|
|

Развитие фармацевтического рынка не только в Украине, но и в мире происходит в значительной степени за счет генерических лекарственных препаратов. Проблеме регуляции оборота этих препаратов посвящено множество публикаций. Однако подходы к решению вопроса о необходимости проведения клинических испытаний для оценки их эффективности и безопасности определены еще не полностью и остаются дискутабельными. В совершенствовании этих подходов заинтересованы как регуляторные органы, так и владельцы регистрационных свидетельств (торговых лицензий).
В последние годы для установления методологии и организации проведения клинических испытаний генерических препаратов принят ряд руководств, на которые мы ссылаемся в данной публикации.
Инновационный препарат. ВОЗ: препарат, впервые получивший разрешение на маркетинг (как правило, патентованный препарат) на основании документов, подтверждающих его эффективность, безопасность, качество (в соответствии с требованиями, установленными на момент получения разрешения). Если лекарственное вещество присутствует на рынке в течение многих лет, определить его оригинальный препарат не всегда представляется возможным [1]. ЕМЕА: лекарственный препарат, разрешенный к использованию на рынке на основании полного досье (включающего химические, биологические, фармацевтические, фармакологические, токсикологические и клинические данные). «Препарат сравнения» должен быть «инновационным препаратом» [2].
Генерические препараты. ВОЗ: термин генерический препарат может иметь различные значения (в зависимости от особенностей законодательства). Поэтому его употребления стараются избегать, используя термин воспроизведенные (непатентованные, многоисточниковые) лекарственные препараты. Генерические препараты могут реализовываться либо под общепринятыми непатентованными, либо под новыми торговыми (патентованными) названиями. Они могут поступать на рынок в лекарственной форме и/или с содержанием активного вещества, отличающимися от таковых инновационных препаратов. Если термин генерический препарат все же употребляется, то подразумевают лекарственный препарат, используемый в медицинской практике взаимозаменяемо с инновационным (оригинальным) препаратом, производящийся, как правило, без лицензии от компании-разработчика и реализуемый после истечения срока действия патента или других исключительных прав [1]. ЕМЕА: лекарственный препарат является по существу аналогичным оригинальному препарату, если он соответствует критериям одного и того же количественного и качественного состава в отношении активных субстанций, имеет одну и ту же лекарственную форму и является биоэквивалентным. Исключение составляют те препараты, для которых в свете научных знаний очевидно, что они отличаются от оригинального препарата в отношении безопасности и эффективности [2].
Фармацевтическая эквивалентность. ЕМЕА: лекарственные препараты являются фармацевтически эквивалентными, если они содержат одинаковое количество одной и той же активной субстанции (субстанций) в одних и тех же лекарственных формах, которые соответствуют требованиям одних и тех же или сопоставимых стандартов [2]. Определение ВОЗ аналогично по сути, но еще содержит указание: «предназначены для введения тем же путем» [1]. ЕМЕА: фармацевтическая эквивалентность не обязательно подразумевает биоэквивалентность, поскольку различия во вспомогательных веществах и/или процессах производства могут привести к более быстрому или более медленному растворению и/или всасыванию [2].
Взаимозаменяемые лекарственные средства ВОЗ: взаимозаменяемое лекарственное средство?— препарат, терапевтически эквивалентный препарату сравнения (тому, с которым проводится сравнение) [1].
Далее мы приводим определения EMEA, которые по сути аналогичны определениям ВОЗ.
Биодоступность. Скорость и степень, с которой активная субстанция или ее активная часть всасывается из лекарственной формы и становится доступной в месте действия [2].
Биоэквивалентность. Два препарата считаются биоэквивалентными, если они фармацевтически эквивалентны или альтернативны, и биодоступности этих препаратов после введения в одной и той же молярной дозе являются одинаковыми в такой степени, что их эффекты в отношении эффективности и безопасности практически одинаковы [2].
Терапевтическая эквивалентность. Лекарственный препарат является терапевтически эквивалентным другому препарату, если он содержит ту же активную субстанцию или терапевтически активный компонент и клинически проявляет такую же эффективность и безопасность, как и препарат, эффективность и безопасность которого установлены [2].
Многоисточниковые (генерические) продукты и взаимозаменяемость. Согласно требованиям ВОЗ, все фармацевтические продукты, включая генерические препараты, могут применяться только после одобрения уполномоченным органом по регулированию лекарственных средств. Уполномоченными органами при оценке генерических фармацевтических продуктов, точно так же, как и оригинальных препаратов, должны применяться требования, относящиеся к:
• производству (выполнению требований GMP — Надлежащей производственной практики) * и контролю качества;
• характеристикам продукта и маркировке;
• терапевтической эквивалентности.
Обычно при оценке клинической эквивалентности требуется проведение исследований in vivo или предоставляется подтверждение, что в отдельном случае нет необходимости в проведении такого исследования. Виды исследований, проводимых in vivo, включают исследования биоэквивалентности, фармакодинамические исследования и сравнительные клинические испытания [1].
Доказательство биоэквивалентности является, как правило, наиболее подходящим методом доказательства терапевтической эквивалентности лекарственных препаратов, являющихся фармацевтически эквивалентными или фармацевтически альтернативными, при условии, что такие препараты содержат вспомогательные вещества, не оказывающие влияния на безопасность и эффективность и соответствующие требованиям к маркировке в отношении вспомогательных веществ. Исследование биоэквивалентности является по существу сравнительным изучением биодоступности, предназначенным для установления эквивалентности между тестируемым препаратом и препаратом сравнения [2]. В документах [1–4] изложен международный, а в [5–8] — украинский регламенты изучения биоэквивалентности.
Обоснование отказа от исследований биодоступности и биоэквивалентности in vivo. Подробному освещению этого вопроса посвящены отдельные документы, один из которых издан ВОЗ [3], другой — FDA [4].
Обоснование отказа, изложенное в этих документах, основано на биофармацевтической классификационной системе (БКС), предназначенной для классификации препаратов (лекарственных субстанций) на основе их растворения в воде и степени проникновения через стенку кишечника. Если дополнительно применяется такой критерий, как растворение лекарственного препарата, то БКС охватывает все основные факторы, определяющие степень и скорость абсорбции лекарственного средства из твердой лекарственной формы немедленного высвобождения. Согласно БКС, лекарственные субстанции характеризуются следующим образом:
Класс 1 — высокая растворимость, высокая степень проникновения
Класс 2 — низкая растворимость, высокая степень проникновения
Класс 3 — высокая растворимость, низкая степень проникновения
Класс 4 — низкая растворимость, низкая степень проникновения
Растворимость. Активный фармацевтический ингредиент считается высокорастворимым, если его самое большое количество в лекарственной форме (сила действия), рекомендованное ВОЗ (когда оно указано в примерном перечне основных лекарственных средств ВОЗ), или самое большое количество действующего вещества в препарате (имеющемся на рынке) растворяется в 250 мл или менее воды при рН от 1,2 до 6,8 [3].
Степень проникновения. Активный фармацевтический ингредиент считается высокопроницаемым, если объем его абсорбции у человека составляет 85% (согласно первым рекомендациям по БКС — 90%) или более на основании определения баланса масс или по сравнению с вводимой внутривенно дозой сравнения.
Отнесение лекарственной субстанции к тому или другому классу БКС позволяет решить вопрос о необходимости проведения исследований биоэквивалентности и биодоступности in vivo [3].
Класс 1 — от изучения биоэквивалентности in vivo можно отказаться
Класс 2 — от изучения биоэквивалентности in vivo можно отказаться при определенных условиях (если активное вещество — это слабая кислота и многоисточниковый препарат растворяется в количестве 85% или более при pH 6,8 за 30 мин или менее, а его профиль растворения подобен профилю референтного препарата при pH 1,2; 4,5; 6,8 при обычном тесте «растворение»)
Класс 3 — от изучения биоэквивалентности можно отказаться при определенных условиях (если многоисточниковый и референтный препараты — очень быстро растворимые — 85% или более в течение 15 мин или меньше при pH 1,2; 4,5; 6,8)
Класс 4 — исследования биоэквивалентности проводятся всегда.
Согласно требованиям ЕМЕА [2], исследования биоэквивалентности необходимы для следующих лекарственных форм:
а) немедленного высвобождения с системным действием для перорального применения (таблетки, капсулы, суспензии для перорального применения). Для обоснования отказа от таких исследований должны быть представлены необходимые данные**. В тех случаях, когда раствор для перорального применения тестируется в сравнении с лекарственной формой немедленного высвобождения для перорального применения, требуется сравнительное исследование биоэквивалентности, если только не может быть обосновано его отсутствие**;
б) немедленного высвобождения не для перорального применения с системным действием
в) местного применения с системным действием;
г) модифицированного высвобождения и трансдермальные. Требования по исследованиям биоэквивалентности согласно специальному руководству;
д) фиксированные комбинации лекарственных средств. Комбинации лекарственных средств обычно следует оценивать в отношении биодоступности и биоэквивалентности отдельных активных субстанций раздельно (в случае новой комбинации) или как существующую комбинацию. Критерии** следует применять в отношении отдельных компонентов. Исследование новой комбинации необходимо проводить таким образом, чтобы можно было определить возможность межлекарственных взаимодействий, сказывающихся на фармакокинетике отдельных компонентов;
е) местного применения (для перорального, назального, ингаляционного, глазного, накожного, ректального, вагинального и др. путей введения), предназначенных для действия без системного всасывания, определение биоэквивалентности на основании системных измерений не применяется, и в принципе требуются фармакодинамические или сравнительные клинические исследования. Их отсутствие должно быть обосновано. В тех случаях, если системное воздействие, вызванное местным применением местнодействующего лекарственного средства, влечет за собой риск системных побочных действий, системное действие должно быть оценено.
Положения ВОЗ [1] практически те же, но в них отдельно выделяют случаи, когда применяют тест растворения in vitro:
а) Лекарственные средства, для которых не требуются исследования in vivo (см. выше);
б) Различное содержание активных веществ в составе лекарственных форм многоисточниковых препаратов, если лекарственные средства производятся одним и тем же производителем на одном и том же предприятии, если:
• качественный состав в препаратах с разным содержанием активных веществ практически одинаков;
• отношение активных ингредиентов и вспомогательных веществ в препаратах с разным содержанием активных веществ практически одинаково, или, в случае препаратов с разным содержанием активных веществ, соотношение между вспомогательными веществами одинаково;
• выполнено соответствующее исследование эквивалентности по крайней мере одного препарата с одним содержанием активного вещества из числа составов лекарственной формы с разным содержанием активного вещества;
• в случае, если фармакокинетика в отношении системной доступности имеет линейный характер в пределах диапазона терапевтических доз.
Согласно руководству ВОЗ [1], исследования эквивалентности не являются необходимыми для лекарственных форм, которые:
а) применяются парентерально в виде водного раствора и содержат ту(те) же активную(ые) субстанцию(и) в той же концентрации и те же вспомогательные вещества в сравнимых концентрациях;
б) являются растворами для перорального применения, содержащими активную субстанцию в той же концентрации и не содержащими вспомогательное вещество, которое предположительно может нарушать прохождение активной субстанции через пищеварительный тракт или ее абсорбцию;
в) являются газом;
г) являются порошком для приготовления раствора, и раствор соответствует критерию (а) или критерию (б);
д) являются ушными/глазными лекарственными средствами в виде водного раствора, содержащего те же активные вещества в тех же концентрациях и практически те же вспомогательные вещества в сравнимых концентрациях;
е) являются препаратами для местного применения в виде водных растворов и содержат те же активные вещества в тех же концентрациях и практически те же вспомогательные вещества в сравнимых концентрациях;
ж) являются препаратами для ингаляционного применения или назальными спреями, тестируемыми в отношении введения посредством практически одинаковых устройств или без них, приготовленными как водные растворы, содержащие те же активные вещества в тех же концентрациях и практически те же вспомогательные вещества в сравнимых концентрациях.
Положения п. а–в соответствуют требованиям руководства ЕМЕА [2].
Необходимость проведения испытаний генерических препаратов. Согласно статье 8(3) (і) Директивы 2001/83/ЕС, в общем случае заявка на получение торговой лицензии сопровождается результатами таких испытаний [9]:
• физико-химических, биологических или микробиологических;
• токсикологических и фармакологических;
• клинических.
Статья 10(1) этой же Директивы в порядке ограничения статьи 8(3) (і) не требует от заявителя предоставления результатов токсикологических, фармакологических и/или клинических испытаний, если он может представить одно из доказательств:
• во-первых, лекарственный препарат является по существу аналогичным лекарственному препарату, уже лицензированному в государстве — члене ЕС, в котором подается заявка, и владелец торговой лицензии на оригинальный лекарственный препарат согласен с тем, что при изучении заявки на аналогичный лекарственный препарат будут использованы данные фармакологических, токсикологических и/или клинических исследований, содержащиеся в лицензионной документации на оригинальный лекарственный препарат;
• во-вторых, медицинское применение вещества или веществ, входящих в состав лекарственного препарата, хорошо изучено, признаны их эффективность и удовлетворительная степень безопасности (посредством предоставления подробных библиографических ссылок на опубликованные научные данные);
• в-третьих, лекарственный препарат является по существу аналогичным лекарственному препарату, который был лицензирован в Сообществе не менее чем за 6 лет до этого и находится в продаже в том государстве – члене ЕС, куда подается заявка. Указанный минимальный срок лицензирования может быть увеличен до 10 лет в отношении тех лекарственных препаратов, которые получены методами высоких технологий.
Если лекарственный препарат — новая комбинация известных ингредиентов:
• необходимо предоставить результаты токсикологических, фармакологических или клинических испытаний, относящихся к этому сочетанию;
• нет необходимости приводить ссылки, относящиеся к каждому ингредиенту в отдельности.
Установление эффективности и безопасности препаратов на основании результатов клинических испытаний. В части 4 Приложения 1 той же Директивы в разделе «Клиническая эффективность и безопасность» отмечается, в частности, что
• клинические испытания проводятся в виде контролируемых клинических испытаний и, если возможно, рандомизированных;
• в протоколе испытаний следует тщательно описать статистические методы, которые будут применяться, документировать меры, принятые во избежание систематической ошибки, особенно методы рандомизации;
• в экспертном отчете необходимо обсудить, в какой мере различные испытания позволяют оценить безопасность, а также значимость методов ее оценки;
• если клинические отчеты по эффективности и безопасности лекарственного препарата недостаточно обоснованы с научной точки зрения, они не могут быть приняты в качестве достоверных доказательств;
• значимость данных об эффективности и безопасности лекарственного препарата возрастает, если они получены несколькими независимо работающими компетентными исследователями.
|
|
О проведении клинических испытаний генерических препаратов в Украине
Согласно приказу МЗ Украины № 281 от 01.11.2000, для проведения клинического испытания после определения клинических баз заказчик подает в ГП «Государственный фармакологический центр» МЗ Украины следующие материалы:
• Протокол клинического испытания лекарственного средства
• Брошюру исследователя или Инструкцию для медицинского применения лекарственного средства
• Информацию для исследуемого и/или форму информированного письменного согласия
• Индивидуальную регистрационную форму (в случае необходимости).
Наиболее часто применяемыми дизайнами клинических испытаний генерических препаратов являются следующие:
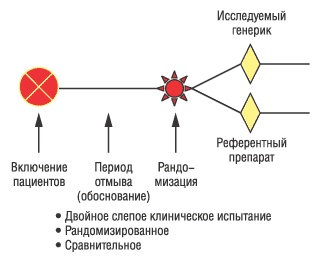
•?Параллельный дизайн клинического испытания (рис. 1). Такой вид «параллельной» модели считается наиболее оптимальным для определения эффектов лечения и формулирования выводов на основе полученных результатов
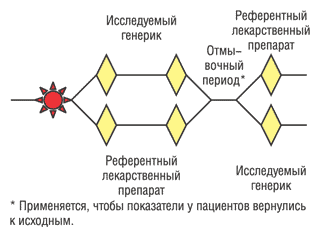
•?Перекрестный дизайн испытания (рис. 2). В отличие от планов исследований в параллельных группах, «перекрестные» модели позволяют оценить эффекты как изучаемых лекарственных препаратов, так и сравнительных курсов лечения у одних и тех же испытуемых. Может использоваться, например, для сравнения анальгетиков при лечении пациентов с хронической болью.
Заключение. В последние годы мировым сообществом были предприняты серьезные шаги по интеграции в области регуляторных требований к регистрации лекарственных препаратов, в том числе — формирование единых требований к организации, объему и структуре клинических испытаний. Подходы к исследованию генерических препаратов отличаются от таковых, применяемых в отношении новых лекарственных препаратов. Эти дифференцированные подходы в настоящее время на этапе обсуждения и пересмотра.
В Украине среди лекарственных препаратов, направленных на клинические испытания, преобладают генерические (89% общего количества препаратов, изученных за период 01.2003–11.2004). Это накладывает определенные обязательства на всех участников этого процесса, прежде всего, на заказчиков (спонсоров) клинических испытаний. По нашему мнению, пришло время создания при крупных отечественных предприятиях отделов по организации и контролю (мониторинг и аудит) за проведением клинических испытаний с обязательным выделением сотрудников, отвечающих за мониторинг побочных реакций как при медицинском применении лекарственных препаратов, так и при проведении клинических испытаний. n
|
ЛИТЕРАТУРА |
|
|
* Принципы и правила GMP лекарственных средств, в том числе исследуемых лекарственных средств, изложены в Директиве 2003/94/ЕС.
** Для обоснования освобождения лекарственного препарата от исследования биоэквивалентности in vivo прежде всего принимаются во внимание критерии, основанные на БКС, то есть высокая растворимость, высокая степень проникновения у активной субстанции и высокая степень растворения лекарственного продукта. Также принимаются во внимание следующие характеристики:
а) относящиеся к активной субстанции:
• риск «терапевтической неудачи» или побочных реакций на лекарственное средство;
• риск бионеэквивалентности;
• растворимость: если для активной субстанции характерна высокая растворимость в воде, продукт вообще может быть освобожден от исследований биоэквивалентности, за исключением того, если, с учетом других характеристик, освобождение может повлечь потенциальный риск;
• фармакокинетические свойства: линейная или полная абсорбция свидетельствует о высокой проницаемости и уменьшает возможность снижения биодоступности.
б) характеристики, относящиеся к лекарственному продукту:
• быстрое растворение: в случае освобождения от исследований биоэквивалентности, данные in vitro должны демонстрировать похожие профили растворения у тестируемого препарата и препарата сравнения в каждом из трех буферных растворов в диапазоне рН 1–8 при 37 °С (предпочтительно с рН, составляющими 1,0; 4,6; 6,8). В случаях, когда свыше 85% активной субстанции растворяется в течение 15 мин, аналогичность профилей растворимости может считаться доказанной;
• вспомогательные вещества: вещества, входящие в состав лекарственного препарата, хорошо известны и их воздействие на фармакокинетику активной субстанции не ожидается. При использовании необычно большого количества известного вспомогательного вещества или нового вспомогательного вещества должна быть предоставлена дополнительная документация;
• производство: метод производства готовой продукции в отношении критических физико-химических свойств активной субстанции (например, размеры частиц, полиморфизм) должны быть адекватно приняты во внимание и внесены в документацию при разработке фармацевтического раздела досье.
Владимир Мальцев, ГП «Государственный фармакологический центр» МЗ Украины
Фото Игоря Кривинского
ТРЕБОВАНИЯ К ПРОИЗВОДСТВУ И МАРКИРОВКЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ КЛИНИЧЕСКИХ ИСПЫТАНИЙ
|
ДИРЕКТИВА КОМИССИИ 2003/94/ЕС
ОТ 8 ОКТЯБРЯ 2003 г.
ОБ УСТАНОВЛЕНИИ ОСНОВНЫХ ПРИНЦИПОВ И ПРАВИЛ НАДЛЕЖАЩЕЙ ПРОИЗВОДСТВЕННОЙ ПРАКТИКИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ ЧЕЛОВЕКА И ИССЛЕДУЕМЫХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ ЧЕЛОВЕКА**
КОМИССИЯ ЕВРОПЕЙСКИХ СООБЩЕСТВ,
принимая во внимание Договор об образовании Европейского сообщества,
принимая во внимание Директиву 2001/83/ЕС Европейского Парламента и Совета от 6 ноября 2001 г. о своде законов Сообщества в отношении лекарственных препаратов для человека (?1), в последней редакции, с поправками, внесенными Директивой Комиссии 2003/63ЕС (?2), и, в частности, статью 47 Директивы,
поскольку:
(1) Все лекарственные препараты для человека, произведенные в странах Сообщества или импортируемые, включая лекарственные препараты, предназначенные для экспорта, должны быть произведены в соответствии с принципами и правилами Надлежащей производственной практики.
(2) Эти принципы и правила установлены Директивой Комиссии 91/356/ЕЕС от 13 июля 1991 г. об установлении принципов Надлежащей производственной практики лекарственных средств для человека (?3).
(3) Статья 13(3) Директивы 2001/20/ЕС Европейского Парламента и Совета от 4 апреля 2001 г. о сближении законов, правил и административных норм государств-членов в отношении выполнения Надлежащей клинической практики при проведении клинических испытаний лекарственных препаратов для применения у человека (?4) требует составления в соответствии с руководством по Надлежащей производственной практике, подробного руководства по аспектам, которые необходимо учитывать при оценке продукции в целях выпуска серий на территории Сообщества.
(4) Следовательно, необходимо расширить и адаптировать положения Директивы 91/356/ЕЕС для того, чтобы охватить требованиями Надлежащей производственной практики исследуемые лекарственные препараты.
(5) В связи с тем, что большинство положений Директивы 91/356/ЕЕС должно быть исправлено, то ради ясности эту Директиву необходимо заменить.
(6) Для обеспечения соблюдения принципов и правил Надлежащей производственной практики необходимо установить подробные положения в отношении инспекций, проводимых уполномоченными органами, и определенных обязанностей производителя.
(7) Все производители должны использовать эффективную систему управления качеством своих производственных операций, для чего требуется внедрение системы обеспечения качества лекарственных средств.
(8) Принципы и правила Надлежащей производственной практики должны быть установлены в отношении управления качеством, персонала, помещений и оборудования, документации, технологического процесса, работ по контракту, рекламаций и отзыва продукции, а также самоинспекции.
(9) Для защиты людей, участвующих в клинических испытаниях, и для гарантии отслеживаемости исследуемых лекарственных препаратов необходимо установить особые требования к маркировке этих препаратов.
(10) Меры, которые предусмотрены в данной Директиве, соответствуют мнению Постоянного Комитета по лекарственным средствам для человека, учрежденного согласно статье 121 Директивы 2001/83/ЕС,
ПРИНЯЛА СЛЕДУЮЩУЮ ДИРЕКТИВУ:
Статья 1
СФЕРА ДЕЙСТВИЯ
Настоящая Директива определяет принципы и правила Надлежащей производственной практики лекарственных препаратов для человека, производство которых требует лицензирования согласно статье 40 Директивы 2001/83/ЕС и в отношении исследуемых лекарственных средств для человека, производство которых требует лицензирования согласно статье 13 Директивы 2001/20/ЕС.
Статья 2
ОПРЕДЕЛЕНИЯ
В настоящей Директиве используются следующие определения:
1. ” Лекарственный препарат” (medicinal product) – любой продукт, ссответствующий определению, приведенному в статье 1(2) Директивы 2001/83/ЕС;
2. “Исследуемый лекарственный препарат” (investigational medicinal product) – любой продукт, соответствующий определению, приведенному в статье 2(d) Директивы 2001/20/ЕС;
3. “Производитель” (manufacturer) – любое лицо, занимающееся любыми видами деятельности, для которых необходима лицензия, согласно статье 40(1) и (3) Директивы 2001/83/ЕС или лицензия, согласно статье 13(1) Директивы 2001/20/ЕС;
4. “Уполномоченное лицо” (qualified person) – должностное лицо, предусматриваемое статьей 48 Директивы 2001/83/ЕС или статьей 13(2) Директивы 2001/20/ЕС;
5. “Обеспечение качества лекарств” (pharmaceutical quality assurance) – совокупность всех организационных мероприятий, предпринятых в целях обеспечения того, что лекарственные препараты или исследуемые лекарственные препараты удовлетворяют требованиям качества, определяемым их предназначением;
6. “Надлежащая производственная практика” (good manufacturer practice) – часть мероприятий по обеспечению качества, гарантирующих, что продукцию постоянно производят и контролируют по стандартам качества, соответствующим ее предназначению;
7. “Кодирование” (blinding) – намеренное сокрытие названия исследуемого лекарственного средства в соответствии с инструкциями спонсора;
8. “Раскодирование” (unblinding) – раскрытие названия закодированного продукта.
Статья 3
ИНСПЕКЦИИ
1. Посредством периодических инспекций согласно статье 111(1) Директивы 2001/83/ЕС и посредством инспекций, указанных в статье 15(1) Директивы 2001/20/ЕС, государства – члены ЕС должны обеспечить соблюдение производителями принципов и правил Надлежащей производственной практики, установленных настоящей Директивой. Государства – члены ЕС должны также принять во внимание публикуемый Комиссией сборник по процедурам Сообщества в отношении инспекций и обмена информацией.
2. Для интерпретации принципов и правил Надлежащей производственной практики производители и представители уполномоченных органов должны обратиться к детализированным правилам, предусмотренным во втором абзаце статьи 47 Директивы 2001/83/ЕС. Эти правила опубликованы Комиссией в “Руководстве по Надлежащей производственной практике лекарственных препаратов и исследуемых лекарственных препаратов”.
Статья 4
СООТВЕТСТВИЕ НОРМАМ НАДЛЕЖАЩЕЙ ПРОИЗВОДСТВЕННОЙ
ПРАКТИКИ
1. Производитель должен обеспечивать проведение всех производственных операций в соответствии с нормами Надлежащей производственной практики и лицензией на производство. Это положение также должно применяться к лекарственным препаратам, предназначенным только для экспорта.
2. В отношении лекарственных препаратов и исследуемых лекарственных препаратов, импортируемых из третьих стран, импортер должен гарантировать, что продукция произведена в соответствии со стандартами Надлежащей производственной практики, требования которых по крайней мере не ниже установленных Сообществом.
Кроме того, импортер лекарственных препаратов должен гарантировать, что эти продукты произведены производителями, которые должным образом лицензированы для подобной деятельности. Импортер исследуемых лекарственных препаратов должен гарантировать, что продукция была произведена тем производителем, о котором уведомлены уполномоченные органы и который признан ими для выполнения этой задачи.
Статья 5
ЛИЦЕНЗИРОВАНИЕ
1. Производитель должен гарантировать, что все производственные операции в отношении лекарственных препаратов, для которых необходима торговая лицензия***, выполняются в соответствии с той информацией, которая представлена в заявке на получение торговой лицензии, принятой компетентными уполномоченными органами.
В случае исследуемых лекарственных препаратов производитель должен гарантировать, что все производственные операции осуществляются в соответствии с информацией, содержащейся в заявке, поданной спонсором в уполномоченные органы в соответствии со статьей 9(2) Директивы 2001/20/ЕС.
2. Производитель должен регулярно совершенствовать методы производства в соответствии с достижениями научно-технического прогресса и разработкой исследуемого лекарственного средства.
Если необходимо внесение изменений в лицензионное досье или поправки в заявку, указанную в статье 9(2) Директивы 2001/20/ЕС, то заявка на изменения должна быть подана в уполномоченные органы.
Статья 6
СИСТЕМА ОБЕСПЕЧЕНИЯ КАЧЕСТВА
Производитель должен создать и внедрить эффективную систему обеспечения качества лекарств при активном участии администрации и персонала различных задействованных служб.
Статья 7
ПЕРСОНАЛ
1. В целях обеспечения качества лекарств производитель на каждом производственном участке должен иметь достаточное количество компетентного и квалифицированного персонала.
2. Обязанности управляющего и контролирующего персонала, включая Уполномоченных Лиц, ответственных за внедрение и выполнение стандартов Надлежащей производственной практики, должны быть изложены в должностных инструкциях. Иерархические отношения должны быть отражены в организационной схеме. Организационные схемы и должностные инструкции следует утверждать в соответствии с внутренним распорядком производителя.
3. Персонал, о котором идет речь в пункте 2, должен быть наделен достаточными полномочиями для правильного выполнения своих обязанностей.
4. Персонал должен получить первоначальное и проходить дальнейшее обучение подтверждаемой эффективности, включающее, в частности, теорию и практику по применению концепции обеспечения качества и Надлежащей производственной практики и, при необходимости, частные требования к производству исследуемых лекарственных препаратов.
5. Должны быть разработаны и соблюдаться программы по гигиене труда, адаптированные к определенным видам деятельности. Эти программы включают методики, относящиеся к здоровью, гигиене и одежде персонала.
Статья 8
ПОМЕЩЕНИЯ И ОБОРУДОВАНИЕ
1. Помещения и производственное оборудование следует располагать, проектировать, конструировать, приспосабливать и эксплуатировать таким образом, чтобы они соответствовали процедурам, для выполнения которых предназначены.
2. Помещения и производственное оборудование следует располагать, проектировать и эксплуатировать таким образом, чтобы свести к минимуму риск ошибок, а также обеспечить эффективную очистку и эксплуатацию, которые позволяют избежать контаминации, перекрестной контаминации и вообще любого неблагоприятного воздействия на качество продукции.
3. Помещения и оборудование, предназначенные для производственных операций, влияющих на качество продукции, должны быть соответствующим образом квалифицированы и пройти оценку.
Статья 9
ДОКУМЕНТАЦИЯ
1. Производитель должен создать и поддерживать такую систему документации, в основе которой лежат спецификации, производственные рецептуры, инструкции по обработке и упаковке, методики и протоколы, распространяющиеся на описанные в них различные производственные операции. Документы должны быть четко изложены, не содержать ошибочных сведений и своевременно пересматриваться. Следует заранее разрабатывать методики производственных операций и условий общего характера наряду со специальными документами для производства каждой серии. Этот комплект документов должен давать возможность проследить историю производства каждой серии и изменения, внесенные во время разработки исследуемого лекарственного препарата.
В случае с некоторым лекарственным препаратом документация на всю серию должна храниться не менее одного года после истечения срока годности этой серии или не менее пяти лет после сертификации согласно статье 51(3) Директивы 2201/83/ЕС в зависимости от того, какой срок больше.
В случае с некоторым исследуемым лекарственным препаратом документация на всю серию должна храниться не менее пяти лет после окончания или официального прерывания последнего клинического испытания, в котором были использованы лекарственные препараты данной серии. Спонсор или владелец торговой лицензии, если они разные, должны нести ответственность относительно сохранности документации, как это требуется для торговой лицензии согласно приложению 1 Директивы 2001/83/ЕС, если потребуется для последующего получения торговой лицензии.
2. Если вместо документов в письменной форме используются электронные, фотографические или другие системы обработки данных, то производитель сначала должен провести валидацию этих систем, чтобы доказать, что данные будут соответствующим образом сохраняться в течение всего требуемого периода хранения. Данные, хранящиеся в электронных системах, должны быть доступны в удобной для чтения форме и предоставляться уполномоченным органам по их требованию. Данные, хранящиеся в электронных системах, должны быть защищены от утери или повреждения информации с использованием таких методов, как дублирование, создание резервных копий или перенос в другую систему хранения; также необходимо поддерживать файлы, отслеживающие движение документации.
Статья 10
ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС
1. Различные операции технологического процесса должны проводиться по заранее разработанным инструкциям, методикам и в соответствии с Надлежащей производственной практикой. Для проведения контроля в процессе производства следует иметь соответствующие средства и в достаточном количестве. Все отклонения от процесса производства и дефекты лекарственных препаратов следует документировать и тщательно расследовать.
2. Во избежание перекрестной контаминации и путаницы должны быть приняты соответствующие технические или организационные меры. В случае исследуемых лекарственных препаратов особое внимание должно уделяться обращению с препаратами во время и после операций по кодированию.
3. В случае с лекарственными препаратами любое новое производство или важное изменение процесса производства лекарственного препарата должны пройти валидацию. Важные этапы производственного процесса следует повторно валидировать.
4. В случае с исследуемыми лекарственными препаратами производственный процесс должен быть валидирован как можно полнее, учитывая стадию разработки препарата. По меньшей мере, наиболее важные стадии процесса, такие, как стерилизация, должны пройти валидацию. Все этапы создания и разработки производственного процесса должны быть задокументированы в полном объеме.
Статья 11
КОНТРОЛЬ КАЧЕСТВА
1. Производитель должен организовать и поддерживать систему контроля качества; она должна быть независимой от процесса производства и возглавляться лицом с необходимой квалификацией.
Это лицо должно иметь в своем распоряжении или иметь доступ к одной или нескольким лабораториям по контролю качества, имеющих соответствующий штат и оборудование для проведения необходимых проверок и испытаний сырья, упаковочных материалов, промежуточной и готовой продукции.
2. Относительно лекарственных препаратов, в том числе импортируемых из третьих стран, можно использовать контрактные лаборатории, если это разрешено в соответствии со статьей 12 данной Директивы и пунктом (b) статьи 20 Директивы 2001/83/ЕС.
Относительно исследуемых лекарственных препаратов спонсор должен убедиться, что контрактная лаборатория соответствует информации, содержащейся в заявке, поданной в соответствии со статьей 9(2) Директивы 2001/20/ЕС в уполномоченный регуляторный орган. Если препараты импортируются из третьих стран, аналитический контроль не является обязательным.
3. Во время проведения заключительного контроля готовой продукции перед ее выпуском для реализации или дистрибьюции, или для использования в клинических испытаниях, системе контроля качества необходимо учесть, в дополнение к аналитическим результатам, всю основную информацию: условия изготовления, результаты контроля в процессе производства, проверку производственной документации и соответствие продукции ее спецификациям (включая окончательную упаковку).
4. Образцы готовой продукции каждой серии следует сохранять не менее одного года после истечения срока годности.
В случае с некоторым исследуемым лекарственным препаратом необходимо достаточное количество образцов каждой серии нерасфасованной продукции и основных компонентов упаковки, используемых для каждой серии готовой продукции, сохранять по крайней мере в течение двух лет после окончания или официального прерывания последнего клинического испытания, в котором использовалась данная серия, в зависимости от того, какой период продолжительнее.
Если в государстве – члене ЕС, в котором осуществляется производство, не требуется по закону более длительный период, образцы используемого в процессе производства сырья (кроме растворителей, газов и воды) необходимо хранить не менее двух лет после выпуска продукции. Этот период может быть сокращен, если срок их стабильности, указанный в соответствующей спецификации, меньше. Все эти образцы должны быть доступны для уполномоченных органов.
По соглашению с уполномоченным органом может быть установлен другой порядок отбора и условий хранения проб, сырья и определенных лекарственных препаратов, производящихся в единичных или малых количествах, или если их хранение может требовать особых условий.
Статья 12
РАБОТА ПО КОНТРАКТУ
1. Любая производственная операция или операция, связанная с производством, которая осуществляется по контракту, регулируется письменным контрактом.
2. В контракте должна быть ясно определена ответственность сторон и, в частности, соблюдение Надлежащей производственной практики исполнителем, а также оговорено, каким образом Уполномоченное Лицо, ответственное за выдачу разрешения на реализацию каждой серии, должно осуществлять свои обязанности.
3. Исполнитель не должен заключать субподряды на любую работу, переданную ему заказчиком, без письменного разрешения на то заказчика.
4. Исполнитель должен соблюдать принципы и правила Надлежащей производственной практики; его должны инспектировать компетентные уполномоченные органы в соответствии со статьей 111 Директивы 2001/83/ЕС и статьей 15 Директивы 2001/20/ЕС.
Статья 13
РЕКЛАМАЦИИ, ОТЗЫВ
ПРОДУКЦИИ
И РАСКОДИРОВАНИЕ
В ЭКСТРЕННЫХ СЛУЧАЯХ
В случае с лекарственными препаратами производитель должен создать систему протоколирования и проверки рекламаций наряду с эффективной системой отзыва, быстрого и в любое время, лекарственных препаратов из сети распределения. Любая рекламация о дефекте должна быть запротоколирована и исследована производителем. Производитель должен проинформировать уполномоченный орган о любом дефекте, который может привести к отзыву продукции или ограничению поставки. При возможности следует указать страну назначения поставки.
Любой отзыв должен быть проведен в соответствии с требованиями статьи 123 Директивы 2001/83/ЕС.
2. В случае исследуемых лекарственных препаратов производитель совместно со спонсором должны создать систему протоколирования и проверки рекламаций наряду с эффективной системой отзыва, быстрого и в любое время, исследуемых лекарственных средств, которые уже поступили в сеть дистрибьюции. Производитель должен запротоколировать и исследовать все рекламации о дефекте и информировать уполномоченный орган о каждом дефекте, который может привести к отзыву продукции или ограничению его поставки.
В случае с исследуемыми лекарственными препаратами должны быть указаны все клинические базы исследования и, если возможно, страны назначения поставок.
В случае исследуемого лекарственного препарата, на который выдана лицензия, производитель исследуемого лекарственного препарата должен вместе со спонсором информировать владельца торговой лицензии о каждом дефекте, который может относиться к лицензированному исходному препарату.
Спонсор должен создать систему быстрого раскодирования закодированных препаратов, когда это необходимо для их быстрого отзыва, в соответствии с параграфом 2. Спонсор должен гарантировать, что процедура идентифицирует кодированные продукты настолько, насколько это необходимо.
Статья 14
САМОИНСПЕКЦИЯ
Производитель должен проводить регулярные самоинспекции, составляющие часть системы обеспечения качества, чтобы следить за выполнением и добросовестным отношением к Надлежащей производственной практике и предлагать все необходимые корректирующие меры. Следует вести протоколы таких самоинспекций и любых последующих корректирующих действий.
Статья 15
МАРКИРОВКА
В случае с исследуемыми лекарственными препаратами маркировка должна гарантировать защиту испытуемого и отслеживаемость для обеспечения идентификации препарата и испытания, а также для обеспечения надлежащего применения исследуемого лекарственного препарата.
Статья 16
ОТМЕНА ДИРЕКТИВЫ 91/356/ЕЕС
Директива 91/356/ЕЕС отменена.
Ссылки на отмененную Директиву должны считаться ссылками на настоящую Директиву.
Статья 17
ПЕРЕНЕСЕНИЕ В НАЦИОНАЛЬНОЕ ЗАКОНОДАТЕЛЬСТВО
1. Государствам – членам ЕС следует ввести в действие законы, постановления и принять административные меры, необходимые для выполнения настоящей Директивы, не позднее 30 апреля 2004 г. Следует немедленно передать Комиссии тексты этих положений и корреляционные таблицы между этими положениями и положениями настоящей Директивы.
Принимая эти положения, государства – члены ЕС должны сделать ссылку на настоящую директиву или сопроводить такой ссылкой их официальную публикацию. Процедура для такой ссылки должна быть принята государствами – членами ЕС.
2. Страны – члены ЕС должны сообщить Комиссии тексты основных положений национального законодательства, которые приняты в области, регулируемой настоящей Директивой.
Статья 18
ВСТУПЛЕНИЕ В СИЛУ
Данная Директива вступает в силу на 20-й день после ее опубликования в Официальном журнале Европейского Союза.
Статья 19
АДРЕСАТЫ
Директива адресована странам – членам ЕС.
Составлена в Брюсселе 8 октября 2003 г.
От имени Комиссии ЕС член Комиссии Erkki LIIKANEN
1 Official Journal 311, 28.11.2001, p. 67.
2 Official Journal 159, 27.6.2003, p. 46.
3 Official Journal 193, 17.7.1991, p. 30.
4 Official Journal 121, 1.5.2001, p. 34.
* Official Journal L262, 14.10.2003, p. 22.
** Перевод выполнен редакцией «Еженедельника АПТЕКА» и не является официальным.
*** В документах ВОЗ эквивалентом понятия «торговая лицензия» служит термин «разрешение на маркетинг». — Прим. ред.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим