|

|

Следует отметить, что работа, пожалуй, именно этой секции привлекла больше всего внимания со стороны производителей лекарственных средств (ЛС), и это естественно, потому что происходил живой обмен мнениями между ведущими отечественными экспертами со стороны Государственного фармакологического центра (ГФЦ) МЗ Украины и представителями отечественных фармацевтических компаний.
Участники заседания услышали доклад на очень актуальную тему «Как обеспечить качество и регуляторную приемлемость исследований биоэквивалентности » зарубежного специалиста — д-ра Лембита Раго, координатора по качеству, безопасности, эффективности препаратов Департамента основных лекарственных средств штаб-квартиры ВОЗ (Швейцария).
Сегодня во многих странах, отметил докладчик, исследования биоэквивалентности проводят все чаще и шире используют их результаты в качестве одного из критериев, на основании которых проводят регистрацию препаратов. При этом нередко сталкиваются с рядом проблем, среди которых — отсутствие специально разработанных нормативных документов, необходимого опыта и ресурсов у предприятий отрасли, а также нехватка опытных кадров и финансовых средств у регуляторных органов. Гармонизация требований национальных документов с международными — только первый этап работы, подчеркнул д-р Раго, на последующих необходимо добиваться их реального применения.
Докладчик рассказал, что в рамках программы ООН по предварительной оценке (преквалификации) ЛС (для лечения ВИЧ/СПИДа, малярии, туберкулеза и заболеваний репродуктивной системы) на соответствие стандартам качества, безопасности и эффективности, руководство которой осуществляется ВОЗ (prequalification programme), было выявлено, что многие препараты не соответствуют требованиям качества ().
В рамках программы также выполняют инспекционные проверки (силами команд из 2 инспекторов ВОЗ и 1 национального — в качестве наблюдателя) соблюдения требований GCP контрактными исследовательскими организациями (contract research organization — CRO). В результате выявлено много недостатков общего характера: неправильное составление протоколов, недостаточное обеспечение защиты испытуемых, отсутствие определения обязанностей исследователя, распределения обязанностей между спонсором и CRO и т.д. Нередко отмечали ошибки в биоаналитической части исследования (использование невалидированных методик, аппаратуры и т.п.), некорректность проведения статистического анализа. Часто наблюдали неправильное ведение документации (не указан спонсор, методы анализа фармакокинетических параметров, обоснование применения статистических методов), ошибки в сохранении данных, трудность доступа к ним.
Выяснялось, что иногда спонсоры не проводили мониторинг исследования, необходимый аудит; возникали сомнения в аутентичности результатов лабораторных и инструментальных методов исследований (запись электрокардиограммы одного человека представляли как данные нескольких). Кроме того, нередко исходные данные вообще невозможно было найти.
ВОЗ разработаны, а в последнее время обновлены несколько руководств по проведению исследований биоэквивалентности. Все вместе они собраны в сериях технических отчетов ВОЗ (WHO Technical Report Series — TRS), которые готовят группы экспертов по различным научным и техническим вопросам. В частности, в 40-м отчете Экспертного комитета по спецификациям для лекарственных средств (WHO Expert committee on specifications for pharmaceutical preparations) (TRS 937) можно найти такие важные с практической точки зрения документы:
- Многоисточниковые (генерические) лекарственные средства: руководство по регистрационным требованиям для определения взаимозаменяемости (Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability) (Annex 7);
- Предложение об отказе от соблюдения требований (по доказательству) биоэквивалентности in vivo для препаратов из Примерного перечня основных лекарственных средств ВОЗ в твердых лекарственных формах с немедленным высвобождением действующего вещества (Proposal to waive in vivo bioequivalence requirements for WHO Model List of Essential Medicines immediate-release, solid oral dosage forms) (Annex 8);
- Дополнительное руководство по организации проведения исследований биоэквивалентности in vivo (Additional guidance for organizations performing in vivo bioequivalence studies) (Annex 9);
- Руководство по выбору препарата сравнения для оценки эквивалентности взаимозаменяемости многоисточникового (генерического) продукта (Guidance on the selection of comparator pharmaceutical products for equivalence assessment of interchangeable multisource (generic) products) (Annex 11).
На сайте ВОЗ можно найти также проект документа «Информация для заявителей по выбору препарата сравнения для проекта преквалификации» (Note to applicants on the choice of comparator products for the prequalification project). Большой интерес представляют материалы инспекций CRO, в том числе тех, что проводили исследования биоэквивалентности (WHO Public Inspection reports of CROs) (). На той же интернет-страничке можно найти материалы тренингов и семинаров ВОЗ и советы производителям, желающим участвовать в программе преквалификации.
В завершение доклада д-р Раго отметил, что этическая оценка исследований биоэквивалентности тоже важна, поэтому разработаны соответствующие руководства (Operational Guidelines for Ethics Committees that Review Biomedical Research (TDR/PRD/ETHICS/2000.1) и Surveying and Evaluating Ethical Review Practices: a complementary guideline to the Operational Guidelines for Ethics Committees that Review Biomedical Research (PUB: TDR/PRD/ETHICS/2002.1)).
Отвечая на вопрос слушателей о возможности проведения тех или иных исследований препаратов, при разработке и производстве которых не соблюдены требования GLP, GMP и GCP, д-р Раго подчеркнул, что если препарат не произведен в соответствии с требованиями GMP, исследования биоэквивалентности, как и прочие клинические испытания (КИ), бессмысленны, потому что качество препаратов не сохраняется от серии к серии.
|

Интересный доклад об особенностях клинического изучения биоэквивалентности ЛС представила Людмила Ковтун, руководитель отдела аттестации и инспекции клинических баз ГФЦ МЗ Украины.
Как один из видов КИ, отметила докладчик, исследования биодоступности и биоэквивалентности должны соответствовать требованиям, применяемым в этой области, в том числе, руководствам GCP (Руководство 42-7.0:2005, утвержденное приказом МЗ Украины от 22.06.2005 г. № 373). Кроме того, при планировании и проведении исследований биодоступности и биоэквивалентности ЛС, при составлении регистрационного досье и экспертизе рекомендуется применять Руководство 42-7.1:2005, утвержденное приказом МЗ Украины от 25.04.2005 г. № 191. Положения руководства, составленного на основе соответствующего документа Европейского агентства по лекарственным средствам (European Medicinal Agency — EMEA), содержат рекомендации относительно структуры организации исследований биодоступности и биоэквивалентности.
Исследование биоэквивалентности, отметила Л. Ковтун, по существу представляет собой сравнительное изучение биодоступности, предназначенное для установления эквивалентности между исследуемым и референтным ЛС. Докладчик подчеркнула, что эти исследования являются нетерапевтическими, в них отсутствует непосредственная клиническая польза для человека. Очень важно помнить определения терминов биодоступности и биоэквивалентности согласно руководству, поскольку это облегчает понимание между специалистами, отметила она.
Почему к участию в этих КИ привлекают здоровых добровольцев? Дело в том, что контингент исследуемых должен быть как можно более однородным. Это позволяет свести к минимуму вариабельность и обеспечить возможность определения различий между исследуемыми ЛС, уменьшить разброс полученных данных.
Исследования биоэквивалентности, отметила Л. Ковтун, разделяют на три этапа: 1-й этап — клиническая часть (подбор добровольцев, соответствующих критериям включения, выполнение требований протокола, стандартизация условий проведения исследования, отбор и хранение проб); 2-й — биоаналитическая часть (проводится в соответствии с требованиями GLP — определение активного вещества в биоматериале, валидация методик); 3-й — статистический анализ (обработка полученных данных, их оценка и подготовка заключительного отчета).
Золотым стандартом исследований биоэквивалентности являются такие условия: применение одной дозы исследуемых препаратов у здоровых добровольцев однократно натощак в рамках перекрестного дизайна с достаточным «отмывочным» периодом между периодами приема. Параллельный дизайн исследования может быть применен, если исследуемое ЛС характеризуется очень продолжительным периодом полувыведения и применение перекрестного дизайна нецелесообразно.
Л. Ковтун отдельно остановилась на необходимости соблюдения этических принципов в организации и проведении исследований биоэквивалентности. Как и любые другие КИ, их можно проводить, согласно Хельсинкской декларации, если значимость поставленной цели соразмерима с возможным риском и неудобствами для испытуемых. При этом обязательными условиями являются законность, уважение к людям, а также сведение к минимуму причиняемого вреда и возможности ошибки. Пациент предоставляет свое добровольное информированное согласие и может выйти из исследования на любом этапе.
|

Иногда проведение исследований по биоэквивалентности препарата натощак бывает недостаточным или вообще нежелательным, поэтому целесообразно провести такие же исследования после приема пищи. Основаниями для выбора схемы исследования с приемом препарата после еды являются такие обстоятельства: необходимость избежать влияния на пищеварительный тракт, осложненная фармакокинетика препарата, модифицированное высвобождение действующего вещества, воздействие пищи на лекарственное вещество. При этом выбор дизайна исследования в каждом случае должен быть обоснован.
При проведении исследований по биоэквивалентности предъявляют определенные требования к клинической базе и лаборатории. Обработка полученных данных и подготовка отчета о результатах считаются одним из самых ответственных этапов исследования. От того, насколько ответственно исследователи подойдут к выполнению своих функций, зависит успешность выполнения их основной задачи — доказательство того, что клинически значимая разница между значениями биодоступности испытуемого и референтного препаратов является маловероятной.
Содержательным, хорошо иллюстрированным примерами из деятельности отрасли и регуляторных органов в нашей стране и за рубежом было выступление Владимира Мальцева, руководителя отдела координации и контроля клинических испытаний лекарственных средств ГФЦ, посвященное сравнительным КИ ЛС. Согласно требованиям, принятым ВОЗ и в Европейском Союзе, генерическими считаются препараты, которые могут быть взаимозаменяемы с оригинальными, будучи представлены на рынке после окончания срока действия патента или других эксклюзивных прав на оригинальный препарат. Генерические ЛС соответствуют таким требованиям:
- содержат такой же активный ингредиент, как и оригинальный препарат;
- имеют ту же биодоступность;
- выпускаются в той же лекарственной форме;
- сохраняют качество, эффективность и безопасность, аналогичные таковым у оригинальных;
- не нарушают патентной защиты оригинальных препаратов;
- их стоимость ниже, чем оригинального препарата;
- соответствуют фармакопейным требованиям, производятся в условиях GMP;
- имеют те же показания к применению и меры предосторожности.
Последнее В. Мальцев особенно подчеркнул, отметив, что компании часто стремятся проигнорировать это требование.
В качестве референтного, с которым сравнивают по сути аналогичный препарат для прохождения сокращенной процедуры регистрации, согласно приказу МЗ Украины от 26.08.2005 г. № 426, рассматривают препарат, зарегистрированный в Украине на основании экспертизы полного регистрационного досье (данные химического, биологического, фармацевтического, фармакологического, токсикологического и клинического изучения). ВОЗ предусмотрены еще два критерия, пользуясь которыми всегда можно осуществить подбор референтного препарата, отметил В. Мальцев. Так, можно использовать препарат, лидирующий на рынке по объемам продаж при условии его установленной эффективности, безопасности и качества. В случае, когда вышеперечисленное невозможно, национальный регуляторный орган может определить препарат, находящийся на рынке другой страны, при условии его установленной эффективности, безопасности и качества.
О терапевтической эквивалентности двух препаратов можно говорить при условии наличия фармацевтической эквивалентности или альтернативности и осуществлении доказательства биоэквивалентности одним из следующих путей:
I. Исследование биоэквивалентности на людях;
II. Исследование фармакодинамики на людях;
III. КИ;
IV. Тест на растворимость in vitro (в некоторых случаях).
Выбор метода исследования должен быть адекватным конкретному случаю и основываться на представлениях о лекарственной форме и соответствии дизайна исследования его цели — выявлению различий между ЛС, поступившими к месту своего действия в организме.
|
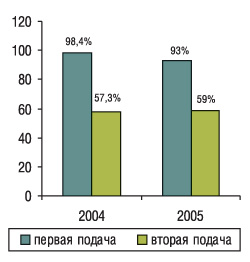
В США, где накоплен большой опыт в этой области, с 1984 г. действует норма, согласно которой производители, желающие получить разрешение на маркетинг генерического препарата, должны представить данные, которые свидетельствовали бы о биоэквивалентности исследуемого препарата инновационному. Отдел генерических препаратов (Office of Generic Drugs — OFD) Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) очень строго подходит к рассмотрению заявок на препараты, и многие заявки бывают отклонены как при первой, так и при повторной подаче (рис. 1).
Какие подходы приняты в развитых странах, в том числе с учетом рекомендаций ВОЗ, в отношении установления биоэквивалентности? Считается, что в случае достаточной концентрации ЛС в оцениваемых биологических жидкостях (плазма крови), могут быть выполнены сравнительные фармакокинетические исследования. В некоторых случаях (с учетом биофармацевтической классификационной системы) проводят испытания in vitro препаратов с немедленным высвобождением действующего вещества. В случаях, если ЛС не достигает достаточной концентрации в доступных биологических жидкостях, сравнительные фармакодинамические исследования являются следующей альтернативой. Сравнительные КИ обычно наименее чувствительны, но в случаях, когда невозможно определить фармакокинетический профиль или найти подходящие фармакодинамические конечные точки, сравнительные КИ могут рассматриваться как приемлемые.
В некоторых случаях исследования биоэквивалентности проводить не требуется. Это касается таких лекарственных форм, как растворы для парентерального введения, растворы для питья, газы, ЛС для местного применения несистемного действия, ингаляционные, назальные спреи в виде водных растворов.
Рассказывая о подходах к организации исследований для определения биоэквивалентности, В. Мальцев привел данные Французского агентства по безопасности лекарственных средств (Agence francaise de securite sanitaire des produits de sante) о результатах инспекций с анализом 78 досье КИ по биоэквивалентности, проводившихся 54 компаниями из 20 стран (Blaye О., 2006 по Ефимцева Т.К., 2006). В результате инспекций недостатки выявлены в 73 файлах! Среди них критические находки — в 40. При этом в 20 досье выявлены данные, свидетельствующие о мошенничестве и фальсификации. Таким образом, проводить испытания биоэквивалентности нужно согласно существующим требованиям, при этом к получаемым данным регуляторные органы должны относиться очень взвешенно.
|
Как один из способов доказательства биоэквивалентности фармакодинамические исследования применяют тогда, когда фармакодинамический ответ легче измеряем или более надежен, чем данные фармакокинетики, или для ЛС местного действия, когда эти данные не имеют значения. При этом измеряемый ответ должен быть фармакологическим или терапевтическим эффектом, который относится к заявленным в отношении эффективности и/или безопасности. Методика должна быть валидирована на правильность, точность, воспроизводимость и специфичность.
Сравнительные КИ используют в тех случаях, когда исследования фармакокинетики и фармакодинамики не дают убедительных доказательств. Количество пациентов и прочие особенности дизайна выбираются в зависимости от особенностей препарата. Следует хорошо понимать, подчеркнул В. Мальцев, что сравнительные исследования для доказательства биоэквивалентности проводятся для регистрации препарата по сокращенной программе. Если выбрана полная программа, то потребуются данные всех пре- и клинических (с I по III фазы) испытаний.
Процесс гармонизации требований к проведению КИ в Украине с современными международными стандартами привел к созданию документа «Порядок проведения клинических испытаний ЛС и экспертизы материалов клинических испытаний» (приказ МЗ Украины от 13.02.2006 г. № 66).
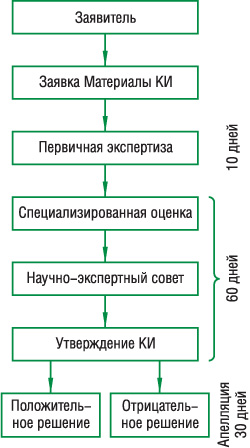
Сравнительные КИ генерических ЛС в Украине проводятся с соблюдением правил GCP и согласно единому Порядку как для сравнительных испытаний, так и для многоцентровых. Испытание начинают только после положительного заключения ГФЦ и одобрения Этического комитета (рассмотрение проводят параллельно в течение 60 дней с момента принятия заявки), при условии обязательного страхования пациентов, проведения испытаний на утвержденных МЗ клинических базах (рис. 2).
Как отметил профессор, спонсоры должны уделять большое внимание правильному составлению протокола. Очень тщательно следует подходить к установлению целевых параметров, которые обычно представляют соответствующие клинические конечные точки, на основании которых делают вывод об интенсивности и времени начала ответа.
В каждом конкретном случае, исходя из клинических соображений (естественное течение болезни, эффективность доступного лечения и выбранный целевой параметр), необходимо определять величину приемлемого диапазона показателей, характеризующих действие препарата. Этот выбор не может быть основан на общем консенсусе по всем препаратам, отметил профессор.
При составлении протокола сравнительного КИ необходимо четко описывать условия включения, предусмотреть наличие контрольной группы, существенно не отличающейся от основной по составу и численности, полу и возрасту, тяжести заболевания и исходному базовому лечению. В качестве контроля может быть использован препарат аналогичного действия с доказанной эффективностью и безопасностью, плацебо или стандартное лечение. Предпочтительными являются слепые рандомизированные испытания.
|
|
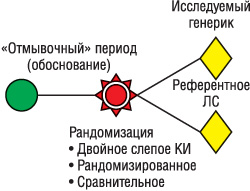
Обычно при проведении сравнительных КИ генерических препаратов применяют параллельный дизайн (рис. 3). Включение плацебо рекомендуют, если препарат не оказывает ярко выраженного клинического действия. Если препарат применяют при тяжелых заболеваниях, участниками исследования становятся пациенты с этой патологией, и препарат назначают на фоне базовой терапии.
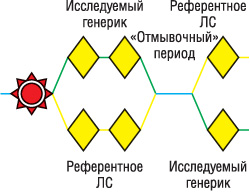
При перекрестном дизайне, который чаще применяют при исследованиях биоэквивалентности, обязательно наличие «отмывочного» периода, чтобы концентрация ЛС в исследуемых биологических жидкостях не превышала 5% максимальной (рис. 4).
|
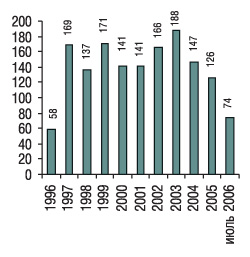
Как отметил В. Мальцев, за 10 лет работы отдела координации и контроля клинических испытаний ГФЦ проведено 1518 сравнительных КИ (рис. 5). Их анализ был представлен в более чем 50 публикациях.
В основном эти КИ организуют и проводят отечественные производители ЛС. Некоторые из них, подчеркнул профессор, создают себе и регуляторным органам ряд проблем, связанных как со сложностями роста, так и с вопросами квалификации, а именно:
- создаются «ножницы» несоответствия внешней атрибутики GMP и качественной разработки ЛС;
- отсутствуют экспериментальные участки производства для наработки опытных образцов ЛС для испытаний;
- немногие фармпредприятия, даже с большими объемами производства, имеют соответствующим образом подготовленные подразделения по вопросам клинической разработки ЛС и фармаконадзору.
Руководитель отдела по клиническим испытаниям ГФЦ обратил внимание участников конференции на острую необходимость создания таких подразделений в составе отечественных компаний.
Среди основных замечаний к составлению протоколов испытаний были названы: неполная или неправильная статистическая обработка данных, отсутствие реальной рандомизации пациентов (добровольцев), выраженное несоответствие объема и содержания брошюры исследователя нормативным требованиям. Очень существенно то, что не проводятся мониторинг и аудит со стороны спонсора. Результаты инспекции ГФЦ свидетельствуют, что заказчики не всегда проверяют работу исследователей, хотя обязанность проводить мониторинг возложена именно на них.
За период 1999–2006 гг. сотрудниками ГФЦ проведены 269 инспекционных проверок этих исследований, по результатам которых 6% КИ приостановлено или остановлено полностью. В. Мальцев подчеркнул, что в дальнейшем требования к исследованиям будут становиться более строгими.
Среди основных недостатков, выявленных во время инспекций, названы: недостаточное знание исследователями протокола и своих обязанностей, недоукомплектация файла исследователя, нарушение маркировки, условий хранения и распределения исследуемого препарата, нарушение процедуры получения информированного согласия, неправильное ведение индивидуальных регистрационных форм и первичной документации, нарушение процедуры предоставления информации о побочных реакциях.
|

Таким образом, отметил В. Мальцев, КИ генерического ЛС должно быть научно обосновано и этически оправдано (определены цели и соотношение польза/риск); дизайн исследования необходимо основывать на знании фармакокинетики, фармакодинамики и терапевтических особенностей активного фармацевтического ингредиента. Кроме того, должна быть представлена информация о процессах производства и данные по выполненным КИ, которые должны подтвердить, что исследуемое ЛС имеет адекватное качество. В завершение выступления профессор подчеркнул, что опыт Украины в проведении сравнительных КИ генерических ЛС свидетельствует о возможностях поступательного внедрения принципов GCP.
Во время заседания секции Ольга Баула, заместитель директора департамента фармацевтической деятельности ГФЦ, представила два доклада, первый из которых назывался «Фармацевтическая разработка — один из основных элементов обеспечения качества лекарственных средств».
Для чего нужна и что собой представляет фармацевтическая разработка? Это комплексные исследования по разработке готового ЛС, которые убедительно демонстрируют, что выбранная лекарственная форма, предлагаемый состав и технология производства препарата в полной мере обеспечивают совокупность свойств, придающих ЛС способность удовлетворять потребителей в соответствии со своим предназначением и отвечать требованиям, установленным законодательством.
Проведение разработки регулируется рядом международных и украинских документов (приказы МЗ Украины от 26.08.2005 г. № 426 и от 13.02.2006 г. № 66). Данные оформляются как обязательный раздел регистрационного досье: часть II «Химическая, фармацевтическая и биологическая документация»; раздел А, п. 4 в формате, принятом ранее в ЕС, либо модуль 3 «Качество», раздел 3.2.Р.2 в формате Общего технического документа (Common Technical Document — CTD).
Согласно ICH Q8 качество целенаправленно «встраивается» в препарат, а не проверяется, и проведение фармацевтической разработки преследует такие цели:
- целенаправленно сформировать качество ЛС;
- гарантировать высокую степень вероятности того, что любая единица каждой серии препарата, производимого в условиях промышленного производства, будет иметь качество, соответствующее предполагаемому применению;
- определить границы, в которых можно изменять процесс после внедрения, когда еще можно выпускать продукт с той эффективностью, безопасностью, качеством, которые были в КИ.
Разработка состава (качественные и количественные характеристики, физико-химические свойства действующего вещества, влияющие на его биодоступность, совместимость компонентов) и выбор лекарственной формы должны быть подтверждены экспериментальными данными, на основании которых определяются критерии приемлемости физико-химических и фармакотехнологических параметров, оказывающих влияние на функциональные характеристики препарата.
Особую значимость при разработке препаратов в твердых лекарственных формах имеет тест «растворение», с помощью которого возможно оптимизировать состав лекарственной формы, оценить поведение действующего вещества при проведении сравнительных исследований in vitro, контролировать стадии технологического процесса и изменения в процессе производства, устанавливать качество готового препарата. Организации и проведению сравнительных исследований in vitro с использованием теста «растворение» при подтверждении эквивалентности ЛС был посвящен второй доклад О. Баулы.
Результаты сравнительных исследований in vitro с использованием теста «растворение» с учетом высокой растворимости и проницаемости действующего вещества на основании биофармацевтической классификационной системы могут рассматриваться как доказательство биоэквивалентности. При этом оформляется запрос в регуляторный орган об исключении исследований in vivo.
Таким образом, результаты исследований по фармацевтической разработке позволяют уменьшить количество вносимых в досье изменений, обусловленных неоптимальными процессами и препаратами; дают инструменты для проведения глубинного анализа возможных отклонений при серийном производстве.
На основании результатов исследований по фармацевтической разработке производитель принимает решение о разработке спецификаций и утверждении поставщиков действующих и вспомогательных веществ, производственной рецептуре, технологическом процессе, спецификации готового ЛС, сроках и условиях его хранения. Больше того, фармацевтическая разработка предоставляет обоснование для выбора метода оценки эффективности и безопасности генерических ЛС.
|

Игорь Зупанец, профессор, заведующий кафедрой клинической фармакологии с фармацевтической опекой Национального фармацевтического университета (НФаУ), рассказал об особенностях проведения КИ на здоровых добровольцах. В этой области в нашей стране одной из основных проблем остается ограниченное количество специализированных клиник для проведения КИ I фазы и исследований биоэквивалентности. Одна клиника может провести не более 5–7 испытаний I фазы в год, поэтому актуальным является создание новых баз, а также расширение коечного фонда и материально-технической базы уже существующих.
Почему для этих видов испытаний нужны отдельные клиники? Госпитализировать в лечебно-профилактическое учреждение здорового добровольца невозможно; кроме того, как определять в таких случаях количество проведенных койко-дней? Необходимо учитывать общественное мнение, беспокойство других пациентов по поводу того, почему в лечебно-профилактических учреждениях находятся здоровые люди.
В условиях специализированной клиники доброволец огражден от нежелательных контактов, соблюдается конфиденциальность его участия и т.п. Кроме того, только там можно учесть особенности диеты добровольцев согласно требованиям протокола (определенный питьевой режим, продолжительность периода голодания, отсутствие в пище красящих веществ, определенных специй, продуктов, содержащих кофеин и т.п.). Одной из специализированных клиник, соответствующих предъявляемым требованиям, является Клинико-диагностический центр НФаУ, отметил И. Зупанец. Профессор поделился опытом проведения нескольких исследований биоэквивалентности и КИ I фазы.
|

В ходе заседания секции выступил и специалист по доклиническому изучению ЛС, профессор Виталий Мамчур. Он отметил, что возглавляемая им кафедра фармакологии, клинической фармакологии и технологии лекарств Днепропетровской государственной медицинской академии, проводящая доклинические исследования, тесно связана с 24 сертифицированными на проведение КИ базами академии.
Среди всех отчетов о доклинических исследованиях, отметил В. Мамчур, можно выделить 3 основные разновидности: генерического ЛС (по острой токсичности и 1–2 показателям специфической активности); оригинального препарата (по острой и хронической токсичности, отдаленным последствиям, 3–5 показателям специфической активности); оригинальной комбинации известных компонентов (по острой и хронической токсичности, 1–2 показателям специфической активности). Документация, касающаяся токсикологических и фармакологических исследований, выполненных на активной субстанции и готовой лекарственной форме препарата, включается в «Резюме доклинических данных» (модуль 2) и «Отчеты о доклинических исследованиях» (модуль 4) досье на ЛС в формате CTD.
Отчеты об исследованиях должны быть представлены в таком порядке: фармакология, фармакокинетика, токсикология. Последний раздел может включать данные по генотоксичности, канцерогенности, репродуктивной и онтогенетической токсичности, местной переносимости, антигенности, иммунотоксичности, лекарственной зависимости и пр.
Проблемы проведения доклинических исследований описаны во многих руководствах и справочниках. Так, приложение к модулю 4 в базе «Eudralex» содержит 24 руководства по доклиническим исследованиям (!). Далеко не каждый фармаколог-экспериментатор знаком с каждым из этих документов, отметил В. Мамчур. Доклинические исследования ЛС описаны в десятках отдельных изданий методических рекомендаций, подготовленных ГФЦ. Под редакцией члена-корреспондента АМН Украины А.В. Стефанова в 2002 г. выпущено почти 600-страничное издание методических руководств.
Для клинициста большой интерес представляют данные о фармакокинетике и фармакодинамике препаратов, а их получают на основании результатов именно доклинических исследований. Чрезвычайно важными являются вопросы видовой и половой чувствительности. Так, при значениях коэффициента видовой чувствительности, близких к 1, ЛС не обладает видовой специфичностью или она слабо выражена. Получение других значений должно заставить исследователей проявить настороженность в отношении возможности экстраполяции данных доклинических исследований на человека. По мнению В. Мамчура, этот показатель является достаточно информативным. На него так же, как на показатели токсичности, исследователь обязательно должен обращать внимание при планировании клинических наблюдений.
С помощью отчетов о доклинических экспериментальных исследованиях можно критически оценить протокол КИ, обоснованность показателей, необходимых для осуществления тех или иных клинико-лабораторных исследований, предусмотреть возможность появления, а также объяснить, предупредить и даже лечить нежелательные проявления действия препарата на пути его становления в качестве ЛС, подчеркнул В. Мамчур.
Большой интерес участников конференции вызвал доклад профессора Николая Яблучанского, заведующего кафедрой внутренних болезней Харьковского национального университета им. В.Н. Каразина, посвященный выбору конечных суррогатных и несуррогатных точек в КИ ЛС. Очень познавательным было сообщение Геннадия Воронова об опыте проведения исследований биоэквивалентности в Республике Беларусь. К настоящему времени в этой стране аккредитовано 46 учреждений, на базе которых проводят КИ ЛС практически всех фармакотерапевтических групп; проводят исследования биоэквивалентности (в 2006 г. проведены 7 и еще 5 — запланированы). Важным практическим опытом работы лабораторий фармакологического анализа поделились руководители отечественных лабораторий Виктория Либина и Тамара Шевченко.
В ходе работы секции были подробно рассмотрены статистические аспекты исследований ЛС, на которых в своих докладах останавливались консультант ГФЦ по применению статистических методов Павел Бабич и заведующий отделением клинической фармакологии и фармакотерапии Института терапии им. Л.Т. Малой АМН Украины Юрий Рудык. Прозвучали интересные сообщения об опыте планирования, организации и проведения КИ, а также производстве исследуемых препаратов отечественными производителями — ЗАО НПЦ «Борщаговский ХФЗ», ООО «Фарма Старт». К сожалению, формат нашей публикации не позволяет отразить все интересные выступления. Надеемся, этому будут посвящены другие статьи. n
Продолжение следует.
Дарья Полякова, фото Игоря Кривинского
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим