Программа конференции охватила очень широкий круг тем; как отмечали организаторы, они старались не оставить без внимания все этапы существования лекарственного средства (ЛС): от создания, то есть рождения, — до применения в практическом здравоохранении. Ведь именно клинические испытания вкупе с фармацевтической разработкой позволяют биологически активному веществу, которое определено как потенциально лекарственное, состояться в этом качестве и стать ЛС. Во многом благодаря клиническим испытаниям закладывается основа качества ЛС, которое согласно Закону Украины «О лекарственных средствах» определяется как совокупность свойств, позволяющих лекарственному средству удовлетворить потребности потребителей в соответствии со своим предназначением и отвечать требованиям, установленным законодательством. Поэтому клинические испытания были и будут одним из главных видов деятельности в фармацевтической отрасли, и интерес к их организации и проведению во всем мире только растет.
Процесс открытия (discovery) и разработки (development) лекарственных средств происходит путем проб и ошибок и является чрезвычайно длительным, превосходящим по этому показателю многие другие отрасли. Так, в автомобилестроении на разработку и внедрение нового продукта необходимо около 2 лет, при создании программного обеспечения — всего 1,5 года. Отсюда — необходимость значительных финансовых вложений (Bouckenooghe A., 2004).
|

В оценке затрат на разработку одного препарата (611 млн дол. в 1995 г.) обычно учитывают стоимость успешной разработки 1 ЛС (170 млн дол.) и сопутствующие потери от нереализованных проектов, то есть ЛС, не доведенных до стадии регистрации (441 млн) (Rang P., 2006). По данным Центра по исследованиям и разработке лекарственных средств университета Тафтс (Tufts CSDD) разработка одного препарата в 2000 г. обходилась в среднем в 802 млн дол., в 2003 г. — в 897 млн дол. (DiMasi J.A. et al., 2003). По другим данным в 2000–2002 гг. она составила 1,7 млрд дол. (Gilbert J. et al., 2003). То, что более 70% средств, инвестированных в R&D, потрачены на открытие биологически активных веществ и разработку препаратов, окончившихся неудачами, свидетельствует о необходимости повышения эффективности открытия новых молекул и обеспечения успешности клинических испытаний (КИ) новых ЛС (Rang H.P., 2006).
|


В последнее время создание и выведение на рынок нового ЛС становится не только более дорогим, но и сложным. Регуляторные агентства требуют проведения более длительных КИ с бoльшим количеством участников (DiMasi J.A., 2003). Сегодня препарат, получивший разрешение на маркетинг в США, проходит около 80 исследований с участием свыше 5 тыс. пациентов (Schmidt A., 2005). Кроме того, увеличившееся количество препаратов в продуктопроводе (pipeline) предприятий фармацевтической отрасли не сказывается на числе новых ЛС, получивших одобрение из-за того, что многие продукты сходят с дистанции во время разработки и испытаний.
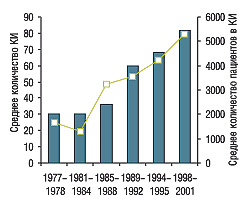
Основные тенденции последних лет в фармацевтической отрасли связаны с увеличением количества исследований и их участников, необходимых для подготовки одной заявки на получение разрешения на маркетинг (New Drug Application – NDA), а также с продолжительностью периода разработки и необходимыми инвестициями (рис. 1). Несмотря на все вышеперечисленное, количество одобренных препаратов с новыми активными фармацевтическими ингредиентами, никогда ранее не получавшими разрешения на маркетинг (новых молекулярных субстанций, new molecular entity — NME), не увеличивается (см. рис. 1).
| Рис. 1. Основные тенденции в фармацевтической отрасли последних лет… (по Schmidt A., «Novartis AG», 2005) | ||||||||
|
|
|||||||
|
|
|||||||
Интересны и другие тенденции последних лет в этой области (PhRMA Survey, 2002; Hindin T.J., 2004; Rang H.P., 2006):
- на фоне резкого повышения стоимости открытия и разработки ЛС прибыль, как правило, начинают получать не ранее, чем через 2 года после лонча;
- уменьшается количество ЛС, получивших разрешение на маркетинг;
- уменьшается доля блокбастеров среди ЛС;
- повышается конкуренция с генериками и me-too* препаратами (например, выведенный на рынок в 1977 г. препарат циметидина (Tagamet) располагал 6-летним периодом рыночной эксклюзивности, тогда как препарат целекоксиба Celebrex (1999) — только 3-месячным);
- растет объем рынка услуг CRO;
- увеличивается количество биотехнологических ЛС (моноклональные антитела, вакцины, цитокины, ферменты и пр.): в 1991–2003 гг. среди зарегистрированных препаратов их было 17%, а в 2002–2003 гг. — уже 30%. В ближайшие годы, как предполагается, их доля увеличится до 50%.
КИ — это тот вид деятельности, который в силу своего общечеловеческого значения всегда вовлекал специалистов и участников из разных стран. Спонсорами КИ помимо фармацевтических компаний все чаще выступает ряд некоммерческих организаций — ВОЗ, Национальный институт здоровья США (National Institutes of Health — NIH), а также частные фонды — Билла и Мелинды Гейтс, «Wellcome Trust» и другие. Хорошо известно, что фармацевтическая отрасль по исследованиям и инновациям движется на Восток и наращивает потенциал в таких странах, как Индия и Китай (INSEAD and Hamilton B.A., 2006). Было бы очень обидно, если бы на этом поприще не смогли вполне реализоваться украинские ученые-исследователи. Пока же проведение первой научно-практической конференции предоставило им возможность обменяться опытом с экспертами ВОЗ, специалистами из России, США, Великобритании, Румынии, Польши, Беларуси, Грузии, Молдовы.
Взаимодействию всех звеньев по обеспечению качества ЛС, одним из которых являются КИ, посвятил свой доклад Виктор Чумак, директор ГФЦ. В Украине за прошедшие годы создана стройная система законодательных и нормативных актов, регулирующих обращение ЛС — сбалансированная правовая пирамида. Благодаря ей обеспечивается доступность препаратов в системе здравоохранения — как физическая (предложение качественных лекарств, наименований которых сегодня на рынке насчитывается 17,8 тыс.), так и экономическая (обеспечивается всей системой здравоохранения и политикой государства в целом: покупательной способностью населения, эффективным использованием имеющихся ресурсов).
ГФЦ через требования к регистрационным документам (что важно в современных условиях — с учетом данных доказательной медицины) влияет на процесс подготовки данных регистрационного досье, то есть на осуществление фармацевтической разработки. При этом обязательно соблюдение требований к активным фармацевтическим ингредиентам для использования при КИ (согласно приложению L к правилам GMP).
Фармацевтическая разработка имеет определяющее значение для того, какими свойствами будет обладать препарат и будут ли они повторяться от серии к серии, — гарантируется это благодаря выполнению требований GMP. На этапе доклинических исследований препарата необходимо соблюдение норм Надлежащей лабораторной практики (GLP). Причем в Украине уже работает сертифицированная на соответствие стандартам GLP лаборатория по доклиническому изучению ЛС, создается экспертный центр по работе в этой области. Наконец, проведение всех КИ, в том числе по изучению биоэквивалентности, должно соответствовать правилам Надлежащей клинической практики (GCP).
Сегодня научно-экспертный совет ГФЦ активно занят решением широкого спектра проблем — от аспектов фармакотоксикологических испытаний до влияния эффекта плацебо на эффективность терапии. Не вызывает сомнений необходимость учета такого фактора, как возраст пациентов, которым предназначен препарат, при его разработке и регистрации. Поскольку испытывают препарат в определенной лекарственной форме и дозировке, не известно, как повлияет на его свойства механическое деление или разрушение лекарственной формы (таблетки, капсулы), — отметил В. Чумак. Задача системы регистрации — принимать во внимание и эти моменты. В настоящее время при составлении инструкций учитывают и аспекты индивидуального взаимодействия ЛС и организма (фармакогеномика), что также требует внимания экспертов.
|
В Украине создана нормативная база, позволяющая регистрировать ЛС в соответствии с самыми высокими мировыми стандартами — в формате общего технического документа (common technical document — CTD). Раздел «фармацевтическая разработка» такого регистрационного досье должен содержать информацию об исследованиях по разработке, проведенных для установления того, что лекарственная форма, состав, производственный процесс, упаковка, микробиологические характеристики и инструкция по медицинскому применению соответствуют цели, указанной в заявке. Это означает, что если разработчик подобрал соответствующую лекарственную форму, содержание вспомогательных веществ и изомерный состав действующего вещества, это практически гарантирует, что генерик будет действовать так же, как его аналог (референтный препарат). В работе системы регистрации очень четко следует разделить те свойства генерического препарата, которые зависят от его разработчика, и те, что изучены компанией-оригинатором и связаны с организмом пациента.
Реальное положение вещей В. Чумак проиллюстрировал рядом примеров. Так, результаты исследований фармакокинетики свидетельствуют, что ее показатели у препаратов нифедипина, представленных на рынке Украины, существенно отличаются. Получается, сколько препаратов — столько и фармакокинетических профилей? Разительные отличия отмечают в скорости высвобождения действующего вещества среди генерических копий препарата Кардикет (изосорбид динитрат в форме таблеток ретард). Почти в 2 раза более быстрое достижение пика концентрации действующего вещества в крови потенциально является причиной повышенной токсичности препаратов. Так же как практически ни один из имеющихся на рынке препаратов диклофенака по своим фармакокинетическим свойствам не соответствует Вольтарену.
В. Чумак отметил, что недостаточно внедрена норма постановления КМУ от 26.05.2005 г. № 376, в соответствии с которой заявитель обязан известить ГФЦ о таких изменениях в производственном процессе:
- замена вспомогательных веществ (таблетки, капсулы);
- замена поставщика субстанции (таблетки, капсулы, суспензии);
- замена основного технологического оборудования;
- замена технологии получения лекарственной формы.
При этом заявителем должна быть представлена в ГФЦ исчерпывающая информация о причинах замены и возможности ее влияния на эффективность, безопасность и качество ЛС и внесены изменения в регистрационные материалы.
|

Уже сегодня, подчеркнул В. Чумак, на сайте ГФЦ представлен проект требований к фармацевтической разработке, и эксперты планируют и дальше работать над методологическим обеспечением этого процесса. Разрабатываются теоретические и практические основы исследований биоэквивалентности методами in vitro, что нашло отражение в нормативных документах (приказы МЗ Украины от 28.06.2005 г. № 426 и от 01.03.2006 г. № 95). Использование такого вида доказательства биоэквивалентности позволяет удешевить разработку препарата, отметил докладчик, тогда как ее подтверждение in vivo, особенно путем сравнительных КИ с обоснованным количеством участников, — намного дороже.
Большие надежды по упорядочиванию работы в этой сфере связывают с деятельностью этических комитетов, которая регламентирована недавно принятым приказом МЗ Украины от 17.07.2006 г. № 485. В частности, следует прояснить целесообразность проведения многих КИ с этической точки зрения. К примеру, ясно, что клинические испытания многих препаратов в форме для внутривенного введения ничего не могут дать ни фармакологу, ни клиницисту. Все актуальные для фармацевтической разработки подобных препаратов вопросы можно решить на доклинической стадии, а проведение КИ с участием пациентов просто неэтично.
Обоснованность проведения тех или иных КИ следует подвергать самой критичной оценке. Например, отечественным производителем освоен выпуск антибактериального средства, проведены соответствующие доклинические и клинические исследования. Но при этом предприятие осуществляет не больше чем фасовку этого препарата. Так что же мы изучаем? Влияние этикетки производителя на эффективность препарата? — с такими словами докладчик обратился к аудитории. Это было бы смешно, если бы не касалось нашей ситуации, — заключил В. Чумак. Если несколько компаний, занимающихся расфасовкой препарата, поставляемого им в форме in bulk, провели его испытания по полной программе, то это может означать, что их целью была вовсе не польза для пациента, а совсем другое, — подчеркнул докладчик.
В разработке дизайна КИ с учетом фармацевтической разработки и технологии получения ЛС приходится решать и куда более сложные вопросы; для проведения такой экспертизы в составе ГФЦ создан отдельный департамент. На основании анализа факторов, которые могут повлиять на эффективность и безопасность препарата, следует формировать перечень критериев включения/исключения пациентов в/из исследование. Еще один важный вопрос: а соответствует ли реальным условиям дальнейшего применения препарата в практике протокол исследования?
Более строго следует подходить к отбору больных. Поскольку все пациенты с точки зрения фармакокинетики отличаются, для проведения настоящих сравнительных КИ выборка должна быть гораздо большей, чем зачастую происходит на практике. Большое внимание сегодня вызывает проблема индивидуальных особенностей взаимодействия препарата с организмом, что наряду с составляющими фармацевтической разработки влияет на концентрацию действующего вещества в плазме крови и, следовательно, терапевтическую эффективность. В зависимости от генетического набора конкретного пациента возможна как повышенная, так и сниженная активность ферментов, отвечающих за биотрансформацию действующего вещества, что может вызвать как снижение терапевтического эффекта, так и развитие побочных эффектов. На малой выборке особенности действия препаратов, связанные с полиморфизмом генов, практически невозможно уловить, так что с точки зрения статистики и доказательности получаемые данные весьма условны. К тому же ряд ЛС вызывает ингибирование или индуцирование активности ферментов, что может сказаться при осуществлении комбинированной терапии.
В завершение своего выступления В. Чумак обратил внимание аудитории на суть прописной (но от этого не менее важной) истины, согласно которой качество и эффективность ЛС являются функцией от принимаемой дозы. На самом деле, не буквально от дозы, а от концентрации в крови, подчеркнул докладчик. Важный аспект: поскольку скорость поступления действующего вещества в кровь зависит от его растворимости, а она, помимо всего прочего, определяется площадью поверхности растворяющегося вещества, увеличение последней ведет к повышению скорости поступления действующего вещества в кровь. Это почти всегда происходит, когда на стадии доклинического изучения нарушают целостность лекарственной формы для приема препарата животными, что нельзя не учитывать разработчикам.
|
Наконец, предметом острых дискуссий являются подходы к подготовке регистрационных материалов препаратов с пролонгированным высвобождением действующего вещества. В таких случаях в состав лекарственной формы входит активный фармацевтический ингредиент в количестве, нередко превышающем минимальный токсикологический уровень. Малейшая ошибка в фармацевтической разработке, — подчеркнул директор ГФЦ, — и вместо эффективного ЛС получится более чем сомнительный с точки зрения безопасности препарат. Такого рода проблемы несомненно должны разрешаться, и сегодня в ГФЦ работают над тем, как наладить преемственность в деятельности фармацевтического департамента и отдела по КИ, отметил В. Чумак. Таким образом, эксперты ГФЦ обладают четким видением того, какие проблемы и в какой последовательности следует решать.
|
Возросшая роль результатов КИ в системе доказательной медицины стала основополагающей в оценке обществом места и значимости новых ЛС, — отметил, выступая на пленарном заседании, Владимир Мальцев, профессор, руководитель отдела координации и контроля клинических испытаний лекарственных средств ГФЦ. Шведский совет по технологии оценки в здравоохранении (Swedish Council on Technology Assessment in Health Care — SBU) и Центр доказательной медицины в Оксфорде (Oxford-Centre for Evidence-Based Medicine) при последовательной поддержке ВОЗ заявили о приоритете рандомизированных двойных слепых КИ среди всех других данных доказательной медицины. Высокой достоверностью (уровень 1А), как указывает оксфордский центр, обладают заключения, основанные на результатах нескольких независимых КИ с совпадением результатов, обобщенных в систематических обзорах. В то же время самым низким уровнем доказательности характеризуются утверждения, базирующиеся на мнении экспертов при отсутствии данных КИ.
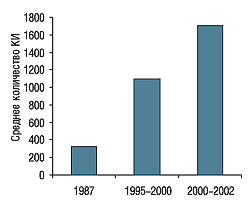
Известно, что проведение КИ — длительный и дорогостоящий процесс, требующий привлечения сотен и тысяч участников (рис. 2). Все компании в настоящее время стремятся к быстрейшему проведению КИ, поскольку ускорение разработки препарата на 1 день экономит 37 тыс. дол. и обеспечивает дополнительный объем продаж составляющий 1,1–1,3 млн. Согласно данным Tufts CSDD компаниями, которые быстрее других разрабатывали лекарственные препараты в 2000–2005 г., были «Bayer AG», «AstraZeneca», «Boehringer-Ingelheim», «Merk&Co». Десятка лидеров по этому показателю уменьшила средний срок разработки ЛС на 20% (с 66,5 мес в 1994–1999 гг. до 53 мес в 2000–2005гг.) (www.centerwatch.org; www.csdd.tufts.edu).
|
|
||||||||
|
|
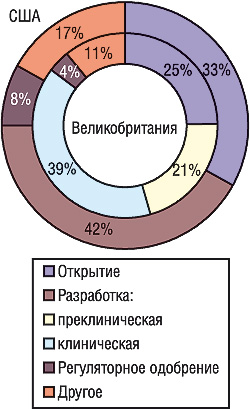
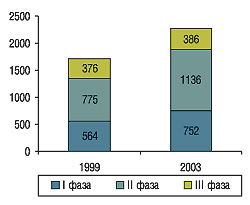
Итак, в разработке новых ЛС главная роль принадлежит КИ. Биофармацевтические компании в 2003 г. потратили на исследования и разработки 36 млрд дол., тогда как в 1982 г. — 5 млрд дол. Через 10 лет каждая из 10 крупнейших компаний расходовала на эти цели в среднем по 2 млрд дол. в год. Сегодня в исследования и разработки реинвестируется 15–20% объема продаж крупнейших компаний — больше, чем в любой другой отрасли. Распределение этих средств представлено на рис. 3.
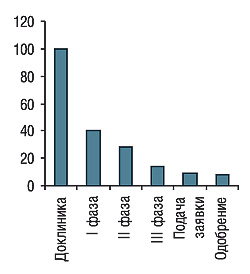
Несмотря на увеличение количества КИ, выводимых на рынок в течение года новых препаратов не становится больше. Во многом это связано с проблемами, возникающими на ранних стадиях разработки, — трудностями оценки значимости результатов, полученных при изучении клеточных, тканевых моделей и животных. Из-за невозможности экстраполяции доклинических данных на людей многие NME не выводятся на рынок. В онкологии 95% разработанных новых молекулярных субстанций (NME) не достигают стадии лонча (Cancer Research UK, 2005). Среди всех препаратов, по данным литературы, успешность разработки не превышает 10%. Так, из каждых 10 тыс. отобранных при скрининге и синтезированных молекул только 50 признают «потенциально лекарственными» и продолжают испытывать in vitro и in vivo, о чем свидетельствуют данные Европейской федерации ассоциаций фармацевтических производителей (European Federation of Pharmaceutical Industries and Associations — EFPIA). Только 10 из них испытывают в КИ I фазы, 3 — II и только 1 становится действующим веществом нового препарата, получившего разрешение на маркетинг (рис. 4).
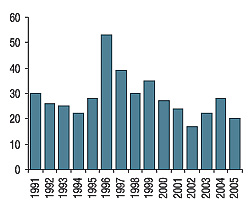
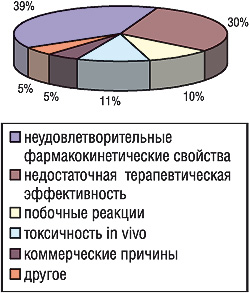
Основными причинами того, что препараты сходят с дистанции, являются неудовлетворительные фармакокинетические свойства (39%) и недостаточная терапевтическая эффективность (30%) (рис. 5). Так, в 2005г. на рынок США было выведено наименьшее количество новых молекулярных субстанций, что связывают с замедлением процесса получения разрешения на маркетинг препаратов.
Испытывая необходимость привлечения к участию в исследованиях все большего количества пациентов, компании тоже сталкиваются с трудностями. Среди факторов, обусловливающих нежелание участвовать в КИ, такие (Taylor S., Leitman R., 2001):
- убеждение, что общепринятая терапия обеспечивает лучшее лечение, чем новые подходы (37%);
- опасение получить плацебо вместо лекарственного средства (31%);
- синдром «морской свинки» («подопытного кролика») (22%);
- необходимость перемещаться к месту проведения КИ (21%) и другие.
Вместе с тем европейцы в целом лояльно относятся к участию в исследованиях: 68% опрошенных готовы согласиться на участие в КИ (2004 International Will&Why Survey; BBK Healthcare по Smit-Marshall P., 2006). Почти 2/3 из них в числе факторов, влияющих на принятие такого решения, называют «доступ к современным научным достижениям». Другие наиболее часто упоминаемые мотивы включают:
- возможность заработать деньги (58%);
- помочь другим людям (57%);
- получить лучшее лечение для себя (48%);
- ускорить свой доступ к лечению (34%).
В 1977 г. FDA предложила правила, определяющие обязанности исследователей и спонсоров, которые вскоре в виде системы Надлежащей клинической практики (Good Clinical Practice — GCP) были внедрены в практику. Европейский Союз приступил к этой работе в 1990 г.
GCP — это стандарт планирования, проведения, выполнения, мониторинга, аудита и документального оформления КИ, а также обработки и представления их результатов, который служит для общества гарантией достоверности полученных данных и защищенности прав, здоровья и анонимности испытуемых. То есть основная ответственность за соблюдение GCP лежит на спонсоре, и для ее внедрения далеко не достаточно обеспечения оборудованием клинических баз, как думают многие.
|
Несмотря на определенные объективные трудности становления КИ ЛС в Украине, за прошедшие 10 лет создана стройная система взаимодействия всех структур, вовлеченных в КИ, отметил В. Мальцев. Еще в 1996г. в составе фармакологического комитета МЗ Украины было создано подразделение, занимающееся вопросами организации и проведения клинических испытаний. Тогда же начаты разработка и внедрение соответствующей нормативной базы и принципов GCP. В последние 5–6 лет разработанные документы активно усовершенствуются. Например, в этом году, отметил докладчик, впервые, с участием отечественных и зарубежных заказчиков испытаний, был разработан перечень лечебно-профилактических учреждений, в которых могут проводиться КИ ЛС, утвержденный приказом МЗ Украины от 11.08.2006 г., № 560. Активно в нашей стране проводят инспекционные проверки (плановая, ретроспективная и целенаправленная). Всего с 1999 г. таких проверок проведено 299,6% из них завершились остановкой испытаний. Это довольно значительная доля, подчеркнул В. Мальцев, что свидетельствует о наличии определенных недостатков в организации работы в этой области. При 396 клинических базах, находящихся в 204 лечебно-профилактических учреждениях, работают этические комиссии.
|

Сегодня в ЕС каждое из государств-членов должно обеспечить вынесение единого заключения, о котором необходимо проинформировать локальные ЭК при клинических базах. Такая же практика сегодня применяется и в Украине, и при проведении многоцентровых КИ заявитель обращается в Центральную комиссию по вопросам этики. Вместе с тем, если испытание было одобрено Центральной этической комиссией, то при наличии действующей локальной комиссии ответственный исследователь информирует ее, хотя она и обязана признать заключение Центральной. Активно проводится мониторинг побочных реакций при проведении исследований. К счастью, за весь период наблюдения произошла остановка только одного исследования в связи с выявлением серьезных побочных реакций — в 2002 г.
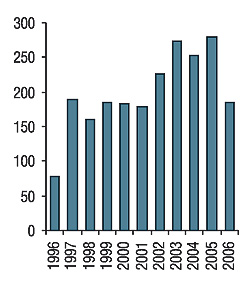
Одним из итогов работы ГФЦ, отметил В. Мальцев, является количество проведенных исследований (рис. 6). Из 2190 испытаний, проведенных за последние 10 лет, преобладают сравнительные КИ по сокращенной программе ЛС отечественных производителей (55%) и международные многоцентровые (25%). Исследования по сокращенной программе препаратов зарубежных производителей составляют 14% проводившихся испытаний.
Увеличивается количество международных многоцентровых исследований: с 36 в 2000 г. до 125 в 2005 г. Их заказчиками в большинстве случаев являются контрактные исследовательские организации (КИО) (90% всех КИ), реже — представительства фармацевтических компаний (10% КИ, отмечается повышение активности). За весь этот период в роли спонсоров выступили 33 КИО (лидеры — «Quintiles» (18%), «PSI» (9%), «Parexel» (8%), «ICON» (5%), «Verum» (3%) и «DHU» (3%)) и 11 фармацевтических компаний. Сегодня насчитывается почти 400 клинических баз, утвержденных приказом МЗ Украины № 560, и работают там более 1500 исследователей. Если ЛПУ не входит в утвержденный МЗ перечень, КИ в нем возможно в случае позитивных выводов ГФЦ по результатам экспертизы, подчеркнул В. Мальцев.
В заключение докладчик обратил внимание на серьезную научно-методическую и образовательную работу, которая проводится ГФЦ, в том числе по вопросам организации и проведения клинических испытаний. Так, в ежегодных семинарах по этим вопросам приняли участие более 2000 исследователей, представителей отечественных и зарубежных заказчиков КИ. n
Продолжение следует.
*Оригинальный препарат, не имеющий существенной терапевтической ценности по сравнению с созданными ранее.
|
Дарья Полякова, фото Игоря Кривинского
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим