инспекции), являющимся его структурным подразделением. Сотрудники отдела, созданного в 1999 г., имеют опыт работы в области организации и проведения клинических испытаний лекарственных средств, а также не зависят от субъектов испытания и не принимают непосредственного участия в проведении клинических испытаний [4].
Инспекционной проверке подлежат все клинические испытания лекарственных средств, включая многоцентровые испытания, которые проводятся в Украине после их официального разрешения Государственным фармакологическим центром МЗ Украины (далее – Центр).
В своей деятельности отдел инспекции руководствуется нормативными документами [4, 5], разработанными на основании Закона Украины «О лекарственных средствах» (1996), а также Стандартными операционными процедурами «Проведение инспекционной проверки клинического испытания в Украине» [6].
Основными задачами инспекций клинических испытаний является проверка:
• соответствия проведения клинических испытаний утвержденному протоколу и действующим нормативным требованиям;
• возможности проведения клинических испытаний на данной клинической базе: включение достаточного количества испытуемых в соответствии с протоколом; проверка оборудования, лаборатории, наличия первичной документации – историй болезней, амбулаторных карт, данных лабораторных и инструментальных методов исследования;
• ознакомленности исследователей и персонала, принимающих участие в клинических испытаниях, с протоколом и другой информацией, касающейся исследуемого препарата;
• защиты прав пациентов: наличие письменного информированного согласия на участие в клиническом испытании;
• соответствия и достоверности записей в индивидуальных регистрационных формах (ИРФ) данным первичной документации;
• документации о получении, хранении, выдаче, распределении и возврате исследуемого лекарственного препарата;
• своевременного предоставления информации о побочных реакциях.
Инспекционная проверка может проводиться в плановом порядке – в ходе клинических испытаний, ретроспективно – по данным архива и целенаправленно – при рассмотрении отдельных вопросов, а именно:
• при возникновении серьезных или непредвиденных побочных эффектов;
• для проверки выполнения рекомендаций, сделанных в ходе предшествующих проверок;
• при одновременном проведении клинических испытаний более 2 препаратов на одной клинической базе;
• при включении в исследования особого контингента: беременных и женщин в период лактации, здоровых добровольцев, ВИЧ-инфицированных, пациентов с психическими расстройствами.
В зависимости от вида проверки в состав инспекционной группы могут включаться эксперты, специализирующиеся в различных отраслях медицины.
Процедура проведения плановой инспекции включает следующие этапы: подготовка, непосредственно проведение проверки на клинической базе, обсуждение результатов, оформление отчета и акта по результатам проверки (рис. 1).
|
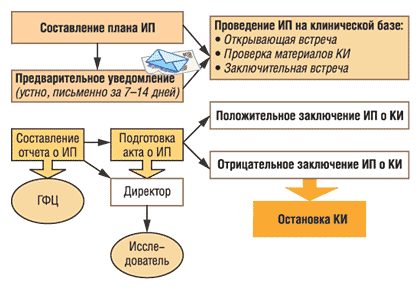
О предстоящей инспекционной проверке руководитель клинического испытания уведомляется в письменной и устной форме за 7-14 дней до ее проведения. В уведомлении указывается:
• цель, дата, время, место, где будет проводиться инспекционная проверка;
• перечень документов, необходимых для проведения проверки;
• перечень помещений, оборудования и лабораторий, которые задействованы в исследовании [7].
Перед началом проведения инспекции составляется план, где определены цель и объем проверки клинических испытаний. План проверки устанавливает лишь минимальный объем, который может быть расширен на основании материалов, полученных в результате инспекции.
Инспектор выполняет работу в строгом соответствии с планом проверки и установленными требованиями (стандартными операционными процедурами).
Лица, проводящие инспекцию и имеющие право прямого доступа к первичной документации, должны принимать разумные меры предосторожности для соблюдения анонимности испытуемых и конфиденциальности информации.
Непосредственно во время инспекционной проверки проверяются:
• комплектация файла исследователя;
• документация по получению, учету и распределению исследуемого лекарственного средства;
• условия хранения исследуемого лекарственного средства;
• документация о возврате или уничтожении (если предусмотрено) неиспользованного лекарственного средства после завершения клинического испытания;
• соответствие данных индивидуальных регистрационных форм данным первичной документации (историям болезней, амбулаторным картам);
• работа лаборатории отделения (кабинетов) функциональной диагностики;
• состав и деятельность комиссии по вопросам этики, действующей при клинической базе.
Обычно осмотр лабораторий осуществляется как часть проверки места проведения клинических испытаний; кроме лаборатории, инспектор может посетить и аптеку, если она включается в процедуру «движения» исследуемого лекарственного средства; документацию по транспортировке или утилизации препарата.
Во время инспекции заполняются проверочные листы, которые по завершении проверки подписывают инспекторы и руководитель клинического испытания. Инспекционная проверка завершается встречей с исследователями, на которой обсуждаются выявленные нарушения и выносятся рекомендации по их устранению.
После окончания инспекционной проверки составляются отчет и акт, которые подписывает руководитель Центра. Акт, в котором указываются результаты проверки и срок, необходимый для устранения выявленных замечаний, отправляется руководителю клинического испытания и заказчику (спонсору). Результаты инспекционных проверок докладываются и обсуждаются на заседаниях Научно-экспертного совета Центра.
В тех случаях, когда в процессе проведения клинических испытаний выявляются серьезные нарушения, клинического испытания могут быть остановлены. К таким случаям относятся:
• угроза жизни или здоровью испытуемых;
• нарушение этических норм проведения испытания;
• выявление серьезных побочных эффектов, отсутствие или недостаточная эффективность лекарственных средств.
Если такие нарушения выявляются при проведении инспекционной проверки, то в Центр предоставляется подробный отчет о выявленных нарушениях. После рассмотрения отчета на заседании Научно-экспертного совета Центра принимается решение об остановке клинических испытаний или отдельного его этапа. Дальнейшее возобновление клинических испытаний возможно после устранения всех выявленных нарушений в установленном порядке.
Анализ инспекций клинических испытаний лекарственных средств в Украине
В Украине инспекции лекарственных средств проводятся с 1999 г. За этот период было проведено 230 инспекций клинических испытаний (рис. 2), среди них – 10 международных многоцентровых исследований (в 8 случаях проводилась инспекционная проверка клинических баз, в 2 – офиса заказчика). Основные замечания:
• неукомплектованный файл исследователя;
• нарушение маркировки исследуемого лекарственного средства;
• нарушение процедуры получения информированного согласия;
• замечания по ведению ИРФ;
• замечания по ведению первичной документации;
• несвоевременное предоставление информации о побочных реакциях.
|
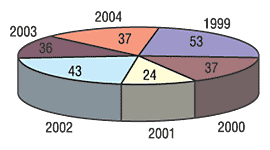
С 1999 по 2004 г. восемь клинических испытаний было приостановлено вследствие выявленных нарушений, связанных с несоблюдением условий протокола и нормативных требований проведения клинические испытания. После исправления недостатков и проведения повторных проверок клинические испытания были продолжены. За тот же период было остановлено два клинических испытания. Одно из них – в связи с развитием серьезных побочных реакций у большинства испытуемых, а другое – в связи с нарушением требования к маркировке исследуемого лекарственного средства.
Избежать большинства выявленных недостатков можно было в случае проведения мониторинга клинического испытания со стороны заказчика (спонсора), поскольку гарантия качества испытания зависит не только от регуляторных органов, но и от взаимодействия спонсора и исследователей.
Центр ежегодно проводит семинары для исследователей по вопросам клинических испытаний. В программу семинаров включаются вопросы проведения инспекционных проверок, рассматриваются и обсуждаются выявленные во время инспекций недостатки и ошибки, а также оговариваются рекомендации по их предупреждению.
В Украине создана нормативная база для проведения инспекционных проверок клинических испытаний. Это важный и необходимый этап контроля за проведением испытаний лекарственных средств со стороны регуляторных органов для подтверждения эффективности и безопасности изучаемых лекарственных средств, которые впоследствии будут разрешены для медицинского применения, а также гарантия защиты прав испытуемых, участвующих в клинических испытаниях. Кроме того, это накладывает ответственность за качество испытаний и на заказчиков (спонсоров), которые со своей стороны должны контролировать их, проводя мониторинг и аудит. Для исследователей инспекция клинических испытаний – это не только контролирующий, но и обучающий процесс. Опыт, приобретенный в процессе проведения клинических испытаний и инспектирования, будет использоваться исследователями в других проектах. n
|
ЛИТЕРАТУРА |
|
|
Людмила Ковтун,
ГП «Государственный фармакологический центр» МЗ Украины,
Дарья Полякова
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим