МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ
НАКАЗ
від 04.01.2013 р. № 3
Зареєстровано в Міністерстві
юстиції України
15 березня 2013 р.
за № 425/22957
Про внесення змін до наказу Міністерства охорони здоров’я України від 26 серпня 2005 року № 426 та визнання такими, що втратили чинність, деяких наказів Міністерства охорони здоров’я України з питань реєстрації лікарських засобів
Відповідно до пункту 2 Порядку державної реєстрації (перереєстрації) лікарських засобів, затвердженого постановою Кабінету Міністрів України від 26 травня 2005 року № 376, НАКАЗУЮ:
1. Унести зміни до Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення, затвердженого наказом Міністерства охорони здоров’я України від 26 серпня 2005 року № 426, зареєстрованого в Міністерстві юстиції України 19 вересня 2005 року за № 1069/11349, виклавши його в новій редакції, що додається.
2. Затвердити Положення про Кваліфікаційну комісію Міністерства охорони здоров’я України, що додається.
3. Визнати такими, що втратили чинність:
наказ Міністерства охорони здоров’я України від 01 листопада 2001 року № 443 «Про реєстрацію деяких видів лікарських засобів», зареєстрований у Міністерстві юстиції України 20 листопада 2001 року за № 975/6166;
Порядок проведення державної реєстрації (перереєстрації) медичних імунобіологічних препаратів в Україні, затверджений наказом Міністерства охорони здоров’я України від 06 грудня 2001 року № 486, зареєстрований у Міністерстві юстиції України 28 лютого 2002 року за № 204/6492;
наказ Міністерства охорони здоров’я України від 17 квітня 2007 року № 190 «Про затвердження Порядку проведення додаткових випробувань лікарських засобів при проведенні експертизи реєстраційних матеріалів», зареєстрований у Міністерстві юстиції України 17 серпня 2007 року за № 949/14216;
Порядок проведення експертизи реєстраційних матеріалів на лікарські засоби, що виробляються згідно із затвердженими прописами, затверджений наказом Міністерства охорони здоров’я України від 17 грудня 2008 року № 754, зареєстрований у Міністерстві юстиції України 21 січня 2009 року за № 45/16061;
, зареєстрований у Міністерстві юстиції України 13 лютого 2010 року за № 156/17451;
наказ Міністерства охорони здоров’я України від 17 березня 2010 року № 236 «Про затвердження Порядку проведення перевірки виробництва лікарських засобів, що подаються на державну реєстрацію», зареєстрований у Міністерстві юстиції України 13 травня 2010 року за № 323/17618.
4. Начальнику Управління лікарських засобів та медичної продукції (Коношевич Л.В.) забезпечити подання цього наказу на державну реєстрацію до Міністерства юстиції України у встановленому порядку.
5. Цей наказ набирає чинності з дня його офіційного опублікування.
6. Контроль за виконанням цього наказу покласти на заступника Міністра — керівника апарату Богачева Р.М.
| В.о. Міністра | О. Толстанов |
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 26.08.2005 р. № 426
(у редакції наказу Міністерства
охорони здоров’я України від 04.01.2013 р. № 3)
Зареєстровано в Міністерстві юстиції України
15 березня 2013 р. за № 425/22957
ПОРЯДОК
проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
І. Загальні положення
1.1. Цей Порядок розроблено відповідно до Законів України «Про лікарські засоби» та «Про захист населення від інфекційних хвороб», Порядку державної реєстрації (перереєстрації) лікарських засобів і Розмірів збору за державну реєстрацію (перереєстрацію) лікарських засобів, затверджених постановою Кабінету Міністрів України від 26 травня 2005 року № 376.
1.2. Цей Порядок проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення (далі — Порядок) поширюється на активні фармацевтичні інгредієнти, продукцію in bulk, готові лікарські засоби, у тому числі медичні імунобіологічні препарати, речовини або комбінації речовин, що містяться у засобах медичного призначення, які у процесі використання надходять до системного кровотоку, а також засоби, які за визначенням Міністерства охорони здоров’я України (далі — МОЗ) реєструються як лікарські засоби (далі — лікарські засоби), такі як:
засоби для дезінфекції шкіри та слизових оболонок носової і ротової порожнин та зовнішніх статевих органів пацієнта перед діагностичними, оперативними маніпуляціями чи лікувальними процедурами;
засоби для дезінфекції шкіри рук медичного персоналу до і після проведення діагностичних та оперативних маніпуляцій і контакту з інфікованим та/або потенційно інфікованим матеріалом;
матеріали перев’язувальні клейкі та інші (тампони, марля, серветки), просочені антисептичними або дезінфекційними засобами, іншими лікарськими засобами та упаковані для роздрібного продажу;
засоби для знищення комах та кліщів, які мають епідеміологічне значення та призначені для нанесення на шкіру людини та її придатки, а також репелентні засоби для безпосереднього нанесення на шкіру людини та її придатки;
лікарські косметичні засоби, які разом з косметичною сировиною містять окремі діючі речовини або їх суміші, що використовуються для профілактики та/або лікування захворювань шкіри людини та її придатків, слизової оболонки носової і ротової порожнин, статевих органів пацієнта і виробляються у формі крему, молочка, олії, маски, бальзаму, гелю, порошку, лосьйону, шампуню, помади, екстракту для ванн, антиперспіранту, зубної пасти, еліксиру тощо;
речовини або комбінації речовин, що містяться у засобах медичного призначення, які у процесі використання надходять до системного кровотоку, зокрема допоміжні засоби для гемотрансфузій тощо.
1.3. Цей Порядок не поширюється на радіофармацевтичні лікарські засоби, які виготовлені відповідно до інструкцій виробника, під час використання у акредитованих закладах охорони здоров’я, виключно із ліцензованих джерел радіонуклідів, радіонуклідних наборів, прекурсорів радіонуклідів.
1.4. Державну реєстрацію (перереєстрацію) лікарського засобу здійснює МОЗ на підставі результатів експертизи реєстраційних матеріалів (реєстраційного досьє) на такий засіб та контролю його якості, проведених Державним експертним центром МОЗ (далі — Центр).
ІІ. Визначення термінів
У цьому Порядку наведені нижче терміни вживаються у таких значеннях:
активний фармацевтичний інгредієнт (лікарська речовина, діюча речовина, субстанція) (далі — АФІ або діюча речовина) — будь-яка речовина чи суміш речовин, що призначена для використання у виробництві лікарського засобу і під час цього використання стає його активним інгредієнтом. Такі речовини мають фармакологічну чи іншу безпосередню дію на організм людини. У складі готових форм лікарських засобів їх застосовують для лікування, діагностики чи профілактики захворювання, для зміни стану, структур або фізіологічних функцій організму, для догляду, обробки та полегшення симптомів.
АФІ або діюча речовина можуть бути: компактні, вкриті оболонкою, гранульовані, здрібнені до певного ступеня або оброблені іншим шляхом та під різними назвами й формами (зокрема у формах пелет, гранул тощо та інших формах випуску);
безпека лікарського засобу — характеристика лікарського засобу, яка базується на порівняльній оцінці користі від його застосування та потенційної шкоди, яка може бути завдана пацієнту при застосуванні ним цього лікарського засобу;
біовейвер — процедура проведення порівняльних досліджень in vitro на підставі біофармацевтичної системи класифікації (далі — БСК) з використанням тесту «Розчинення» для підтвердження еквівалентності генеричного та референтного лікарських засобів системної дії у твердій дозованій формі для орального застосування з негайним вивільненням з метою реєстрації генеричного лікарського засобу без проведення досліджень in vivo;
біодоступність — швидкість і ступінь, з якими діюча речовина або її активний компонент абсорбується (всмоктується) з лікарської форми і стає доступною у місці дії;
біоеквівалентність — два лікарських засоби біоеквівалентні, якщо вони є фармацевтично еквівалентними або фармацевтично альтернативними і якщо їхня біодоступність після введення в одній і тій самій молярній дозі подібна до такого ступеня, що ефекти цих лікарських засобів щодо ефективності та безпеки будуть по суті однаковими;
біологічні лікарські засоби — лікарські засоби, що містять діючі речовини біологічного походження, отримані шляхом виробництва з біологічного джерела (тваринного, людського, рослинного, мікробного або біотехнологічного);
біофармацевтична система класифікації — наукова система класифікації діючих речовин на основі їх розчинності у водних розчинах та ступеня кишкового проникнення;
виробник лікарських засобів — суб’єкт господарювання, який здійснює хоча б один із етапів виробництва лікарських засобів та має ліцензію (дозвіл) на виробництво лікарських засобів (якщо останнє передбачено національним законодавством країни, на території якої знаходяться виробничі потужності виробника);
виробництво лікарських засобів — діяльність, пов’язана із серійним випуском лікарських засобів, яка включає всі або хоча б одну зі стадій технологічного процесу, у тому числі закупівлю матеріалів і продукції, фасування, пакування та/або маркування, зберігання, відповідний контроль, видачу дозволу на випуск (реалізацію), а також оптову торгівлю (дистрибуцію) продукцією власного виробництва;
висновок Державного експертного центру МОЗ щодо ефективності, безпеки та якості лікарського засобу — узагальнений результат експертизи (первинної, та/або попередньої, та/або спеціалізованої) матеріалів реєстраційного досьє на лікарський засіб з наданням рекомендацій щодо його державної реєстрації (перереєстрації), внесення змін до матеріалів реєстраційного досьє на лікарський засіб протягом дії реєстраційного посвідчення чи його нової реєстрації або щодо відмови в реєстрації (перереєстрації), внесенні змін до матеріалів реєстраційного досьє на лікарський засіб протягом дії реєстраційного посвідчення;
високотехнологічні (біотехнологічні) лікарські засоби — лікарські засоби, що містять діючі речовини, отримані за допомогою методів біотехнології, таких як: генно-інженерна технологія, клітинна інженерія, гібридомні технології, інженерна ензимологія та інженерна імунологія тощо;
вторинна упаковка — упаковка, у яку вкладається лікарський засіб у первинній упаковці і яка виконує захисну функцію по відношенню до лікарського засобу та первинної упаковки;
генеричний лікарський засіб (генерик, взаємозамінний) (далі — генерик) — лікарський засіб, який має такий самий кількісний та якісний склад діючих речовин і таку саму лікарську форму, що й референтний препарат, та чия взаємозамінність з референтним препаратом доведена відповідними дослідженнями. Різні солі, прості та складні ефіри, ізомери, суміші ізомерів, комплекси або похідні діючої речовини вважаються однією і тією самою діючою речовиною за умови, що вони суттєво не відрізняються з точки зору безпеки та ефективності. Різні лікарські форми орального застосування з негайним вивільненням діючої речовини вважаються однією і тією самою лікарською формою;
генно-інженерні лікарські засоби — лікарські засоби, які отримані шляхом технологій рекомбінантних ДНК;
гомеопатичний лікарський засіб — будь-який лікарський засіб, виготовлений із продуктів, субстанцій або складових, які називаються гомеопатичною сировиною, відповідно до процедури виготовлення гомеопатичного лікарського засобу, описаної в Державній фармакопеї України (далі — ДФУ) або Європейській фармакопеї або — в разі відсутності такого опису — у Німецькій гомеопатичній фармакопеї (GHP), Гомеопатичній фармакопеї США (HPUS), Британській гомеопатичній фармакопеї (BHP), Гомеопатичній фармакопеї Швабе.
Гомеопатичний лікарський засіб також може містити більше однієї діючої речовини;
державна перереєстрація лікарського засобу — процедура, яка проводиться відповідно до вимог чинного законодавства та цього Порядку з метою продовження строку, протягом якого лікарський засіб дозволяється до медичного застосування в Україні;
державна реєстрація лікарського засобу — процедура, яка проводиться відповідно до вимог чинного законодавства та цього Порядку з метою допуску лікарського засобу до медичного застосування в Україні та внесення його до Державного реєстру лікарських засобів України;
джерело радіонуклідів — будь-яка система, яка містить фіксований первинний радіонуклід, з якого утворюються вторинні радіонукліди, які витягуються шляхом елюювання або в інший спосіб та використовуються в радіофармацевтичному лікарському засобі;
діючі речовини (біологічні агенти) — біологічно активні речовини, у тому числі біологічні агенти (мікроорганізми, віруси, клітини та тканини людини, тварин і їх компоненти), які можуть змінювати стан і функції організму або мають профілактичну, діагностичну чи лікувальну дію та використовуються як основний(і) компонент(и) для виробництва готових лікарських засобів;
добре вивчене медичне застосування — медичне застосування діючої(их) речовини(ин), що входить(ять) до складу лікарського засобу, добре вивчене(і), визнані її (їх) ефективність та прийнятний ступінь безпеки (що підтверджено докладними бібліографічними посиланнями на опубліковані дані щодо післяреєстраційних, епідеміологічних досліджень тощо), і минуло не менше 10 років від дати першого систематичного і документованого застосування в Україні діючої(их) речовини(н) у складі лікарського(их) засобу(ів);
додаткові вивчення (випробування) — дослідження, необхідність яких визначається під час проведення спеціалізованої експертизи та які дозволяють забезпечити складання вмотивованих висновків щодо ефективності, безпеки та якості лікарського засобу при його реєстрації та/або внесенні змін до реєстраційних матеріалів;
дослідження еквівалентності — дослідження, яке визначає еквівалентність між генериком та референтним лікарським засобом при використанні досліджень in vivo та/або in vitro;
дослідження еквівалентності in vitro на підставі БСК — комплексні дослідження, які базуються на класифікації діючої речовини згідно з БСК та розчиненні лікарського засобу, а також включають порівняння профілів розчинення генерика та референтного лікарського засобу у трьох середовищах зі значеннями рН 1,2, рН 4,5 і рН 6,8;
експерт з питань реєстрації Центру — фізична особа, яка має відповідний рівень кваліфікації і знань законодавства України, правил і норм Європейського Союзу, рекомендацій ВООЗ у сфері обігу лікарських засобів, положень Міжнародної конференції з гармонізації технічних вимог до реєстрації лікарських засобів для людини. При цьому експерт не повинен виконувати функції, у тому числі за межами Центру, що призводять до конфлікту інтересів;
експертиза матеріалів реєстраційного досьє на лікарський засіб — попередня та спеціалізована експертиза матеріалів реєстраційного досьє та за необхідності матеріалів додаткових вивчень (випробувань) лікарського засобу з метою підготовки вмотивованих висновків та рекомендацій щодо прийняття рішення про можливість його державної реєстрації (перереєстрації), внесення змін до реєстраційних матеріалів або щодо відмови в реєстрації (перереєстрації), внесенні змін до реєстраційних матеріалів на лікарський засіб;
ефективність лікарського засобу — сприятлива діагностична, лікувальна чи профілактична дія лікарського засобу на встановлення характеру захворювання, його перебіг, тривалість або корекцію стану чи фізіологічних функцій організму людини відповідно до показань до застосування, зазначених в інструкції для медичного застосування;
загальноприйнята назва — міжнародна непатентована назва (далі — МНН) діючої речовини, рекомендована Всесвітньою організацією охорони здоров’я (далі — ВООЗ), або за відсутності такої звичайна загальноприйнята назва;
заява інформованої згоди — заява про державну реєстрацію лікарського засобу, аналогічного зареєстрованому, за умови, що власник реєстраційного посвідчення зареєстрованого раніше лікарського засобу дає згоду використовувати його фармацевтичні, доклінічні та клінічні дані для нової реєстрації лікарського засобу з тим самим кількісним та якісним складом діючих речовин у такій самій лікарській формі;
заява на генерик — заява про державну реєстрацію лікарського засобу, який містить ту саму діючу речовину і в тій самій лікарській формі, що й референтний лікарський засіб;
заява на лікарський засіб з продукції in bulk — заява на державну реєстрацію готового лікарського засобу, виробництво якого здійснюється шляхом фасування та/або кінцевого пакування і маркування продукції in bulk;
заява на лікарський засіб, який має добре вивчене медичне застосування, — заява про державну реєстрацію лікарського засобу, який містить відому(і) діючу(і) речовину(и), медичне застосування якої (яких) добре вивчене в Україні протягом щонайменше 10 років та яка має визнану ефективність та прийнятний рівень безпеки, що підтверджується докладними бібліографічними даними;
заява на фіксовану комбінацію — заява про державну реєстрацію лікарського засобу, що містить відомі діючі речовини, які не використовувались раніше в цій комбінації, а також раніше не використовувались у медичній практиці як окремі лікарські засоби у тій самій лікарській формі, дозах та відповідному співвідношенні, що й у комбінованому лікарському засобі;
заявник (власник реєстраційного посвідчення) (далі — заявник) — юридична або фізична особа, яка є відповідальною за ефективність, якість та безпеку лікарського засобу в порядку, визначеному чинним законодавством, та має ресурси для здійснення фармаконагляду в Україні, а також є відповідальною за достовірність інформації, що міститься у наданих нею матеріалах реєстраційного досьє;
зміни, які можуть мати місце протягом дії реєстраційного посвідчення, — запропоновані заявником зміни, які стосуються зареєстрованого (перереєстрованого) лікарського засобу;
інструкція для медичного застосування лікарського засобу (інструкція про застосування лікарського засобу) (далі — інструкція для медичного застосування) — офіційно затверджена інформація про медичне застосування лікарського засобу, викладена відповідно до цього Порядку, що супроводжує готовий лікарський засіб;
конфіденційна реєстраційна інформація — науково-технічна інформація, що міститься у заяві про державну реєстрацію (перереєстрацію) лікарського засобу та заяві про внесення змін до матеріалів реєстраційного досьє протягом строку дії реєстраційного посвідчення та додатках до них, включаючи частини I, II, III та IV або модулі 1, 2, 3, 4 та 5 матеріалів реєстраційного досьє (за винятком інформації, яка є загальнодоступною, зокрема про назву лікарського засобу, склад діючих речовин, силу дії, які наводяться в межах інструкції для медичного застосування, пакування, про заявника та/або виробника лікарського засобу, інструкції для медичного застосування, інформації щодо небезпечних властивостей лікарського засобу, які можуть завдати шкоду пацієнту під час застосування);
лікарський засіб рослинного походження — будь-який лікарський засіб, що містить виключно діючу(і) речовину(и) з однієї або більше рослинних субстанцій, або один або більше рослинних препаратів, або одну або більше рослинних субстанцій у комбінації з одним або більше рослинним препаратом;
лікарський засіб, що виробляється згідно із затвердженим прописом, — лікарський засіб, до якого входить діюча речовина з добре вивченим медичним застосуванням, з визначеною ефективністю, задовільним ступенем безпеки, пропис на який затверджено МОЗ;
лікарські засоби вітчизняного виробництва — лікарські засоби, що виготовляються локальним (українським) виробником; не вважаються лікарськими засобами вітчизняного виробництва такі лікарські засоби, що пройшли тільки стадії фасування та/або кінцевого пакування і маркування;
лікарські засоби, які одержують з крові або плазми людини, — лікарські засоби на основі компонентів крові; такі лікарські засоби включають, зокрема, альбумін, фактори згортання крові та імуноглобуліни людського походження;
локальний (український) виробник — суб’єкт господарювання, який здійснює усі технологічні етапи виробництва субстанцій та/або готових лікарських засобів на виробничих дільницях, розташованих на території України, та має ліцензію на виробництво лікарських засобів;
медичні імунобіологічні препарати (далі — МІБП) — алергени, антигени, вакцини (анатоксини), цитокіни, імуномодулятори бактерійного походження, а також ті, що створені на основі органів і тканин, препарати, які одержують з крові та плазми людини, імунні сироватки, імуноглобуліни (включаючи моноклональні антитіла), пробіотики, інтерферони, інші лікарські засоби, призначені для використання в медичній практиці з метою лікування, специфічної профілактики, діагностики стану імунітету (in vivo).
МІБП одержують шляхом культивування штамів мікроорганізмів і клітин еукаріотів, екстракції речовин з біологічних тканин, включаючи тканини людини, тварин і рослин (алергени), застосування методів генної інженерії, гібридомної технології, репродукції живих агентів в ембріонах чи тваринах;
назва лікарського засобу — назва, дана лікарському засобу, яка може бути як вигаданою заявником (виробником), так і загальноприйнятою або науковою, що може супроводжуватися назвою торгової марки або назвою заявника (виробника);
незалежний експерт — фізична особа, яка має відповідний рівень кваліфікації, спеціальні знання і на замовлення заявника здійснює наукову чи науково-технічну експертизу та відповідає перед заявником за достовірність і повноту аналізу, обґрунтованість рекомендацій відповідно до вимог завдання заявника на проведення експертизи і не є автором, дослідником об’єкта експертизи; яка не працює експертом у закладах або організаціях, що офіційно визначені як експертні органи об’єктів наукової і науково-технічної діяльності щодо обігу лікарських засобів; або іншим чином пов’язана з офіційними експертними органами, центральним органом виконавчої влади, що забезпечує формування державної політики у сфері охорони здоров’я, або вповноваженими ним органами; яка визначається виключно заявником;
неінтервенційне дослідження — дослідження, у якому лікарські засоби призначаються звичайним способом відповідно до затвердженої інструкції для медичного застосування. Залучення пацієнта в групу з визначеним методом лікування в протоколі клінічного дослідження заздалегідь не передбачено, а призначення лікарського засобу диктується сучасною практикою і не залежить від рішення включити пацієнта у випробування. Не застосовують додаткових діагностичних або моніторингових процедур щодо пацієнтів, а для аналізу зібраних даних використовують епідеміологічні методи;
неправомірне використання реєстраційної інформації відносно безпеки та ефективності лікарського засобу — посилання або інше використання інформації про ефективність та безпеку лікарського засобу, зареєстрованого за повною і незалежною заявою, раніше ніж за 5 років з дати реєстрації такого лікарського засобу в Україні, для подання заяви про державну реєстрацію відповідного генеричного лікарського засобу, що подається для державної реєстрації за відповідною заявою, за винятком випадків, коли заявник відповідно до закону одержав право посилатися та/або використовувати реєстраційну інформацію референтного/оригінального лікарського засобу, або подав власну повну реєстраційну інформацію, що відповідає вимогам до реєстраційної інформації референтного/оригінального лікарського засобу.
Ці вимоги не перешкоджають суб’єкту здійснювати в цей період відповідну розробку лікарського засобу, у тому числі проводити дослідження з еквівалентності між генеричним та референтним лікарськими засобами, для отримання реєстраційного посвідчення після виповнення п’яти років з дати реєстрації в Україні референтного лікарського засобу, визначеного в попередньому абзаці;
оригінальний (інноваційний) лікарський засіб — лікарський засіб, що був уперше у світі зареєстрований на основі повної документації щодо його ефективності, безпеки та якості (повного реєстраційного досьє);
патентований лікарський засіб — лікарський засіб, який надходить в обіг під власною назвою виробника (заявника), право на виробництво (виготовлення), реалізацію та застосування якого охороняється законодавством України про охорону прав інтелектуальної власності;
первинна упаковка — індивідуальна упаковка, яка безпосередньо контактує з лікарським засобом та сприяє збереженню його основних властивостей;
повна і незалежна заява (автономна заява) — заява на державну реєстрацію лікарського засобу, до якого входить нова або відома АФІ або діюча речовина, що зареєстрована у складі лікарського засобу в іншій лікарській формі;
подібний біологічний лікарський засіб (біосиміляр) — біологічний лікарський засіб, подібний щодо ефективності, безпеки та якості до зареєстрованого референтного біологічного препарату, період патентного захисту якого закінчився. Подібність терапевтичної ефективності, безпеки і якості такого лікарського засобу до референтного має бути доведена відповідними порівняльними доклінічними та клінічними дослідженнями;
представник заявника (уповноважена особа, що виступає від імені заявника) (далі — представник заявника) — юридична або фізична особа, якій на підставі відповідного доручення заявник надав право представляти його інтереси при проведенні процедур реєстрації, перереєстрації та/або внесення змін до реєстраційних матеріалів на території України та яка є відповідальною за достовірність інформації, що міститься у матеріалах реєстраційного досьє, наданих ним до Центру;
прекурсор радіонукліда — будь-який радіонуклід, призначений для введення радіоактивної мітки до іншої речовини перед її застосуванням;
препарат обмеженого застосування (препарат-сирота) — лікарський засіб, що призначений для діагностики, профілактики чи лікування рідкісного захворювання, тобто захворювання, що загрожує життю чи призводить до втрати працездатності не більше 5 осіб з кожних 10000 жителів на дату подання заяви про державну реєстрацію;
продукція in bulk — будь-який лікарський засіб, що пройшов усі стадії технологічного процесу, за винятком стадії фасування та/або кінцевого пакування і маркування;
пропис (монографія) — інформація про склад, технологію виробництва, контроль якості та застосування лікарського засобу;
радіонуклідний набір — будь-який лікарський засіб, який має бути поєднаний або змішаний з радіонуклідами у готовому радіофармацевтичному лікарському засобі, як правило, перед його застосуванням;
радіофармацевтичний лікарський засіб — будь-який лікарський засіб, який у готовому для застосування стані містить один або декілька радіонуклідів (радіоактивних ізотопів), уведених до нього з медичною метою;
регулярно оновлюваний звіт з безпеки лікарського засобу (регулярний звіт) — письмовий звіт, який містить регулярно оновлювану інформацію з безпеки лікарського засобу відповідно до Порядку здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування, затвердженого наказом Міністерства охорони здоров’я України від 27 грудня 2006 року № 898, зареєстрованого у Міністерстві юстиції України 29 січня 2007 року за № 73/13340;
реєстраційна інформація — науково-технічна інформація в будь-якій формі й вигляді, збережена на будь-яких носіях, у тому числі ілюстрації (карти, діаграми, органограми, малюнки, схеми тощо), фотографії та будь-які інші документовані відомості, що містяться в заяві про державну реєстрацію (перереєстрацію) лікарського засобу, заяві про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення та в реєстраційних досьє і визначена як конфіденційна згідно з абзацом тридцять дев’ятим цього розділу;
реєстраційне посвідчення на лікарський засіб — документ, у якому міститься інформація про лікарській засіб, зареєстрований в Україні та внесений до Державного реєстру лікарських засобів України та міжвідомчої бази даних про зареєстровані в Україні лікарські засоби;
реєстраційне посвідчення на лікарський засіб (медичний імунобіологічний препарат) — документ, у якому міститься інформація про медичний імунобіологічний препарат, зареєстрований в Україні та внесений до Державного реєстру лікарських засобів України та міжвідомчої бази даних про зареєстровані в Україні лікарські засоби;
реєстраційний номер — кодова позначка, яка присвоюється лікарському засобу при державній реєстрації і зберігається за лікарським засобом незмінною на весь період перебування лікарського засобу на фармацевтичному ринку України;
реєстраційні матеріали (матеріали реєстраційного досьє) — комплект документів, що додаються до заяви про державну реєстрацію (перереєстрацію, внесення змін до реєстраційних матеріалів) лікарського засобу, на підставі яких можна зробити обґрунтований висновок щодо його ефективності, безпеки та якості;
референтний лікарський засіб — лікарський засіб, з яким має порівнюватися генеричний лікарський засіб і який насамперед є оригінальним (інноваційним) лікарським засобом з доведеною ефективністю, безпекою та якістю;
рослинні препарати — препарати, одержані у результаті обробки рослинних субстанцій шляхом витягування, дистиляції, віджимання, подрібнення, очищення, концентрації та ферментації. Сюди входять потовчені або порошкоподібні рослинні субстанції, настойки, екстракти, ефірні олії, віджаті соки та оброблені витяжки;
рослинні субстанції — цілі, подрібнені або порізані рослини, частини рослин, водоростей, грибів, лишайників у необробленій, зазвичай засушеній формі, іноді свіжі. Певні витяжки з рослин (наприклад, смоли), не призначені для лікування, також вважаються рослинними субстанціями. Рослинні субстанції чітко визначаються морфологічною частиною рослини, що використовується, та її ботанічною назвою відповідно до біномної системи (рід, вид, різновид та джерело);
сила дії лікарського засобу — уміст активної(их) речовини (речовин) у кількісному вираженні на одиницю дози, або одиницю об’єму, або одиницю маси відповідно до лікарської форми;
терапевтична еквівалентність — два лікарські засоби вважаються терапевтично еквівалентними, якщо вони є фармацевтично еквівалентними або фармацевтично альтернативними лікарськими засобами і після введення пацієнтам одним і тим самим шляхом, в однаковій молярній дозі їх ефекти щодо безпеки й ефективності за суттю аналогічні. Це можна довести відповідними дослідженнями: порівняльними фармакокінетичними дослідженнями, порівняльними фармакодинамічними дослідженнями, порівняльними клінічними дослідженнями або дослідженнями еквівалентності in vitro;
традиційний лікарський засіб — лікарський засіб, зокрема рослинного походження, який відповідає таким умовам:
лікарський засіб відповідно до його складу та призначення передбачений для застосування без нагляду лікаря з метою діагностики, без приписання або рецепта або без спостереження за процесом лікування;
лікарський засіб застосовується у певних концентрації та дозуванні;
лікарський засіб призначений для орального, зовнішнього або інгаляційного застосування;
є документальне підтвердження того, що лікарський засіб застосовувався у медичній практиці не менше 30 років у світі та не менше 10 років в Україні;
є досить даних щодо традиційного застосування лікарського засобу (безпека застосування при звичайних умовах, доведена ефективність);
узагальнюючий звіт — письмовий звіт, який узагальнює інформацію з безпеки лікарського засобу, що міститься у двох або більше регулярно оновлюваних звітах з безпеки лікарського засобу;
фармацевтична еквівалентність — лікарські препарати є фармацевтично еквівалентними, якщо вони мають однакову лікарську форму, вводяться тим самим шляхом, містять ту саму кількість тієї самої діючої речовини (тих самих діючих речовин) у тих самих дозованих формах, відповідають вимогам тих самих або порівнюваних стандартів. Фармацевтична еквівалентність передбачає терапевтичну еквівалентність, якщо лікарська форма препарату унеможливлює вплив на біодоступність таких факторів, як відмінності у допоміжних речовинах та/або технології виробництва, або інші коливання, які можуть впливати на швидкість розчинення та/або абсорбції діючої речовини лікарського засобу;
фармацевтично альтернативні лікарські засоби — препарати, що містять однакову молярну кількість тієї самої діючої речовини, але відрізняються за лікарською формою (наприклад, таблетки порівняно з капсулами) та/або хімічною формою (наприклад, інші солі, інші ефіри). Альтернативні лікарські засоби доставляють ту саму діючу речовину тим самим шляхом введення, але не є фармацевтично еквівалентними. Вони можуть бути або можуть не бути біоеквівалентними або терапевтично еквівалентними референтному препарату;
якість лікарського засобу — сукупність властивостей, які надають лікарському засобу здатність задовольняти споживачів відповідно до свого призначення і відповідають вимогам, встановленим законодавством.
Інші терміни, що використовуються у цьому Порядку, вживаються у значеннях, наведених у законодавстві.
ІІІ. Порядок проведення експертизи реєстраційних матеріалів на лікарський засіб, що подається на державну реєстрацію (перереєстрацію)
3.1. Проведенню експертизи за бажанням заявника може передувати надання Центром безкоштовних консультацій з питань державної реєстрації (перереєстрації) лікарських засобів.
3.2. Для проведення експертизи заявник подає до МОЗ за принципом «єдиного вікна»:
заяву про державну реєстрацію лікарського засобу, у тому числі МІБП згідно з додатком 1 до цього Порядку (у разі надання заяви на багатокомпонентні лікарські засоби в декількох варіаціях доз діючих речовин, які змінюються непропорційно, заява розглядається як на різні лікарські засоби), а у випадку традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами, подається заява про державну реєстрацію (перереєстрацію) лікарського засобу за формою, наведеною у додатку 2 до цього Порядку;
заяву про державну реєстрацію (перереєстрацію) АФІ або діючих речовин за формою, наведеною у додатку 3 до цього Порядку;
заяву про державну перереєстрацію лікарського засобу за формою, наведеною у додатку 4 до цього Порядку, а у випадку традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами, подається заява про державну реєстрацію (перереєстрацію) лікарського засобу за формою, наведеною у додатку 2 до цього Порядку.
На бажання заявника може проводитись одночасно реєстрація готового лікарського засобу та діючої речовини, що входить до складу цього лікарського засобу, із наданням реєстраційних посвідчень про державну реєстрацію АФІ, діючої речовини та готового лікарського засобу за умови, що матеріали реєстраційного досьє подаються у форматі загального технічного документа (далі — ЗТД), структура якого наведена у додатку 5 до цього Порядку.
Інформація про подану до МОЗ заяву вноситься до електронної бази даних.
Збір та обробка персональних даних здійснюються відповідно до вимог Закону України «Про захист персональних даних».
3.3. Проведення експертизи лікарського засобу, що подається на державну реєстрацію (перереєстрацію), включає такі етапи:
первинна експертиза заяви про державну реєстрацію лікарського засобу з точки зору його належності до заборонених до застосування в Україні та кваліфікації типу такої заяви з метою визначення обсягу реєстраційних матеріалів відповідно до вимог цього Порядку;
попередня експертиза комплектності матеріалів реєстраційного досьє лікарського засобу відповідно до типу заяви та вимог, визначених у пункті 3.5 цього розділу;
спеціалізована експертиза матеріалів реєстраційного досьє лікарського засобу, що проводиться з метою складання вмотивованого висновку щодо ефективності, безпеки та якості лікарського засобу. Складовою спеціалізованої експертизи є (за потреби) додаткове вивчення (додаткові випробування).
3.4. Порядок проведення первинної експертизи.
3.4.1. За результатами розгляду заяви МОЗ протягом трьох робочих днів надсилає до Центру лист-направлення разом із копією заяви (за типом заяви, визначеним заявником).
Лист-направлення на первинну експертизу вноситься до електронної бази даних.
Після отримання заяви та листа-направлення від МОЗ експерти з питань реєстрації Центру у строк до семи робочих днів здійснюють первинну експертизу заяви щодо можливості реєстрації лікарського засобу з точки зору його належності до заборонених до застосування в Україні та кваліфікації типу такої заяви з метою визначення обсягу реєстраційних матеріалів відповідно до вимог цього Порядку.
За результатами первинної експертизи заяви Центр протягом трьох робочих днів направляє до МОЗ відповідні рекомендації, що оформляються висновком Центру щодо первинної експертизи заяви, які протягом 5 робочих днів розглядаються кваліфікаційною комісією МОЗ відповідно до Положення про Кваліфікаційну комісію Міністерства охорони здоров’я України, затвердженого наказом Міністерства охорони здоров’я України від 04 січня 2013 року № 3. Висновок щодо первинної експертизи заяви вноситься до електронної бази даних.
3.4.2. За результатами розгляду кваліфікаційною комісією МОЗ висновку Центру щодо первинної експертизи заяви МОЗ протягом трьох робочих днів надає заявнику письмову відповідь та у разі встановлення можливості реєстрації (перереєстрації) заявленого лікарського засобу оформляє лист-направлення на попередню та спеціалізовану експертизу до Центру і рекомендує заявнику надати матеріали реєстраційного досьє до Центру для здійснення їх експертизи. Лист-направлення МОЗ на попередню та спеціалізовану експертизу вноситься до електронної бази даних.
На підставі укладеного між Центром і заявником договору проводиться експертиза наданих матеріалів реєстраційного досьє.
Якщо МОЗ за результатами кваліфікації заяви встановив, що заявник має надати іншу заяву, йому рекомендується разом з матеріалами реєстраційного досьє надати нову заяву, визначену у пункті 3.2 цього розділу і наведену у відповідних додатках до цього Порядку, яка направляється до Центру для початку попередньої експертизи відповідно до пункту 3.5 цього розділу.
У разі незгоди заявника щодо рекомендованого типу заяви заявник має право звернутися до МОЗ разом із додатковими матеріалами або оскаржити рішення МОЗ у встановленому законодавством порядку.
3.4.3. Первинна експертиза заяви на лікарський засіб, поданий з метою його державної перереєстрації, не здійснюється.
3.5. Порядок проведення попередньої експертизи.
3.5.1. Після отримання листа-направлення МОЗ на проведення попередньої та спеціалізованої експертизи матеріалів реєстраційного досьє лікарського засобу заявником після оплати державного збору за реєстрацію (перереєстрацію) лікарського засобу та вартості експертних робіт подаються до Центру матеріали реєстраційного досьє з урахуванням рекомендацій кваліфікаційної комісії МОЗ щодо типу заяви та вимог:
розділів ІХ та Х цього Порядку та додатка 5 до цього Порядку — для реєстрації;
розділу ХІІ цього Порядку — для гомеопатичних лікарських засобів;
розділів ХІІІ та ХІV цього Порядку — для традиційних лікарських засобів або лікарських засобів, які виробляються згідно із затвердженим прописом;
розділів ІХ, Х, ХІ цього Порядку та додатка 6 до цього Порядку — у разі реєстрації АФІ або діючої речовини.
Реєстраційні матеріали подаються відповідно до вимог розділу VІ цього Порядку.
У разі перереєстрації надається комплект матеріалів реєстраційного досьє відповідно до додатка 7 до цього Порядку. Для традиційних лікарських засобів або лікарських засобів, які виробляються згідно із затвердженим прописом, надається комплект матеріалів реєстраційного досьє відповідно до додатка 8 до цього Порядку.
Реєстраційні матеріали для перереєстрації можуть подаватися відповідно до додатка 5 або 9 до цього Порядку з урахуванням вимог розділів VII, VIIІ, ІХ та Х цього Порядку (залежно від обраного заявником формату матеріалів реєстраційного досьє).
У разі державної реєстрації (перереєстрації) лікарських засобів, які належать до групи лікарських засобів «Медичні гази», до МОЗ подається заява згідно з додатком 2 до цього Порядку, матеріали реєстраційного досьє подаються відповідно до переліку, наведеного у додатку 6 до цього Порядку, доповненого інструкцією для медичного застосування, з урахуванням вимог розділу ХVI цього Порядку.
Представник заявника має гарантувати достовірність інформації, наведеної у наданих матеріалах реєстраційного досьє.
Якщо під час експертизи матеріалів реєстраційного досьє, поданих на державну перереєстрацію, виникає потреба внесення додаткових змін, заявник надає гарантійний лист щодо сплати вартості експертизи таких змін.
За наявності зауважень Центру щодо внесення змін до матеріалів реєстраційного досьє генерика, поданого на реєстрацію (перереєстрацію), зокрема інструкції для медичного застосування, з метою приведення її у відповідність до затвердженої інструкції для медичного застосування оригінального (референтного) лікарського засобу, допускається відстрочення оплати змін, що запропоновані Центром, без зупинки експертних робіт.
При цьому заявник має або надати обґрунтування відсутності необхідності внесення запропонованих Центром змін, або сплатити вартість проведення експертних робіт за запропоновані зміни протягом 30 календарних днів з дати надання Центром зауважень щодо необхідності внесення змін до інструкції для медичного застосування генеричного лікарського засобу.
Якщо Центром раніше здійснювалася експертиза доклінічних та/або клінічних матеріалів, заявник оплачує тільки експертні роботи щодо додаткових матеріалів, якщо такі є.
У разі коли заявник протягом 3 місяців з дати офіційного отримання рекомендації кваліфікаційної комісії МОЗ не подає до Центру відповідних матеріалів реєстраційного досьє, Центр надає МОЗ рекомендації щодо зняття заяви з розгляду. МОЗ протягом 5 робочих днів розглядає надані рекомендації та приймає рішення щодо відмови у державній реєстрації, що затверджується наказом МОЗ. Про прийняте рішення МОЗ у строк до 10 робочих днів письмово повідомляє заявника. При цьому реєстраційний збір та вартість проведення експертних робіт заявникові не повертаються, але за бажанням заявника можуть бути використані для оплати інших його експертних робіт у Центрі.
Надалі така заява може бути подана до МОЗ в установленому порядку.
3.5.2. Попередня експертиза матеріалів реєстраційного досьє на лікарський засіб, що подається на державну реєстрацію (перереєстрацію), починається з дати отримання матеріалів реєстраційного досьє, сформованих відповідно до обраного заявником та рекомендованого МОЗ типу заяви. Інформація про це вноситься до електронної бази даних.
Попередню експертизу матеріалів реєстраційного досьє лікарського засобу здійснюють експерти з питань реєстрації Центру.
Термін відстрочення надання засвідченої копії документа, що підтверджує відповідність умов виробництва лікарського засобу вимогам до виробництва лікарських засобів в Україні (належної виробничої практики), не входить до встановлених законодавством строків проведення експертизи реєстраційних матеріалів. Попередня експертиза реєстраційних матеріалів здійснюється протягом 14 робочих днів. За результатами попередньої експертизи Центр надає заявнику письмову відповідь та заносить відповідну інформацію до електронної бази даних.
3.5.3. При негативних результатах попередньої експертизи Центр письмово повідомляє заявника про те, що матеріали реєстраційного досьє не можуть бути направлені на спеціалізовану експертизу, докладно зазначивши підстави з посиланням на номери розділів, глав, статей, пунктів цього Порядку, або одноразово запитує у заявника додаткові дані та/або інформацію, необхідні для забезпечення відповідності матеріалів реєстраційного досьє вимогам, встановленим цим Порядком. Копія такого листа направляється до МОЗ та вноситься до електронної бази даних.
Заявник має надати додаткові дані та/або інформацію згідно із зауваженнями Центру у строк до 90 робочих днів у разі реєстрації або в строк до 60 робочих днів у разі перереєстрації або листа з обґрунтуванням термінів, необхідних для їх доопрацювання. Ця інформація вноситься протягом 3 робочих днів до електронної бази даних.
Час, потрібний для підготовки та подання додаткових даних та/або інформації, не входить до строку проведення попередньої експертизи.
Центр має прийняти доопрацьовані заявником матеріали реєстраційного досьє протягом 3 робочих днів після звернення заявника та внести отриману інформацію до електронної бази даних.
Якщо заявник не надає Центру додаткові дані та/або інформацію або надає їх у неповному обсязі, а також якщо надані заявником додаткові дані та/або інформація не забезпечують відповідності матеріалів реєстраційного досьє встановленим вимогам (за винятком засвідченої копії документа, що підтверджує відповідність умов виробництва лікарського засобу вимогам до виробництва лікарських засобів в Україні (належної виробничої практики)), то Центр надає рекомендації МОЗ щодо відмови в державній реєстрації (перереєстрації) лікарського засобу.
МОЗ протягом 5 робочих днів розглядає таку рекомендацію на кваліфікаційній комісії і про прийняте рішення повідомляє заявника в письмовій формі у строк до 10 робочих днів. Рішення МОЗ про відмову в державній реєстрації (перереєстрації) лікарського засобу може бути оскаржене у встановленому законодавством порядку.
При цьому реєстраційний збір та вартість проведених експертних робіт заявникові не повертаються. Надалі на бажання заявника матеріали з урахуванням зауважень Центру подаються на державну реєстрацію (перереєстрацію) лікарського засобу в установленому порядку.
3.5.4. У разі відповідності матеріалів реєстраційного досьє лікарського засобу відповідно до типу заяви та вимог розділів Порядку та додатків до цього Порядку, визначених у підпункті 3.5.1 цього пункту, встановлених за результатами попередньої експертизи матеріалів реєстраційного досьє, вони направляються на спеціалізовану експертизу, про що Центр письмово повідомляє заявника та вносить відповідну інформацію до електронної бази даних.
3.6. Порядок проведення спеціалізованої експертизи.
3.6.1. Спеціалізовану експертизу матеріалів реєстраційного досьє лікарського засобу здійснюють експерти з питань реєстрації Центру із залученням позаштатних експертів та консультантів, метою якої є складання вмотивованого висновку щодо його ефективності, безпеки та якості, у тому числі експертиза методів аналізу, які містяться у проекті методів контролю якості (далі — МКЯ), щодо дотримання вимог ДФУ та/або Європейської фармакопеї, а за відсутності вимог у цих фармакопеях — інших провідних фармакопей (Британська фармакопея, фармакопея США, фармакопея Японії).
Якщо заявник керується вимогами національної фармакопеї або інших фармакопей, стандарти яких відповідають стандартам ДФУ, Європейської фармакопеї, то посилання на такі фармакопеї є прийнятними та не потребують внесення змін до матеріалів реєстраційного досьє з метою приведення у відповідність до ДФУ або Європейської фармакопеї. Ця інформація має бути відображена експертом з питань реєстрації Центру, що надає рекомендації МОЗ у частині якості лікарського засобу, у своєму експертному висновку.
Якщо лікарський засіб, на який подано заяву про державну реєстрацію (перереєстрацію), був зареєстрований регуляторними органами держав, які керуються високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ, та надані матеріали реєстраційного досьє були оцінені у рамках наведеної процедури такими регуляторними органами, від заявника не вимагається вносити зміни до МКЯ з посиланням на стандарти, що застосовуються в Україні. При цьому будь-які відмінності у специфікації лікарського засобу з фармакопейними вимогами заявник має обґрунтувати.
3.6.2. Під час проведення спеціалізованої експертизи матеріалів реєстраційного досьє з метою одержання додаткових даних щодо ефективності, безпеки та якості лікарського засобу Центр може дворазово запитати у заявника необхідні додаткові матеріали з посиланням на номери розділів, глав, статей, пунктів цього Порядку, при цьому не допускається нових запитів щодо матеріалів, які вже розглядалися експертом, у тому числі під час складання висновків за результатами експертизи. Зауваження до матеріалів реєстраційного досьє, що викладаються у запитах Центру до заявника, вносяться до електронної бази даних.
Центр має прийняти доопрацьовані матеріали протягом 3 робочих днів після звернення заявника та внести отриману інформацію до електронної бази.
Якщо заявник протягом 90 робочих днів після другого запиту Центру щодо додаткових матеріалів не надсилає цих матеріалів, або не обґрунтовує інший строк надання матеріалів, або надає їх у неповному обсязі (за винятком засвідченої копії документа, що підтверджує відповідність умов виробництва лікарського засобу вимогам до виробництва лікарських засобів в Україні (належної виробничої практики)), Центр надає рекомендації МОЗ щодо відмови в державній реєстрації (перереєстрації) лікарського засобу.
МОЗ протягом 5 робочих днів розглядає таку рекомендацію на кваліфікаційній комісії і про прийняте МОЗ рішення, що затверджується наказом, повідомляє заявника в письмовій формі у строк до 10 робочих днів. Рішення про відмову в державній реєстрації (перереєстрації) лікарського засобу може бути оскаржене у встановленому законодавством порядку.
При цьому реєстраційний збір та вартість проведення експертних робіт заявникові не повертаються. Надалі, на бажання заявника, матеріали реєстраційного досьє з урахуванням зауважень Центру подаються на державну реєстрацію (перереєстрацію) лікарського засобу в установленому порядку.
3.6.3. Під час проведення спеціалізованої експертизи матеріалів реєстраційного досьє Центр може рекомендувати МОЗ направити лікарський засіб на додаткові випробування та/або додаткову експертизу згідно з розділами ХІХ та ХХ цього Порядку. Кваліфікаційна комісія МОЗ протягом 5 робочих днів розглядає надані матеріали та надає відповідні рекомендації МОЗ. Про прийняте рішення МОЗ у строк 10 робочих днів письмово повідомляє заявника.
Додаткові випробування можуть включати випробування щодо: фармацевтичної розробки лікарського засобу, доклінічних досліджень, ефективності та/або безпеки лікарського засобу, біодоступності та еквівалентності генеричних лікарських засобів.
Додаткові випробування здійснюються після оплати їх вартості, установленої договором між заявником та закладом охорони здоров’я/установою, що проводить такі випробування.
Результати додаткових випробувань подаються до Центру для проведення їх спеціалізованої експертизи.
Час, потрібний для проведення додаткових випробувань, та їх вартість не входять до строку і вартості експертних робіт.
Про завершення спеціалізованої експертизи Центр інформує заявника письмово.
3.7. Після завершення проведення спеціалізованої експертизи Центр протягом 5 робочих днів складає та направляє до МОЗ умотивовані висновки щодо ефективності, безпеки та якості лікарського засобу, до яких додає засвідчену копію документа, що підтверджує відповідність умов виробництва лікарського засобу, поданого на реєстрацію (перереєстрацію), вимогам до виробництва лікарських засобів в Україні, виданого Державною службою України з лікарських засобів, і рекомендує або не рекомендує лікарський засіб до державної реєстрації (перереєстрації).
Інформація про складання та направлення до МОЗ умотивованих висновків щодо ефективності, безпеки та якості лікарського засобу, а також самі висновки вносяться до електронної бази даних.
Центр також рекомендує МОЗ до затвердження інструкцію для медичного застосування на лікарський засіб за формою, наведеною у додатку 10 до цього Порядку, фармакопейну статтю або методи контролю якості, що містять текст маркування упаковки, та рекомендує до погодження технологічний регламент або технологію виробництва. За бажанням заявника Центр може рекомендувати МОЗ до затвердження коротку характеристику на лікарський засіб.
На підставі поданих Центром висновків та рекомендацій МОЗ у місячний строк після їх отримання приймає рішення, що затверджується наказом МОЗ про державну реєстрацію (перереєстрацію) лікарського засобу або про відмову в державній реєстрації (перереєстрації) лікарського засобу, і розміщує цей наказ на своєму офіційному сайті.
Рішенням про державну реєстрацію затверджуються фармакопейна стаття або методи контролю якості лікарського засобу, що містять текст маркування первинної та вторинної (за наявності) упаковок, інструкція для медичного застосування лікарського засобу, здійснюється погодження технологічного регламенту або технології виробництва лікарського засобу, а також лікарському засобу присвоюється реєстраційний номер, який вноситься до Державного реєстру лікарських засобів та міжвідомчої бази даних про зареєстровані в Україні лікарські засоби. До Державного реєстру заноситься інформація щодо можливості рекламування лікарського засобу.
Зберігання документів, наведених у цьому Порядку та додатках до нього, які надаються заявником з метою державної реєстрації (перереєстрації), здійснюється Центром.
Видача затверджених МОЗ при реєстрації (перереєстрації) лікарського засобу документів на лікарський засіб здійснюється МОЗ за принципом «єдиного вікна».
3.8. Лікарський засіб не може бути рекомендований до державної реєстрації, якщо за результатами спеціалізованої експертизи не підтвердились висновки щодо його ефективності, безпеки та якості, а саме:
лікарський засіб шкідливий для здоров’я людини (переважання ризику від застосування лікарського засобу над очікуваною користю) та/або терапевтична ефективність засобу відсутня за умов застосування згідно з інструкцією для медичного застосування;
склад лікарського засобу не відповідає зазначеному в матеріалах реєстраційного досьє;
матеріали реєстраційного досьє, що додаються до заяви, не відповідають вимогам цього Порядку та додатків до нього;
якщо під час проведення спеціалізованої експертизи матеріалів реєстраційного досьє на лікарський засіб, що містить ту саму діючу речовину, що й референтний/оригінальний лікарський засіб, було виявлено, що використано або посилались на інформацію щодо ефективності та безпеки цього референтного/оригінального лікарського засобу, зареєстрованого в Україні вперше на підставі поданої в повному обсязі (повної) реєстраційної інформації, у період раніше ніж за 5 років з дня першої реєстрації референтного/оригінального лікарського засобу в Україні. Зазначена вимога не поширюється на випадки, коли заявник відповідно до закону одержав право посилатися та/або використовувати реєстраційну інформацію референтного/оригінального лікарського засобу або подав власну повну реєстраційну інформацію, що відповідає вимогам до реєстраційної інформації референтного/оригінального лікарського засобу, або в інших випадках, передбачених статтею 9 Закону України «Про лікарські засоби»;
якщо внаслідок такої реєстрації будуть порушені захищені патентом чинні майнові права інтелектуальної власності, у тому числі при виробництві, використанні, продажу лікарських засобів. Відомості про факт такого порушення надаються МОЗ та Центру.
3.9. Центром у разі наявності підстав щодо неможливості державної перереєстрації лікарського засобу подаються відповідні висновки та рекомендації до кваліфікаційної комісії МОЗ. За відсутності таких підстав Центр укладає договір із заявником щодо експертизи матеріалів реєстраційного досьє про перереєстрацію та проводить попередню та спеціалізовану експертизи відповідно до вимог цього Порядку.
3.10. Лікарський засіб не може бути рекомендований до державної перереєстрації, якщо під час проведення експертизи матеріалів реєстраційного досьє за результатами фармаконагляду було встановлено, що лікарський засіб шкідливий для здоров’я людини (переважання ризику від застосування лікарського засобу над очікуваною користю) та/або терапевтична ефективність засобу відсутня за умови застосування згідно з інструкцією для медичного застосування.
У перереєстрації також може бути відмовлено, якщо матеріали реєстраційного досьє не відповідають вимогам, встановленим цим Порядком.
Про прийняте рішення щодо відмови у перереєстрації МОЗ у строк до 10 робочих днів надсилає заявнику письмову мотивовану відповідь.
3.11. За наявності об’єктивних та обґрунтованих причин і у випадках, викладених у пункті 4.7 глави 4 розділу VII цього Порядку та у главі 5 розділу Х цього Порядку, після консультацій із заявником лікарський засіб може бути рекомендований до державної реєстрації за умови додержання заявником таких зобов’язань:
продовжувати дослідження щодо лікарського засобу після реєстрації;
повідомляти про побічні реакції на лікарський засіб.
3.12. У разі незгоди з результатами попередньої та/або спеціалізованої експертизи заявник може оскаржити рішення Центру до МОЗ протягом 30 робочих днів з дати його одержання. Відповідні матеріали для обґрунтування оскарження мають бути надані заявником до МОЗ протягом 30 робочих днів після подання такого оскарження.
Центр за направленням МОЗ здійснює спеціалізовану експертизу наданих заявником додаткових матеріалів у строк до 60 робочих днів з дати їх одержання та надає відповідні обґрунтовані рекомендації МОЗ. МОЗ у місячний строк приймає відповідне рішення, про що інформує письмово заявника.
3.13. За бажанням заявника Центр надає заявнику доступ до окремих розділів електронної бази даних у частині лікарських засобів, поданих за направленням МОЗ на експертизу до Центру. Умови доступу заявника до окремих розділів електронної бази визначаються умовами договору на проведення експертизи, укладеного між заявником та Центром.
IV. Порядок проведення експертизи матеріалів про внесення змін до матеріалів реєстраційного досьє на лікарський засіб
4.1. Проведенню експертизи за бажанням заявника передує надання Центром безкоштовних консультацій з питань внесення змін до реєстраційних матеріалів на лікарський засіб.
4.2. Заявник зобов’язаний повідомити МОЗ про будь-які зміни, наведені у додатку 11 до цього Порядку та в підпунктах 4.2.2-4.2.4, 4.4.4 цього розділу, щодо зареєстрованого лікарського засобу з наданням вичерпної інформації про причини цих змін та їх можливий вплив на ефективність, безпеку, якість лікарського засобу і внести відповідні зміни до матеріалів реєстраційного досьє.
4.2.1. За характером зміни класифікують на:
зміни типу IА — незначні зміни, що виявляють незначний вплив або не виявляють впливу на якість, безпеку та ефективність лікарського засобу, які стосуються внесення поправок до змісту матеріалів реєстраційного досьє, поданих на момент прийняття рішення про реєстрацію (перереєстрацію) лікарського засобу, і не потребують його нової реєстрації;
зміни типу ІБ — незначні зміни, які не можуть бути змінами типу ІА та типу ІІ і не потребують нової реєстрації;
зміни типу II — будь-які зміни до матеріалів реєстраційного досьє, що не потребують нової реєстрації лікарського засобу та можуть виявляти значний вплив на його якість, безпеку та ефективність, але не можуть розглядатися як зміни типу IА та ІБ.
4.2.2. Термінові зміни, що стосуються безпеки лікарського засобу, — термінові тимчасові обмеження, пов’язані з безпекою використання лікарського засобу, які впроваджуються заявником у разі виявлення ризику для здоров’я людини при застосуванні зареєстрованого (перереєстрованого) лікарського засобу. Про кожну причину, характер і дату передбачуваного введення в дію тимчасових термінових обмежень заявник повідомляє негайно Центр. У разі якщо протягом 24 годин з моменту отримання такої інформації в Центру не виникає заперечень, заявник вводить у дію ці термінові обмеження і одночасно не пізніше ніж через 48 годин від моменту прийняття рішення щодо введення термінових змін подає заяву на внесення таких змін згідно з цим Порядком.
У разі якщо заявнику стало відомо про виявлені небезпечні властивості лікарського засобу для здоров’я або життя людини, що стали підставою для прийняття заявником рішення про введення обмежень до його застосування, заявник повідомляє Центр у будь-який спосіб та подає заяву про зміни, пов’язані з виявленими проблемами з безпеки, разом з відповідною документацією щодо цих виявлених проблем з безпеки негайно, але не пізніше ніж протягом 15 календарних днів від дати отримання цієї інформації. Запропоновані зміни затверджуються відповідним наказом МОЗ, після чого лікарський засіб має застосовуватись відповідно до зміненої інструкції для медичного застосування.
У разі якщо Центр отримав обґрунтовану інформацію про виявлені небезпечні властивості лікарського засобу для здоров’я або життя людини, що стали підставою для прийняття відповідного рішення про введення обмежень для застосування лікарського засобу в установленому законодавством порядку, Центр повідомляє про це заявника у будь-який спосіб негайно, але не пізніше ніж протягом 15 календарних днів з дати прийняття такого рішення.
У разі згоди заявника з прийнятим рішенням він повинен негайно, але не пізніше ніж протягом 15 календарних днів з дати отримання цієї інформації подати до МОЗ заяву про внесення змін щодо безпеки лікарського засобу до реєстраційних матеріалів. Запропоновані зміни затверджуються відповідним наказом МОЗ, після чого лікарський засіб повинен вироблятися та застосовуватись відповідно до зміненої інструкції для медичного застосування.
У разі незгоди заявника з прийнятим рішенням та його відмови від унесення змін щодо безпеки лікарського засобу до матеріалів реєстраційного досьє без наданих обґрунтувань або відсутності його реагування стосовно цього рішення МОЗ приймає рішення про заборону (тимчасову заборону) застосування лікарського засобу шляхом припинення дії реєстраційного посвідчення у порядку, передбаченому чинним законодавством.
У разі якщо 15-й календарний день припадає на вихідний або святковий день, інформація повинна бути надана в перший після нього робочий день.
4.2.3. Технічна помилка — невідповідності, допущені у затверджених при реєстрації (перереєстрації) документах. Це не потребує проведення експертизи експертами з питань реєстрації Центру та виявляється при порівнянні затверджених при реєстрації (перереєстрації) документів з матеріалами реєстраційного досьє заявника. До технічних помилок належать:
а) помилки, пов’язані з орфографічними та/або граматичними помилками, в тому числі з транслітерацією;
б) помилки, пов’язані з некоректним перекладом на українську та/або іншу мову адміністративних даних, таких як форма власності, найменування, місцезнаходження заявника/виробника лікарського засобу;
в) невідповідність у МКЯ, що містить маркування первинної та вторинної (за наявності) упаковок лікарського засобу, короткій характеристиці лікарського засобу, інструкції для медичного застосування лікарського засобу;
г) помилки у МКЯ, пов’язані з перекладом або перенесенням інформації щодо:
назви, кількісного та якісного складу лікарського засобу та/або пакувальних матеріалів, терміну придатності, умов зберігання, найменування та місцезнаходження виробника/заявника тощо;
помилки у графічному зображенні первинної, вторинної упаковок, зокрема штрих-коді, місцезнаходженні, номері серії, даті виробництва, терміні придатності тощо; орфографічні та/або граматичні помилки;
помилки у специфікації та/або методах контролю, зокрема цифрові, та помилки, пов’язані з перекладом; помилки в розрахункових формулах, що виникли при скороченні розгорнутих формул;
помилки при посиланні на загальні статті та монографії фармакопеї, нормативні документи за умови, що основна інформація розділу містить вищезазначені посилання;
ґ) невідповідність інформації (різночитання) у межах одного документа;
д) орфографічні та/або граматичні помилки у короткій характеристиці лікарського засобу та інструкції для медичного застосування лікарського засобу за умови, що вони не впливають на зміст;
е) наявність розбіжностей у короткій характеристиці лікарського засобу та інструкції для медичного застосування лікарського засобу, які були затверджені одночасно, щодо викладення інформації, яка стосується медичного застосування лікарського засобу; зазначення доз препарату в одиницях лікарської форми, детальніше викладення інформації в одному з документів;
є) внесення до затверджених під час реєстрації (перереєстрації) лікарського засобу документів назви лікарського засобу або МНН за умови, що така інформація наявна хоча б в одному з таких документів;
ж) орфографічні та/або граматичні помилки у реєстраційному посвідченні на лікарський засіб; наявність розбіжностей між інформацією, наведеною у реєстраційному посвідченні на лікарський засіб, та в затверджених короткій характеристиці, та/або інструкції для медичного застосування, та/або МКЯ.
Виправлення технічної помилки затверджується наказом МОЗ.
4.2.4. Зміни, що потребують нової реєстрації лікарського засобу:
4.2.4.1. Зміни активних речовин:
а) заміна активної речовини на іншу сіль, ефір, похідну тощо за умови, що характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих або використання інших ізомерів, або зміна ізомерного складу, або незначні зміни молекулярної структури речовин біологічного походження, за винятком зміни у АФІ або діючій речовині вакцин для профілактики грипу;
б) зміна вектора, який використовується для виробництва антигена або вихідного матеріалу, включаючи новий головний банк клітин від іншого джерела, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
в) новий ліганд або механізм з’єднання для радіофармацевтичного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
г) застосування інших екстрагентів або співвідношення рослинна лікарська сировина/рослинний препарат для рослинного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих.
4.2.4.2. Зміни сили дії, лікарської форми та способу застосування:
а) зміна біодоступності;
б) зміна фармакокінетики;
в) зміна або введення додаткового дозування лікарського засобу (додаткова доза);
г) зміна або додання нової лікарської форми;
ґ) зміна або додання нового шляху введення (для парентеральних форм у зв’язку з відмінностями в ефективності та безпеці лікарського засобу при внутрішньоартеріальному, внутрішньовенному, внутрішньом’язовому та інших шляхах уведення).
При таких змінах заявник разом з обґрунтуванням потреби внесення змін подає до Центру відповідні розділи матеріалів реєстраційного досьє, які обґрунтовують указані зміни і є достатніми для експертизи лікарського засобу.
4.3. Експертизі підлягає кожна конкретна зміна навіть за умови одночасного їх внесення.
4.4. Для проведення експертизи матеріалів про внесення змін заявник подає до МОЗ:
для змін типу IА, ІБ або II — заяву згідно з додатком 12 до цього Порядку;
для змін, що потребують нової реєстрації, — заяву згідно з додатком 1 до цього Порядку;
для виправлення технічної помилки — лист у довільній формі, у якому зазначені помилка та обґрунтування її виправлення;
для технічних змін — лист у довільній формі, у якому зазначено обґрунтування змін.
До заяви додаються:
копія чинного реєстраційного посвідчення;
підтвердження затвердження запропонованих змін уповноваженим органом країни виробника/заявника (за необхідності).
За результатами розгляду заяви МОЗ надсилає до Центру лист-направлення разом із копією заяви для внесення змін. Інформація про подану до МОЗ заяву вноситься до електронної бази даних.
Після надходження до Центру листа-направлення від МОЗ Центр протягом 3 робочих днів інформує про це заявника і надає рахунок на сплату згідно з договором між заявником та Центром. Заявник здійснює оплату та подає до Центру матеріали реєстраційного досьє, що обґрунтовують запропоновані зміни. Центр приймає матеріали реєстраційного досьє у строк, що не перевищує 3 робочі дні з дати звернення заявника.
У разі ненадання заявником матеріалів реєстраційного досьє протягом 3 місяців з дати інформування Центром заявника Центр надає МОЗ рекомендації щодо зняття заяви з розгляду. МОЗ протягом 5 робочих днів розглядає надані рекомендації та приймає відповідне рішення. Про прийняте рішення МОЗ письмово повідомляє заявника. При цьому вартість проведення експертних робіт заявнику не повертається. Надалі така заява може бути подана до МОЗ за процедурою, установленою цим Порядком. Відповідна інформація вноситься до електронної бази даних.
4.4.1. У разі внесення змін типу IА або IБ до реєстраційного досьє одного лікарського засобу заява повинна стосуватися лише однієї зміни типу IА або IБ. У разі одночасного внесення декількох змін типу IА або IБ окрема заява подається щодо кожної зміни, яка повинна містити посилання на інші заяви про внесення змін.
У разі якщо зміна типу IА призводить до інших послідовних змін типу IА, одна заява може включати всі послідовні зміни разом з описом відповідностей між такими послідовними змінами типу IА.
У разі якщо зміна типу IБ призводить до інших послідовних змін типу IА або IБ, одна заява може включати всі послідовні зміни разом з описом відповідностей між такими послідовними змінами типу IА або ІБ.
Якщо зміни потребують послідовної перевірки короткої характеристики, інструкції для медичного застосування лікарського засобу, МКЯ, що містить маркування первинної, вторинної (за наявності) упаковок, це вважається частиною змін.
4.4.2. У разі внесення значних змін типу II заява повинна стосуватися лише однієї зміни типу II. Якщо необхідно внести декілька змін типу II, окрема заява повинна подаватися щодо кожної зміни; кожна така заява повинна містити посилання на іншу заяву.
У разі якщо зміна типу II призводить до інших послідовних змін типу II, одна заява може включати всі послідовні зміни разом з описом відповідностей між такими послідовними змінами типу II.
Якщо зміни потребують послідовної перевірки короткої характеристики, інструкції для медичного застосування, МКЯ, що містить маркування первинної, вторинної (за наявності) упаковок, це вважається частиною змін.
4.4.3. У разі внесення технічних змін, що стосуються зміни найменування виробника та/або заявника, що вносяться заявником для приведення у відповідність з нормативно-правовими актами України, зокрема Законом України «Про акціонерні товариства», або стосуються, наприклад, зміни контактної інформації, що призводить до необхідності оновлення інформації у відповідних розділах матеріалів реєстраційного досьє, такі зміни не потребують проведення експертизи експертами з питань реєстрації Центру. Перевірка матеріалів реєстраційного досьє здійснюється Центром на безоплатній основі та у строк до 10 робочих днів з дати надходження листа-направлення від МОЗ.
4.4.4. У випадку відсутності в додатку 11 до цього Порядку класифікації змін, що вносяться до матеріалів реєстраційного досьє під час дії реєстраційного посвідчення, ця зміна зазначається як підпункт «Інші зміни» у відповідному пункті додатка 12 до цього Порядку.
Дані зміни за класифікацією можуть бути як зміни типу ІА, ІБ, так і зміни ІІ типу.
До змін типу ІА відносять:
а) зміни виключно адміністративного характеру:
зміна найменування та/або місцезнаходження заявника (власника реєстраційного посвідчення);
зміна найменування та/або місцезнаходження виробника готового лікарського засобу;
зміна найменування та/або місцезнаходження виробника субстанції;
б) виключення будь-якої виробничої дільниці для таких стадій виробництва готового лікарського засобу: виробництво АФІ або діючої речовини, виробництво проміжного продукту чи готового лікарського засобу, пакування, випуск серії, контроль серії;
в) зміни у МКЯ АФІ або діючої речовини або вихідної сировини/проміжних продуктів або реагентів, що використовуються у виробничому процесі, за умови, що не змінюється сама методика (наприклад, змінюється довжина хроматографічної колонки, але не її тип), проведена валідація зміненого методу контролю, яка показує відповідність попередньому методу, та не змінюються межі, встановлені у специфікації;
г) зміни, що вносяться у зв’язку зі змінами у ДФУ, або Європейській фармакопеї, або іншій фармакопеї (якщо субстанція не описана у ДФУ чи Європейській фармакопеї), або приведення у відповідність до цих фармакопей;
ґ) зміни в упаковці лікарського засобу, що не контактує безпосередньо з лікарським засобом та не впливає на доставку, застосування, безпеку або стабільність лікарського засобу;
д) зміни у специфікаціях на лікарський засіб, АФІ або діючу речовину, вихідні матеріали, проміжні продукти, допоміжні речовини, що призводять до звуження меж, за умови, що нові параметри залишаються у рамках заявленого діапазону та не є результатом непередбачених обставин у процесі виробництва.
До змін типу ІІ відносять:
а) зміни, пов’язані з введенням нового показання до застосування або змінами вже існуючого;
б) значні зміни у короткій характеристиці лікарського засобу, зокрема, на підставі нових відомостей щодо якості, результатів доклінічних досліджень та клінічних випробувань, нових повідомлень про безпеку лікарського засобу, включаючи термінові зміни, що стосуються безпеки;
в) зміни, пов’язані зі змінами поза діапазоном затверджених специфікацій, меж та критеріїв прийнятності, якщо це не пов’язано із приведенням у відповідність до вимог ДФУ чи Європейської фармакопеї;
г) зміни, що стосуються суттєвих змін у виробничому процесі, складі лікарського засобу, специфікаціях, складі домішок у АФІ або діючій речовині чи готового лікарського засобу, які можуть мати істотний вплив на його якість, безпеку та ефективність;
ґ) зміни, пов’язані зі змінами у виробничому процесі або дільниці для виробництва АФІ або діючої речовини біологічного походження;
д) зміни, пов’язані з введенням нового проектного простору або розширенням затвердженого, який був розроблений відповідно до Європейських та міжнародних наукових керівництв;
е) зміни заявника, крім одночасних змін заявника та місцезнаходження виробництва.
До змін типу ІБ відносять незначні зміни, які не можуть бути змінами типу ІА та типу ІІ і не потребують нової реєстрації.
4.5. Експертиза пропозицій щодо внесення змін до матеріалів реєстраційного досьє проводиться у Центрі на підставі листа-направлення МОЗ після отримання матеріалів реєстраційного досьє, що підтверджують запропоновані зміни, та оплати її вартості, установленої договором між заявником та Центром.
4.6. При внесенні змін типу ІА, ІБ до Центру на експертизу подаються матеріали згідно з додатком 11 до цього Порядку.
При внесенні змін ІІ-го типу заявник повинен надати Центру:
матеріали, які обґрунтовують унесення змін;
відповідні матеріали з поправками, унесеними відповідно до заяви;
доповнені або оновлені існуючі експертні звіти (огляди, висновки) з урахуванням змін (за необхідності);
якщо зміни призводять до змін у МКЯ, що містять маркування первинної, вторинної (за наявності) упаковок лікарського засобу, інструкції для медичного застосування, примірники цих документів для затвердження, викладені відповідно до вимог розділів XVI та XVІII цього Порядку та додатка 10 до цього Порядку.
У разі внесення змін в інструкцію для медичного застосування, які не стосуються та не впливають на клінічну інформацію (назва лікарського засобу, склад, лікарська форма, основні фізико-хімічні властивості, виробник, місцезнаходження, умови зберігання, термін придатності тощо), у складі інших матеріалів реєстраційного досьє разом із заявою заявник подає зміни до інструкції для медичного застосування лікарського засобу за формою, наведеною у додатку 13 до цього Порядку.
За бажанням заявника матеріали реєстраційного досьє можуть подаватися у вигляді змін до певних розділів реєстраційного досьє у форматі ЗТД.
4.7. Експертиза матеріалів про внесення змін складається з таких етапів:
попередньої експертизи матеріалів реєстраційного досьє, метою якої є перевірка їх відповідності заявленому типу змін, яка триває 5 робочих днів з дати їх надходження до Центру;
спеціалізованої експертизи матеріалів реєстраційного досьє про внесення змін.
Розгляд виправлення технічних помилок здійснюється у рамках попередньої експертизи (протягом 20 днів з дати надання заявником листа-звернення та необхідних документів, що їх обґрунтовують) шляхом порівняння виправлених матеріалів реєстраційного досьє з матеріалами оригінального реєстраційного досьє заявника.
4.8. У разі відповідності матеріалів реєстраційного досьє лікарського засобу заявленому типу змін, встановлених за результатами попередньої експертизи матеріалів реєстраційного досьє, вони направляються на спеціалізовану експертизу, а у випадку виправлення технічних помилок та внесення технічних змін Центром надаються рекомендації МОЗ щодо їх затвердження, про що Центр письмово повідомляє заявника та вносить відповідну інформацію до електронної бази даних.
При процедурі виправлення технічних помилок та внесенні технічних змін спеціалізована експертиза матеріалів реєстраційного досьє не проводиться.
4.9. У разі невідповідності матеріалів реєстраційного досьє лікарського засобу заявленому типу змін, встановлених за результатами попередньої експертизи матеріалів реєстраційного досьє, Центр повідомляє про це заявника письмово і надає відповідне обґрунтування з посиланням на номери розділів, глав, статей, пунктів цього Порядку або одноразово запитує у заявника додаткові дані та/або інформацію, необхідні для забезпечення відповідності наданих матеріалів реєстраційного досьє вимогам. Відповідна інформація вноситься до електронної бази даних.
Заявник має надати додаткові дані та/або інформацію згідно із зауваженнями Центру, або листа з обґрунтуванням термінів, необхідних для їх доопрацювання, у строк до 30 робочих днів. Час, потрібний для доопрацювання, не входить до строку проведення експертизи. Центр має прийняти доопрацьовані матеріали протягом 3 робочих днів після звернення заявника. Відповідна інформація вноситься до електронної бази даних.
Якщо заявник не надає Центру додаткових даних та/або інформації, а також якщо надані заявником додаткові дані та/або інформація не забезпечують відповідності матеріалів реєстраційного досьє встановленим вимогам, то Центр надає МОЗ рекомендації щодо відмови у внесенні змін до матеріалів реєстраційного досьє лікарського засобу.
Для змін типу ІА додаткові матеріали та/або інформація у заявника не запитуються. У разі невідповідності матеріалів реєстраційного досьє лікарського засобу заявленому типу змін, вимоги до яких викладено в додатку 11 до цього Порядку, Центр надає МОЗ рекомендації щодо відмови у внесенні змін до матеріалів реєстраційного досьє лікарського засобу.
МОЗ у строк до 10 робочих днів розглядає надані рекомендації та приймає відповідне рішення, що затверджується наказом. Про прийняте рішення МОЗ письмово повідомляє заявника. При цьому вартість проведення експертних робіт заявнику не повертається. Відповідна інформація вноситься до електронної бази даних.
Надалі на бажання заявника матеріали про внесення змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення з урахуванням зауважень Центру подаються в установленому порядку.
4.10. Під час проведення спеціалізованої експертизи матеріалів реєстраційного досьє про внесення змін з метою одержання додаткових даних щодо впливу змін на ефективність, безпеку та якість лікарського засобу Центр може одноразово запитати у заявника додаткові дані та/або інформацію з посиланням на номери розділів, глав, статей, пунктів цього Порядку. Час, потрібний для їх підготовки, не входить до строку проведення експертних робіт.
Після отримання запитаних додаткових даних та/або інформації Центр здійснює їх експертизу у строк, що не перевищує 30 календарних днів від дати отримання Центром додаткових матеріалів.
У разі невідповідності наданих матеріалів встановленим вимогам Центр надає МОЗ рекомендації щодо відмови у внесенні змін. Відповідна інформація вноситься до електронної бази даних. МОЗ у строк до 10 робочих днів розглядає надані рекомендації та приймає відповідне рішення, що затверджується наказом МОЗ. Про прийняте рішення МОЗ письмово повідомляє заявнику.
Якщо заявник протягом 60 робочих днів не надсилає запитаних додаткових даних та/або інформації або листа з обґрунтуванням строків, необхідних для їх підготовки, або надає їх у неповному обсязі, Центр в установленому порядку надає МОЗ рекомендації щодо відмови у внесенні змін. Відповідна інформація вноситься до електронної бази даних.
Для змін типу ІА додаткові матеріали та/або інформація у заявника не запитуються. У разі невідповідності матеріалів реєстраційного досьє лікарського засобу заявленому типу змін, вимоги до яких викладено в додатку 11 до цього Порядку, Центр надає МОЗ рекомендації щодо відмови у внесенні змін до матеріалів реєстраційного досьє лікарського засобу.
МОЗ у строк до 10 робочих днів розглядає надані рекомендації та приймає відповідне рішення, що затверджується наказом. Про прийняте рішення МОЗ письмово повідомляє заявника. При цьому вартість проведення експертних робіт заявнику не повертається. Надалі на бажання заявника матеріали про внесення змін можуть бути подані в установленому порядку.
4.11. За результатами експертизи Центр надає рекомендації та висновки до МОЗ щодо внесення змін до матеріалів реєстраційного досьє або змін, що потребують нової реєстрації лікарського засобу, враховуючи при цьому строк, зазначений заявником у заяві, протягом якого заявлені зміни мають бути введені в дію, що відображається в наказі МОЗ щодо затвердження відповідних змін. Відповідна інформація вноситься до електронної бази даних.
Про прийняте рішення МОЗ письмово повідомляє заявника.
У випадку, якщо зміни стосуються інструкції для медичного застосування лікарського засобу, МКЯ, Центр також рекомендує МОЗ до затвердження оновлену інструкцію для медичного застосування або зміни неї, зміни до МКЯ, що включають текст, що наводиться у маркуванні (за необхідності).
На підставі поданих Центром висновків та рекомендацій МОЗ у місячний строк після їх отримання приймає рішення про внесення змін або про відмову в них і розміщує таке рішення на сайті МОЗ у вигляді наказу МОЗ.
Після підписання МОЗ наказу щодо внесення змін видача затверджених МОЗ примірників документів на лікарський засіб, що підтверджують факт затвердження зміни, здійснюється МОЗ за принципом «єдиного вікна».
4.12. У разі незгоди з рекомендаціями Центру за результатами експертизи щодо змін, які потребують нової реєстрації, заявник може оскаржити це рішення до МОЗ протягом 30 робочих днів з дати його одержання. Відповідні матеріали для обґрунтування оскарження мають бути надані заявником до МОЗ протягом 30 робочих днів після подання такого оскарження.
Центр за направленням МОЗ здійснює експертизу наданих заявником матеріалів у строк до 60 робочих днів з дати їх одержання та надає відповідні обґрунтовані рекомендації МОЗ. МОЗ у місячний строк приймає відповідне рішення, про що інформує письмово заявника.
V. Строки проведення експертизи
Для експертизи встановлюються такі строки її проведення:
5.1. Не більше 210 робочих днів, починаючи з дати офіційного надходження до Центру матеріалів реєстраційного досьє на державну реєстрацію, триває експертиза матеріалів щодо лікарського засобу, який подається на державну реєстрацію за повною і незалежною заявою (автономною заявою), а також щодо МІБП, поданого на державну реєстрацію.
Не більше 90 робочих днів, починаючи з дати офіційного надходження до Центру матеріалів реєстраційного досьє, триває експертиза матеріалів щодо лікарських засобів, зазначених у підпунктах 6.10.1 та 6.10.2 пункту 6.10 розділу VI цього Порядку.
5.2. Не більше 90 робочих днів, починаючи з дати офіційного надходження до Центру матеріалів реєстраційного досьє, триває експертиза матеріалів щодо:
лікарських засобів, які подаються на державну реєстрацію відповідно до інших типів заяв, визначених у додатку 1 до цього Порядку;
АФІ або діючих речовин;
лікарських засобів, що подаються на державну перереєстрацію;
традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами;
змін, що потребують нової реєстрації.
5.3. Не більше 60 робочих днів після надходження до Центру відповідних матеріалів реєстраційного досьє триває експертиза матеріалів про внесення змін до матеріалів реєстраційного досьє.
Цей період може бути скорочений через терміновість питання, якщо зміни стосуються безпеки застосування лікарського засобу.
5.4. Датою завершення експертизи матеріалів реєстраційного досьє, зазначених у пунктах 5.1-5.3 цього розділу, вважається дата підписання керівником Центру висновків експертів з питань реєстрації за результатами проведеної експертизи, а саме:
умотивованих висновків щодо ефективності, безпеки та якості лікарського засобу та рекомендацій його до державної реєстрації (перереєстрації);
рекомендацій щодо відмови у державній реєстрації (перереєстрації);
висновків щодо внесення змін до матеріалів реєстраційного досьє чи нової реєстрації лікарського засобу в установленому порядку;
рекомендацій щодо відмови у внесенні змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення на лікарській засіб.
Висновки та рекомендації оформлюються Центром у вигляді переліків лікарських засобів, рекомендованих до реєстрації, перереєстрації, внесення змін у реєстраційні матеріали та внесення до Державного реєстру лікарських засобів, до яких додаються експертні висновки щодо ефективності, безпеки та якості лікарського засобу та примірники інструкцій для медичного застосування лікарського засобу, фармакопейних статей або МКЯ, що містять маркування первинної, вторинної (за наявності) упаковок, із штампом Центру «контрольний примірник»; переліків лікарських засобів, не рекомендованих до реєстрації, перереєстрації, внесення змін у реєстраційні матеріали разом із експертними висновками. Ці матеріали направляються до МОЗ для прийняття рішення.
Одночасно з направленням до МОЗ переліків лікарських засобів, рекомендованих до реєстрації, перереєстрації, внесення змін у реєстраційні матеріали та внесення до Державного реєстру лікарських засобів та лікарських засобів, яким рекомендовано відмовити у реєстрації, перереєстрації, внесенні змін у реєстраційні матеріали, зазначені переліки розміщуються Центром на своєму сайті.
Зберігання матеріалів реєстраційного досьє, наведених у цьому Порядку, здійснюється Центром.
Видача документів заявнику, зокрема інструкції для медичного застосування або змін до неї, короткої характеристики (за необхідності), фармакопейної статті або МКЯ лікарського засобу або змін до них, що містять маркування первинної, вторинної (за наявності) упаковок, здійснюється МОЗ за принципом «єдиного вікна».
5.5. До строків експертних робіт, зазначених у пунктах 5.1-5.3 цього Порядку, не входить час, коли матеріали були на доопрацюванні в заявника, час, необхідний на отримання відповідей від третіх осіб (у т.ч. — уповноважених органів України та/або інших країн) на запити Центру, пов’язані із проведенням експертизи, а також час проведення додаткових вивчень (випробувань).
VI. Основні вимоги до матеріалів реєстраційного досьє
6.1. Матеріали реєстраційного досьє, що надаються на експертизу, повинні містити:
6.1.1 для державної реєстрації — інформацію, яка зазначена у розділах ІХ та Х цього Порядку (готові лікарські форми, продукція in bulk, продукція із in bulk), розділі ХІІІ цього Порядку (традиційні лікарські засоби рослинного походження), розділі ХІV цього Порядку (лікарські засоби, що виробляються за затвердженими прописами) або розділі ХІ цього Порядку (діючі речовини або АФІ, зокрема у формах пелет, гранул та інших формах випуску);
6.1.2 для державної перереєстрації — інформацію, яка зазначена у додатку 7 або додатку 8 до цього Порядку (традиційні лікарські засоби або лікарські засоби, які виробляються згідно із затвердженим прописом);
6.1.3 для внесення змін до матеріалів реєстраційного досьє — інформацію, яка зазначена у додатку 11 до цього Порядку, або інформацію, необхідну для експертизи для внесення змін ІІ типу з урахуванням вимог пункту 4.6 розділу IV цього Порядку, та змін, що потребують нової реєстрації з урахуванням вимог додатка 5 до цього Порядку та розділів ІХ та Х цього Порядку.
При підготовці матеріалів реєстраційного досьє заявник має керуватися вимогами, викладеними у розділах VII та VIII цього Порядку (тільки при перереєстрації), або при підготовці матеріалів реєстраційного досьє у форматі ЗТД — вимогами розділів ІХ та Х цього Порядку, з урахуванням вимог розділів XVI, XVII, XVIIІ цього Порядку та додатків 10, 14 до цього Порядку, у разі традиційних лікарських засобів рослинного походження та лікарських засобів, що виробляються згідно із затвердженими прописами, — розділів ХІІІ та XIV цього Порядку, гомеопатичних лікарських засобів — розділу ХІІ цього Порядку.
Для перереєстрації лікарських засобів надається комплект матеріалів реєстраційного досьє залежно від типу лікарського засобу відповідно до додатка 7 або 8 до цього Порядку.
Для державної реєстрації лікарських засобів, на які відповідно до чинного законодавства України видано патент, заявник подає копію патенту або ліцензії, якою дозволяється виробництво та продаж зареєстрованого лікарського засобу, а також документ, що підтверджує чинність патенту в Україні. Заявник подає лист, в якому вказується, що права третьої сторони, захищені патентом або передані за ліцензією, не порушуються у зв’язку з реєстрацією лікарського засобу, за формою, наведеною у додатку 15 до цього Порядку.
У разі реєстрації АФІ або діючих речовин можливе їх використання як лікарських засобів в упаковці in bulk, якщо АФІ або діючі речовини частково пройшли технологічні стадії їх переробки у готову лікарську форму (зокрема, у формах: грануляту, пелет, гранул, покритих оболонкою, інших формах випуску). У такому випадку визначається обсяг додаткових матеріалів у форматі ЗТД (додаток 5 до цього Порядку) для підтвердження властивостей, наданих АФІ або діючим речовинам внаслідок зазначеної часткової технологічної переробки (пролонгована або відтермінована дія, стійкість до дії шлункового соку тощо).
6.2. При реєстрації готового лікарського засобу специфікації на діючу речовину (розділи реєстраційного досьє 3.2.S.4 додатка 5 до цього Порядку) можуть містити додаткові показники щодо технологічних параметрів АФІ або діючої речовини, обґрунтовані при фармацевтичній розробці (розділи реєстраційного досьє 3.2.S.2 додатка 5 до цього Порядку), та доповнюють показники специфікацій, що містяться у МКЯ на АФІ або діючу речовину та/або у сертифікаті якості, виданому виробником.
6.3. Вимоги до якості лікарського засобу повинні бути не нижче від встановлених у ДФУ та/або Європейській фармакопеї.
У разі відсутності відповідної монографії на готовий лікарській засіб у ДФУ та/або Європейській фармакопеї вимоги до якості лікарського засобу повинні бути не нижче від встановлених в інших провідних фармакопеях світу (Британська фармакопея, фармакопея США, фармакопея Японії).
У разі відсутності відповідної монографії на готовий лікарській засіб у вищезазначених фармакопеях вимоги до якості генеричного лікарського засобу повинні встановлюватися виробником та відповідним чином обґрунтовуватися на підставі доведення взаємозамінності з референтним препаратом.
У разі внесення змін до загальних або окремих статей ДФУ та/або Європейської фармакопеї та/або Британської фармакопеї та/або фармакопеї США та/або фармакопеї Японії заявник має протягом 6 місяців з дати введення в дію цих змін привести у відповідність до них діючі методи контролю якості зареєстрованого лікарського засобу.
6.4. Від заявника не вимагається надання результатів власних токсикологічних та фармакологічних випробувань або результатів клінічних випробувань, якщо він може надати один з таких доказів:
6.4.1. Лікарський засіб є генериком до референтного лікарського засобу і заявник вже зареєстрованого референтного лікарського засобу згоден з тим, аби при вивченні заяви на генерик були використані дані фармакологічних, токсикологічних та/або клінічних випробувань, які містяться в реєстраційних матеріалах на референтний лікарський засіб (глава 2 розділу Х цього Порядку).
6.4.2. Лікарський засіб є генериком до референтного лікарського засобу, що доведено дослідженнями, проведеними відповідно до розділу ХХ цього Порядку.
Дослідження біодоступності не вимагатимуться від заявника, якщо він може довести відповідність генеричного лікарського засобу критеріям, зазначеним у розділі ХХ цього Порядку.
У разі коли при реєстрації генеричного лікарського засобу, еквівалентність якого має бути доведена фармакокінетичними дослідженнями (біоеквівалентність), результати цих досліджень ще не доступні, а представлені дослідження еквівалентності проведено із застосуванням інших методів, ніж ті, що передбачені розділом ХХ цього Порядку, що належним чином обґрунтовано заявником, то реєстраційне досьє може бути прийняте з урахуванням певних зобов’язань. Ці зобов’язання повинні включати програму досліджень біоеквівалентності генеричного лікарського засобу до референтного згідно з розділом ХХ цього Порядку, які мають бути проведені протягом двох років з дати отримання заявником реєстраційного посвідчення, та результати досліджень мають бути внесені до матеріалів реєстраційного досьє в установленому порядку.
При цьому інструкція для медичного застосування лікарського засобу генеричного лікарського засобу має містити інформацію, ідентичну до тієї, яка наведена в інструкції для медичного застосування референтного лікарського засобу.
Будь-які відмінності інструкції для медичного застосування генеричного лікарського засобу мають обґрунтовуватись результатами, отриманими при багатоцентрових клінічних дослідженнях (глава 2 розділу Х цього Порядку).
6.4.3. Медичне застосування АФІ або діючої речовини, або речовин, які входять до складу лікарського засобу, добре вивчене (абзац двадцять третій розділу ІІ цього Порядку), визнано їх ефективність та задовільний ступінь безпеки (шляхом надання докладних бібліографічних посилань на опубліковані наукові дані) (глава 1 розділу Х цього Порядку).
Якщо надано бібліографічні посилання на опубліковані наукові дані, вони викладаються з урахуванням вимог, наведених у розділах ІХ та Х цього Порядку в обсягах, які дозволяють заявнику не надавати результатів власних токсикологічних та фармакологічних випробувань або результатів клінічних випробувань.
6.5. Якщо лікарські засоби містять у своєму складі відомі АФІ або діючі речовини, які раніше не застосовувались у даному сполученні з терапевтичною метою, то необхідно надати результати токсикологічних, фармакологічних або клінічних випробувань, що належать до цього сполучення; при цьому немає необхідності надавати посилання, які стосуються до кожної складової (глава 4 розділу Х цього Порядку).
Це не стосується випадків, коли комбіновані лікарські засоби містять відомі АФІ або діючі речовини, які раніше використовувались у медичній практиці як окремі лікарські засоби у тих самих лікарській формі, дозах та відповідному співвідношенні для досягнення тієї самої терапевтичної мети. При цьому необхідно надати результати досліджень, що засвідчать як на стадії виробництва, так і протягом терміну зберігання комбінованого лікарського засобу відсутність між складовими будь-якої взаємодії, яка може вплинути на ефективність та безпеку лікарського засобу.
6.6. У випадку реєстрації подібних біологічних лікарських засобів (біосимілярів), які не можуть вважатись генериками через специфічність процесу виробництва, застосовуваної сировини, особливостей молекулярної будови та терапевтичної дії, крім модулів 1, 2, 3 необхідно надати додаткові дані, що підтверджують належний рівень безпеки (токсикологічні чи інші доклінічні вивчення) та ефективності (клінічні випробування) (глава 3 розділу Х цього Порядку).
6.7. Якщо лікарські засоби є гомеопатичними лікарськими засобами, які відповідають умовам розділу ХІІ цього Порядку, чи традиційними лікарськими засобами рослинного походження, які відповідають умовам розділу ХІІІ цього Порядку, або лікарськими засобами, що виробляються згідно із затвердженими МОЗ прописами, які відповідають умовам розділу XIV цього Порядку, від заявника не вимагаються наукові клінічні дані. При цьому матеріали щодо якості лікарського засобу повинні бути надані в повному обсязі, а також у разі виникнення сумнівів щодо його безпеки на запит Центру повинні надаватися дані, необхідні для експертизи безпеки лікарського засобу (глави 10, 11 розділу Х цього Порядку).
6.8. Матеріали щодо державної реєстрації джерела радіонуклідів повинні також містити таку інформацію та характеристики (вимоги викладено у главі 9 розділу Х цього Порядку):
загальний опис системи разом з докладним описом компонентів системи, які можуть вплинути на склад та якість лікарського засобу із вторинних радіонуклідів;
якісні та кількісні характеристики елюату або сублімату.
6.9. Обсяг матеріалів реєстраційного досьє на лікарські засоби, що подаються на державну реєстрацію, визначається типом заяви (додаток 1 або додаток 2 до цього Порядку). Перелік документів для проведення експертизи матеріалів для державної реєстрації АФІ або діючої речовини, медичних газів визначено в додатку 6 до цього Порядку.
6.10. З метою надання необхідної медичної допомоги населенню при лікуванні соціально небезпечних хвороб (туберкульоз, ВІЛ/СНІД, вірусні гепатити, а також рідкісні захворювання) при реєстрації оригінальних лікарських засобів та лікарських засобів, що пройшли процедуру прекваліфікації ВООЗ та включені до переліку ВООЗ прекваліфікованих лікарських засобів, можуть бути застосовані окремі вимоги до матеріалів реєстраційного досьє на такі лікарські засоби:
6.10.1. Оригінальний (інноваційний) лікарський засіб для лікування соціально небезпечних хвороб (туберкульоз, ВІЛ/СНІД, вірусні гепатити, а також лікарський засіб з оригінальною молекулою для лікування рідкісних захворювань) був зареєстрований в країні, регуляторні органи якої застосовують високі стандарти до якості, що відповідають стандартам, рекомендованим ВООЗ, зокрема: Адміністрація з харчових продуктів та лікарських засобів США (FDA); Європейське агентство з медичних продуктів (EMA) (за централізованою процедурою); Агенція з терапевтичних продуктів Швейцарії (Swissmedic); Агентство з лікарських засобів та продуктів медичного призначення Японії (PMDA); Агентство з регулювання лікарських засобів та продуктів медичного призначення Великобританії (MHRA); Австралійська адміністрація лікарських засобів (TGA). Крім того, необхідно надати:
а) офіційний звіт з оцінки лікарського засобу, складений регуляторним органом, завірений печаткою заявника (для фізичної особи — за наявності)/представника заявника;
б) письмове підтвердження від заявника, що заявлений на реєстрацію лікарський засіб відповідає лікарському засобу, що зареєстрований в країні, регуляторний орган якої застосовує високі стандарти до якості, що відповідають стандартам, рекомендованим ВООЗ.
6.10.2. Лікарський засіб для лікування туберкульозу або ВІЛ/СНІДу пройшов процедуру прекваліфікації та включений до переліку ВООЗ прекваліфікованих лікарських засобів (далі — перелік ВООЗ). При цьому поданий на реєстрацію лікарський засіб має вироблятися на виробничій дільниці, що пройшла інспектування ВООЗ у рамках програми прекваліфікації та зазначена у переліку ВООЗ; вид та розмір упаковки заявленого до реєстрації лікарського засобу зазначено в переліку ВООЗ. Крім того, необхідно надати:
а) офіційний звіт ВООЗ з оцінки препарату (WHOPAS(s)), завірений печаткою заявника/представника заявника;
б) офіційний звіт ВООЗ з інспектування виробників та відділів клінічного тестування (WHOPIR(s)), завірений печаткою заявника/представника заявника;
в) офіційне підтвердження від ВООЗ стосовно чинності вищезазначених документів на дату проведення державної реєстрації лікарського засобу в Україні, а також зобов’язання щодо своєчасного інформування Центру у разі анулювання або внесення будь-яких змін до статусу прекваліфікованого лікарського засобу;
г) письмове підтвердження від заявника, що заявлений на реєстрацію лікарський засіб відповідає лікарському засобу, що пройшов прекваліфікацію, та внесений до офіційного переліку ВООЗ на підставі документів, зазначених у підпунктах «а» та «б» цього пункту. При цьому лікарський засіб може реєструватися в Україні під назвою, що відрізняється від назви, під якою він зареєстрований в іншій країні або зазначений у переліку ВООЗ.
6.10.3. У таких випадках заявник може подавати інформацію, що міститься в модулі 1 додатка 5 до цього Порядку (з перекладом на українську або російську мову) та модулі 2 додатка 5 до цього Порядку (з перекладом на українську або російську мову за необхідності), на паперовому носії. Модулі 3, 4 та 5 матеріалів реєстраційного досьє додатка 5 до цього Порядку заявник може подавати в електронному вигляді, а також МКЯ, що містить маркування первинної, вторинної (за наявності) упаковок, інструкцію для медичного застосування лікарського засобу, а за бажанням заявника — коротку характеристику лікарського засобу.
При реєстрації лікарських засобів, визначених у підпунктах 6.10.1 та 6.10.2 пункту 6.10 цього розділу, експертиза реєстраційних матеріалів здійснюється позачергово та без сплати вартості експертних робіт. Передреєстраційний контроль за показниками якості таких лікарських засобів може проводитися за процедурою «Lot Release» (шляхом експертизи матеріалів контролю якості серії лікарського засобу (протоколи контролю якості серії, аналітичні звіти), наданих виробником) (далі — «Lot Release»).
6.11. Матеріали реєстраційного досьє подаються до Центру відповідно до типу заяви у 3-х примірниках. За погодженням з Центром заявник може подавати окремі частини реєстраційного досьє на електронному носії (модуль 5 додатка 5 до цього Порядку).
Матеріали реєстраційного досьє подаються українською, або російською, або англійською мовою. У разі подання реєстраційного досьє англійською мовою заявник має надати також переклад на українську або російську мову модуля 1 додатка 5 до цього Порядку, а на вимогу Центру — модуля 2 додатка 5 до цього Порядку, а також методи контролю якості готового лікарського засобу.
Матеріали реєстраційного досьє, що надаються для експертизи під час державної реєстрації АФІ або діючої речовини, подаються українською, або російською, або англійською мовою. У разі подання реєстраційного досьє англійською мовою заявник має надати також переклад на українську або російську мову модуля 1 додатка 6 до цього Порядку, а на вимогу Центру — модуля 2 додатка 6 до цього Порядку, а також методи контролю якості готового лікарського засобу.
Переклад реєстраційних матеріалів має бути засвідчений підписом особи, яка його здійснила, із зазначенням прізвища, імені, по батькові та посади такої особи, а також скріплений печаткою заявника.
6.12. Перелік документів для проведення експертизи матеріалів для державної перереєстрації лікарського засобу визначено в додатку 7 до цього Порядку або за бажанням заявника — у форматі ЗТД (додаток 5 до цього Порядку), до якого мають додаватися:
регулярно оновлюваний звіт з безпеки лікарського засобу та/або узагальнюючий звіт;
перелік одержаних за останні 5 років в Україні рекламацій на лікарський засіб (у хронологічному порядку) з докладним аналізом причин, що були підставами для видачі рекламацій, та заходів, ужитих заявником для запобігання їм у майбутньому;
перелік гарантій заявника та зобов’язань, наданих після реєстрації/перереєстрації (у хронологічному порядку), із зазначенням характеру, стану виконання (рекомендації щодо післяреєстраційних досліджень, усунення будь-яких недоліків, що надавалися контрольними та іншими органами тощо);
для генериків, у яких на час реєстрації відповідні дані, що підтверджують їх еквівалентність з референтним препаратом, були відсутні, необхідно надати дані, отримані на підставі досліджень відповідно до розділу ХХ цього Порядку. Ці дослідження мають проводитися під час знаходження лікарського засобу на ринку у рамках програми досліджень біоеквівалентності генеричного лікарського засобу до референтного, зазначеної у підпункті 6.4.2 пункту 6.4 цього розділу.
Від заявника не вимагається надання результатів досліджень еквівалентності генеричного лікарського засобу до референтного, отриманих на підставі досліджень відповідно до розділу ХХ цього Порядку, у разі, коли заявлений лікарський засіб застосовується у медичній практиці на території України принаймні протягом 10 років, що підтверджується відповідними бібліографічними даними щодо його ефективності, безпеки та якості.
6.13. Перелік документів для проведення експертизи матеріалів реєстраційного досьє на традиційні лікарські засоби та лікарські засоби, що виробляються згідно із затвердженими прописами, для державної перереєстрації визначено у додатку 8 до цього Порядку.
6.14. Для проведення експертизи матеріалів реєстраційного досьє на АФІ або діючі речовини, які подаються для державної перереєстрації разом із заявою про державну реєстрацію (перереєстрацію) АФІ або діючої речовини, за формою, наведеною у додатку 3 до цього Порядку, додатково подаються:
оновлені методи контролю якості,
оновлені дані про технологію виробництва та інформація щодо відсутності змін у методах контролю якості та технології виробництва АФІ або діючих речовин.
6.15. У разі якщо заявник подає до МОЗ заяву про перереєстрацію лікарського засобу менше ніж за 90 календарних днів до закінчення строку дії реєстраційного посвідчення, заява подається відповідно до додатка 1 до цього Порядку, а для традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами, — до додатка 2 до цього Порядку. Реєстраційні матеріали подаються згідно з додатками 5, 8 до цього Порядку (для традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами), з урахуванням вимог розділів ІХ, Х, ХІІІ, ХІV цього Порядку.
VIІ. Вимоги до матеріалів реєстраційного досьє
(у форматі чотирьох частин)
1. Вимоги до частини I «Резюме досьє» матеріалів реєстраційного досьє
1.1. Адміністративні дані (І.А.):
Лікарський засіб, що є об’єктом заяви про перереєстрацію, ідентифікується за назвою лікарського засобу та за назвою АФІ або діючих речовин разом з лікарською (фармацевтичною) формою, способом введення, силою дії та кінцевою формою випуску готового лікарського засобу, уключаючи упаковку.
Указуються найменування та місцезнаходження заявника разом з найменуванням та місцезнаходженням виробників і виробничих дільниць, пов’язаних з різними стадіями виробництва (уключаючи виробника готового лікарського засобу і виробників діючих речовин), та за необхідності найменування та місцезнаходження імпортера.
Заявник указує кількість томів матеріалів реєстраційного досьє, що супроводжує заяву.
До адміністративних даних належать:
копії ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника (є необхідними як для повного, так і неповного виробництва, а також для різних процесів фасування, пакування і маркування);
засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи;
копія реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ, перелік країн, де даний лікарський засіб зареєстровано/перереєстровано;
копія короткої характеристики лікарського засобу/інструкції для медичного застосування, розробленої та затвердженої відповідно до нормативних вимог країни заявника/виробника, відповідно до встановлених вимог;
перелік країн, у яких було подано заяви на реєстрацію.
1.2. Коротка характеристика лікарського засобу, інструкція для медичного застосування (І.В.):
Копія короткої характеристики лікарського засобу/інструкції для медичного застосування, затвердженої відповідно до нормативних вимог країни заявника/виробника або інших країн, регуляторні органи яких керуються високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ.
Крім того, заявник надає проект маркування на упаковці (первинній, вторинній (за наявності)), інструкцію для медичного застосування лікарського засобу, складені відповідно до вимог розділів ХVI та XVIII цього Порядку.
Заявник пропонує проект короткої характеристики лікарського засобу, складеної відповідно до вимог розділу XVII цього Порядку, структура якої наведена в додатку 14.
1.3. Звіти незалежних експертів (І.С.):
Звіти експертів з документації стосовно хімічних, фармацевтичних та біологічних випробувань лікарського засобу, а також стосовно документації про фармако-токсикологічні та клінічні випробування повинні бути підготовлені експертами-фахівцями, зокрема:
експертом-аналітиком — відомості про те, чи відповідає лікарський засіб заявленому складу, із зазначенням та обґрунтуванням методів контролю, які використовуються у виробництві;
експертом-фармакологом або фахівцем з аналогічною експериментальною компетенцією — дані щодо токсичності лікарського засобу та його фармакологічних властивостей;
експертом-клініцистом — висновки про те, чи зміг він переконатися, що ефекти лікарського засобу в осіб, які його приймали, збігаються з відомостями, наданими заявником відповідно до встановлених вимог; що лікарський засіб добре переноситься; рекомендації відносно дозувань, результати обговорення протипоказань та побічних реакцій.
При цьому за необхідності експерти мають указати підстави для використання бібліографічних посилань на наукову літературу.
Звіти експертів повинні містити змістовну експертизу якості лікарського засобу, досліджень, виконаних на тваринах і за участю людини, а також усі дані, які мають відношення до експертизи. Звіт пишеться таким чином, аби при читанні можна було скласти чітке уявлення про властивості, якість, пропоновані специфікації та методи контролю, про безпеку, ефективність, переваги та недоліки лікарського засобу.
Усі важливі деталі узагальнюються в доповненнях до звітів експертів, за змогою, включаючи дані у формі таблиць та графіків. Звіти експертів та резюме повинні містити чіткі перехресні посилання на інформацію, яка міститься в основній документації.
Кожен експертний звіт повинен готуватися кваліфікованим та досвідченим спеціалістом. Він повинен бути підписаний та датований експертом. До звіту додається коротка інформація про освіту, спеціалізацію та професійний досвід експерта, а також інформація про професійні стосунки між заявником та експертом.
2. Вимоги до частини IІ «Хімічна, фармацевтична та біологічна документація» матеріалів реєстраційного досьє
2.1. Загальні вимоги:
Усі методики випробувань повинні відповідати сучасному науковому рівню, а також перевірені виробником на достовірність. При цьому повинні бути надані результати досліджень з перевірки на достовірність.
Усі процедури випробувань описуються достатньо точно та докладно, аби можна було відтворити їх при проведенні випробування.
Усі спеціальні прилади та устаткування описуються досить докладно та, за змогою, супроводжуються схемою.
За потреби наводяться склади лабораторних реактивів із зазначенням способу їх приготування.
Якщо методики випробувань описані в Державній фармакопеї України або Європейській фармакопеї, то їх опис може бути замінений докладним посиланням на відповідну фармакопею.
2.2. Склад (ІІ.A.):
Матеріали реєстраційного досьє повинні включати опис якісних та кількісних характеристик усіх складових лікарського засобу, а також матеріали щодо фармацевтичної розробки. Зазначені характеристики та матеріали слід подавати відповідно до наведених нижче вимог.
2.2.1. Якісні характеристики:
Під якісними характеристиками усіх складових лікарського засобу мають на увазі зазначення або опис:
діючих речовин;
допоміжної(их) речовини(н) незалежно від її (їх) походження або використовуваної кількості, включаючи барвники, антиоксиданти, консерванти, ад’юванти, стабілізатори, згущувачі, емульгатори, ароматизатори, смакові добавки тощо;
складових речовин оболонок лікарських засобів, призначених для внутрішнього застосування або введення пацієнтам в інший спосіб, — капсули, капсули желатинові, капсули ректальні тощо.
На доповнення до цих характеристик має бути надана будь-яка необхідна інформація про первинну упаковку і, у відповідних випадках, про спосіб її укупорення разом з докладними відомостями про пристрої для застосування або введення лікарського засобу, які будуть постачатися разом з лікарським засобом.
У разі перереєстрації АФІ або діючої речовини у матеріалах реєстраційного досьє, що супроводжують заяву, мають бути наведені такі відомості:
структурна формула, а також обґрунтовані докази структури, наприклад, на основі синтезу та спектральних даних (якщо такі є);
інформація щодо можливої ізомеризації із зазначенням ізомерів або можливості утворення поліморфних модифікацій;
фізико-хімічні та інші властивості, уключаючи біологічну активність;
інформація щодо домішок.
Для радіофармацевтичних лікарських засобів, до яких уводять радіоактивні мітки після постачання виробником, АФІ або діючою речовиною вважається та частина складу лікарської форми, яка призначена для перенесення або зв’язування радіонукліда. Наводяться докладні відомості про джерело радіонукліда. Крім того, указуються усі сполуки, які мають першочергове значення для введення радіоактивної мітки.
У джерелі АФІ або діючими речовинами вважають як первинні, так і вторинні радіонукліди.
2.2.2. Загальноприйнята термінологія, яку використовують при описі складових речовин лікарського засобу, означає таке:
для діючих речовин, які наведені в ДФУ або Європейській фармакопеї, має бути вказана основна назва, яка наведена на початку відповідної монографії, з посиланням на відповідну фармакопею;
для інших діючих речовин має бути наведена МНН, рекомендована ВООЗ, яка може супроводжуватися іншою непатентованою назвою або за її відсутності — точною науковою (хімічною) назвою; для діючих речовин, які не мають МНН або точної наукової назви, мають бути наведені дані про те, яким чином та з чого вони приготовані (за необхідності, ця інформація має бути доповнена відповідними докладними вказівками);
для барвників має бути наведений відповідний код «Е» згідно з Директивою 78/25/ЕЕС «Про зближення законодавчих положень держав-членів щодо барвників, дозволених для застосування у лікарських засобах».
2.2.3. Кількісні характеристики:
а) для того аби навести кількісні характеристики діючих речовин лікарських засобів необхідно, залежно від відповідної лікарської форми, указати масу кожної діючої речовини або кількість одиниць біологічної дії у розрахунку на одиницю дози або на одиницю маси, або об’єму.
Для діючих речовин, які не можна визначити хімічним шляхом, указують одиниці біологічної активності або, якщо такі є, міжнародні одиниці біологічної активності, установлені ВООЗ. Якщо міжнародні одиниці не встановлені, то одиниці біологічної активності повинні бути виражені таким чином, аби надати однозначну інформацію про активність діючих речовин. За змогою має бути вказана біологічна активність на одиницю маси.
Додатково вказуються:
для ін’єкційних форм: маса та кількість одиниць біологічної активності для кожної діючої речовини із розрахунку на об’єм лікарського засобу в одиничній упаковці, ураховуючи у відповідних випадках об’єм лікарського засобу, що використовується після його підготовки для безпосереднього застосування;
для лікарських засобів у формі крапель: маса або кількість одиниць біологічної активності для кожної діючої речовини у кількості крапель, яка відповідає 1 мл або 1 г лікарського засобу;
для сиропів, емульсій, гранульованих та інших лікарських форм, призначених для введення у виміряних кількостях: маса або кількість одиниць біологічної активності для кожної діючої речовини на відмірену кількість;
б) якщо діючі речовини присутні у вигляді складних сполук або похідних, то слід визначити їх у кількісному вираженні, указавши їх сумарну масу, а за необхідності — масу активної частини або частин молекули;
в) кількісний вміст діючої речовини, яка є сіллю або гідратом, завжди виражається у перерахуванні на масу активної частини (частин) молекули;
г) вимога виражати вміст діючих речовин в одиницях маси активних частин, як наведено в підпункті «в» цього пункту, не може бути застосована до радіофармацевтичних лікарських засобів. Для радіонуклідів радіоактивність виражається в бекерелях на певну дату та, якщо потрібно, на певний час із зазначенням часового поясу. Зазначається вид випромінення.
2.3. Фармацевтична розробка (ІІ.А.4.):
Інформація щодо фармацевтичної розробки лікарського засобу повинна містити обґрунтування вибору складу, складових речовин та первинної упаковки, а також пояснення щодо призначення допоміжних речовин у готовому лікарському засобі. Обґрунтування має супроводжуватись науковими даними фармацевтичних досліджень. Необхідно зазначити надлишки в процесі виробництва з їх обґрунтуванням. Крім того, мають бути зазначені характеристики технологічного процесу (критичні параметри), які можуть впливати на відтворюваність від серії до серії, функціональні характеристики та якість лікарського засобу. До цього розділу можуть бути включені публікації літературних джерел.
Повинна бути надана інформація щодо придатності системи упаковка/укупорка з точки зору вибору матеріалів, захисту від вологи та світла, сумісності конструкційних матеріалів з лікарським засобом, уключаючи сорбцію контейнером речовин або видалення речовин з контейнера та/або безпеку матеріалів контейнерів.
Для радіофармацевтичних лікарських засобів має бути додатково наведено обґрунтування хімічної/радіохімічної чистоти та її взаємозв’язок з біорозподілом.
2.4. Метод виготовлення (відомості про технологію виробництва) (ІІ.В.):
Опис способу виробництва лікарського засобу має надаватися у вигляді відомостей про технологію виробництва. Відомості про технологію виробництва лікарського засобу повинні відповідати наведеним нижче вимогам.
Опис способу виробництва складається таким чином, щоб дати відповідний короткий огляд характеру дій, що виконуються під час виробництва лікарського засобу, і повинен містити інформацію про різні стадії виробництва, що дає змогу оцінити, чи можуть процеси, здійснювані при виробництві лікарського засобу, негативно змінювати його компоненти. У разі безперервного виробництва — докладну інформацію про запобіжні заходи, що застосовуються для забезпечення однорідності готової продукції.
З цією метою відомості про технологію виробництва лікарського засобу повинні містити:
а) виробничу формулу (згідно з вимогами, наведеними в пункті 2.4.1 цієї глави);
б) схему стадій технологічного процесу (згідно з вимогами пункту 2.4.2 цієї глави);
в) опис усіх стадій технологічного процесу (згідно з вимогами пункту 2.4.2 цієї глави);
г) інформацію про експериментальні дослідження з валідації виробничого процесу, якщо використовується нестандартний спосіб виробництва або якщо він критичний для продукції;
ґ) докладний опис процесів стерилізації та/або процедур забезпечення асептичних умов (для виробництва стерильних лікарських засобів).
2.4.1. Виробнича формула (рецептура) повинна щонайменше містити таку інформацію:
передбачуваний розмір серії (у разі змінюваного та/або альтернативного розміру серії має бути надане відповідне обґрунтування) та дані, які доводять постійну відповідність готової продукції усім специфікаціям;
назву і кількість усіх використовуваних у ході виробництва речовин, включаючи інгредієнти, які видаляють з продукції під час технологічного процесу, наприклад розчинники. Мають бути також указані речовини, які можуть використовуватися не завжди (наприклад, кислоти і луги для регулювання pH). Для всіх речовин повинні бути надані посилання на стандарти їх якості. Надлишки повинні бути вказані в кількісному вираженні й обґрунтовані в розділі реєстраційного досьє, що описує фармацевтичну розробку (пункт 2.3 цієї глави);
для кожного інгредієнта повинні бути вказані верхня і нижня межі прийнятності для дійсної кількості кожної речовини від номінальної кількості у виробничій рецептурі на серію. Для діючих речовин ці межі прийнятності повинні бути в інтервалі від 95% до 105% від номінальної кількості; для допоміжних речовин межі прийнятності — в інтервалі від 90% до 110% від номінальної кількості є допустимими без додаткового обґрунтування. Можуть допускатися ширші межі прийнятності, які повинні бути обґрунтовані доказом, що серії зі складом, подібним до значень нижніх і верхніх запропонованих меж прийнятності, продовжують відповідати специфікаціям на готову продукцію. Якщо кількість діючої речовини, яка повинна використовуватися, розраховують з урахуванням дійсного кількісного вмісту в серії цієї діючої речовини (факторизація), то це має бути зазначено. Якщо використовують інші речовини для того, щоб загальна маса серії готової продукції дорівнювала масі, передбаченій у виробничій рецептурі на серію, то це також має бути зазначено.
2.4.2. Виробничий процес:
а) схема стадій технологічного процесу повинна містити:
зазначення стадій та операцій технологічного процесу (уключаючи операції пакування готової продукції);
зазначення, де вихідну сировину і матеріали вводять у процес;
зазначення критичних стадій (операцій) технологічного процесу, а також точки проведення контролю процесу, випробувань проміжної продукції або контролю готової продукції;
при перереєстрації АФІ або діючої речовини повинна бути наведена схема синтезу із зазначенням молекулярних формул, маси, діапазонів виходів, хімічної структури вихідних інгредієнтів, проміжної продукції, реактивів, самої АФІ або діючої речовини (включаючи стереохімію), режимів роботи та розчинників (за наявності);
б) опис усіх стадій технологічного процесу має бути достатній для експертизи того, чи можуть ці процеси негативно вплинути на якість лікарського засобу під час виробництва. Опис виробничого процесу, уключаючи упаковку, повинен відображати послідовність стадій і масштаб виробництва. Представлена інформація повинна свідчити про те, що з високим ступенем вірогідності забезпечується відповідність специфікаціям кожної одиниці кожної серії готової продукції. Для цього:
нові (нестандартні) процеси або технології та операції з пакування, які безпосередньо впливають на якість лікарського засобу, мають бути описані більш детально. Має бути вказане обладнання, щонайменше його вид (наприклад, барабанний змішувач, гомогенізатор на лінії) і у відповідних випадках робочі параметри. Надана інформація повинна свідчити про те, що з високою мірою імовірності забезпечується відповідність специфікаціям кожної одиниці кожної серії готової продукції;
для стадій (операцій) процесу необхідно вказати відповідні параметри, такі як час, температура або pH. Числові значення взаємопов’язаних параметрів повинні бути представлені як очікуваний діапазон. Числові діапазони для критичних етапів повинні бути обґрунтовані. У деяких випадках необхідно вказувати умови навколишнього середовища (наприклад, низька вогкість для шипучого лікарського засобу);
наявність альтернативних стадій виробничого процесу (наприклад, два альтернативних способи стерилізації контейнерів) повинна супроводжуватися підтвердженням, що всі процеси, які пропонуються, постійно забезпечуватимуть виробництво готової продукції відповідно до специфікацій;
повинні бути описані точки проведення контролю в процесі виробництва і відповідні межі прийнятності. Так, мають бути надані критерії прийнятності і випробування (з обґрунтуванням, уключаючи експериментальні дані), що виконуються на критичних етапах виробничого процесу для гарантії того, що процес є контрольованим. Потрібно також надати інформацію про якість і контроль проміжної продукції, що виділяється під час виробництва;
має бути наведена інформація про те, яким чином забезпечується однорідність готової продукції;
в) для наборів радіофармацевтичних лікарських засобів опис способу виготовлення включає як докладний опис виробництва набору, так і детальні рекомендації щодо його остаточної обробки для одержання радіоактивного лікарського засобу.
Для радіонуклідів наводяться пов’язані з ними ядерні реакції.
2.5. Методи контролю вихідних матеріалів (ІІ.С.):
2.5.1. Під вихідною сировиною розуміють усі складові речовини лікарського засобу та, за необхідності, компоненти його первинної упаковки.
У разі якщо АФІ або діюча речовина, яка виготовляється не самим заявником, не описана в ДФУ або Європейській фармакопеї або АФІ або діюча речовина описана в ДФУ або Європейській фармакопеї, але її отримують у спосіб, при якому зберігаються домішки, не зазначені в монографії фармакопеї, та монографія не придатна для належного контролю якості АФІ або діючої речовини, то заявник має вжити заходів, аби докладний опис способу виробництва та перевірка правильності процесу були надані безпосередньо виробником АФІ або діючої речовини. Однак у такому разі виробник АФІ або діючої речовини надає заявникові всі дані, які можуть стати необхідними для того, аби заявник міг відповідати за даний лікарський засіб. Виробник надає заявникові документ, у якому вказується, що він гарантує однорідність серій і не вноситиме змін у виробничий процес або специфікації без повідомлення про це заявнику.
Матеріали реєстраційного досьє, які супроводжують заяву на затвердження таких змін, подаються до Центру.
До матеріалів реєстраційного досьє уключаються результати випробувань, пов’язаних з контролем якості всіх складових речовин, що використовуються, включаючи аналіз серій, особливо для АФІ або діючих речовин. Документи мають охоплювати положення, наведені нижче:
а) вихідні речовини, наведені у фармакопеях.
Монографії ДФУ та Європейської фармакопеї застосовуються до всіх діючих речовин, які в них зазначені.
Для усіх складових речовин, що відповідають вимогам ДФУ або Європейської фармакопеї, уважається, що вони відповідають установленим вимогам. У цьому разі опис аналітичних методів може бути замінений докладним посиланням на відповідну фармакопею.
Однак, якщо вихідну речовину, указану в ДФУ або Європейській фармакопеї, одержують у спосіб, при якому залишаються домішки, які не контролюються відповідно до монографій фармакопеї, то слід зазначити ці домішки та їх граничнодопустимий вміст, а також описати методику їх визначення.
Барвники повинні відповідати вимогам чинного законодавства.
Мають бути вказані стандартні тести, які виконуються для перевірки кожної серії вихідної сировини. Якщо тести відрізняються від наведених у фармакопеї, то необхідно надати докази про те, що вихідні речовини відповідають вимогам до їх якості, які пред’являються цією фармакопеєю.
Якщо специфікація, яка міститься у ДФУ або Європейській фармакопеї, не достатня для гарантії якості АФІ або діючої речовини, то Центр може вимагати від заявника надання більш придатних специфікацій.
Центр може надати інформацію вповноваженим органам, відповідальним за відповідну фармакопею. Заявник повинен надати вповноваженим органам цієї фармакопеї докладні відомості про виявлені недоліки та про додаткові специфікації, що застосовуються.
Якщо вихідна речовина не описана ні в ДФУ, ні в Європейській фармакопеї, то припустимим є її відповідність монографії фармакопеї іншої країни; у таких випадках заявник подає копію монографії (за необхідності її переклад) та, за потреби, документи перевірки правильності вказаних у монографії методик випробувань;
б) вихідні речовини, які не наведені у фармакопеях (ДФУ або Європейській фармакопеї).
Якщо складова речовина не наведена у зазначених фармакопеях, то її слід описати у вигляді специфікації, до якої входить така інформація, а саме:
назва речовини, наводяться усі торговельні або наукові синоніми;
опис АФІ або діючої речовини у формі, подібній до застосовуваної у ДФУ або Європейській фармакопеї; її супроводжують необхідними пояснювальними даними, особливо щодо молекулярної структури, а також відповідним описом способу синтезу. Якщо АФІ або діючу речовину можна описати лише за допомогою способу виготовлення, то опис повинен бути достатньо докладним для характеристики АФІ або діючої речовини, постійної як за складом, так і за дією;
методи встановлення тотожності можуть бути описані як у вигляді повного опису методик, які використовуються при виготовленні АФІ або діючої речовини, так і у формі методик для рутинних випробувань;
тести на чистоту описують відносно до сумарної кількості очікуваних домішок, особливо тих, які можуть мати шкідливий вплив, та, за потреби, тих домішок, які при можливому сполученні з іншими АФІ або діючими речовинами, до яких має відношення подана заява, здатні негативно вплинути на стабільність лікарського засобу або викривити результати аналітичних випробувань;
для складних діючих речовин рослинного або тваринного/людського походження необхідно розмежувати випадки, коли наявність потенційних фармакологічних ефектів потребує хімічних, фізичних або біологічних методів контролю основних складових речовин, та випадки, коли діючі речовини містять одну або кілька груп речовин, які мають подібну активність і щодо яких припустиме визначення сумарної кількості;
при використанні речовин тваринного/людського походження повинні бути описані методи, які забезпечують відсутність потенційно патогенних агентів;
для радіонуклідів указуються їх походження, тотожність ізотопу, імовірні домішки, носій, застосування та специфічна активність;
зазначаються особливі застережні заходи, якщо вони необхідні при зберіганні вихідної речовини, та, за необхідності, максимальний період зберігання перед проведенням повторних випробувань;
в) фізико-хімічні властивості, які впливають на біодоступність.
При загальному описі характеристик АФІ або діючих речовин, від яких може залежати біодоступність лікарського засобу, незалежно від того, чи наведені вони у фармакопеях, указуються такі дані:
кристалічна форма та коефіцієнти розчинності;
розмір часток (у відповідних випадках — після їх розпилення);
стан сольватації;
коефіцієнт розподілу масло/вода (за необхідності, якщо ця інформація розглядається як суттєва, додатково можуть бути запитані також значення pK та pH).
Для речовин, що використовуються виключно у формі розчину, необхідно вказувати дані лише за попередньою вимогою;
г) для радіофармацевтичних лікарських засобів до вихідної сировини відносять і речовини, призначені для опромінення;
ґ) при перереєстрації АФІ або діючої речовини повинні бути надані:
перелік усіх матеріалів, що використовуються при виготовленні АФІ або діючої речовини (сировина, вихідні інгредієнти, розчинники, реактиви, каталізатори тощо) із зазначенням стадій (операцій) технологічного процесу, на яких вони використовуються;
інформація щодо якості та контролю цих матеріалів;
інформація (за необхідності), що свідчить про відповідність цих матеріалів вимогам стандартів, які відповідають їх використанню (уключаючи видалення або контроль мікроорганізмів).
2.6. Методи контролю проміжних продуктів (за необхідності) (ІІ.D.):
У матеріалах реєстраційного досьє мають міститися докладні відомості про контрольні випробування продукту на проміжних стадіях виробничого процесу з метою забезпечення сталості технічних характеристик та технологічного процесу.
Ці випробування мають більше значення для контролю відповідності лікарського засобу його складу, коли у виняткових випадках заявник пропонує для випробувань готової продукції аналітичний метод, який не передбачає кількісного визначення усіх діючих речовин (або усіх допоміжних складових речовин, до яких пред’являються ті самі вимоги, що й до діючих речовин). Це застосовується і в разі, коли контроль якості готової продукції залежить від контрольних випробувань, які виконуються в процесі виробництва, особливо якщо лікарський засіб, переважно, характеризують за способом його одержання.
Для лікарських засобів тваринного походження заявник повинен продемонструвати, що лікарський засіб тваринного походження виготовлений в умовах, що зводять до мінімуму ризик передачі губчатої енцефалопатії.
2.7. Методи контролю якості готового лікарського засобу (ІІ.E.):
2.7.1. При контролі готового лікарського засобу вважається, що до його серії належать усі одиниці даної лікарської форми, які були виготовлені з однієї кількості сировини і підлягали однаковій послідовності виробничих операцій та/або стерилізації, або при безперервному технологічному процесі всі одиниці готової продукції, виготовлені за певний час.
У матеріалах реєстраційного досьє повинна міститися специфікація, згідно з якою заявник (виробник) здійснює контроль якості цього лікарського засобу. У специфікації повинні бути зазначені всі випробування, які регулярно виконуються для кожної серії готової продукції. Для випробувань, які виконуються нерегулярно, указується частота їх проведення. Також указуються граничні значення характеристик показників для продукції, що випускається.
Матеріали реєстраційного досьє (специфікації) повинні включати докладні відомості про контрольні випробування готового лікарського засобу, які проводяться на етапі видачі дозволу серії на випуск. Вони повинні відповідати вимогам, наведеним нижче:
загальні характеристики готового лікарського засобу;
ідентифікація та кількісне визначення діючої(их) речовини(н);
ідентифікація та кількісне визначення допоміжних речовин;
випробування безпеки;
вимоги до пакувального матеріалу (внутрішня/зовнішня упаковка) лікарського засобу.
Положення монографій ДФУ або Європейської фармакопеї на лікарські форми та радіофармацевтичні лікарські засоби застосовуються до всіх визначених у них лікарських засобів.
Якщо використовуються методи випробувань та граничнодопустимі величини, відмінні від наведених у монографіях ДФУ або Європейської фармакопеї, то наводиться підтвердження того, що готовий лікарський засіб при випробуванні відповідно до вимог цих монографій відповідає фармакопейним вимогам, які пред’являються до якості відповідної лікарської форми.
2.7.2. Загальні характеристики готового лікарського засобу:
Випробування готового лікарського засобу завжди включають і певні випробування її загальних показників якості. Ці випробування залежно від вимог ДФУ або Європейської фармакопеї пов’язані з контролем середньої маси і максимальних відхилень, механічними, фізичними і мікробіологічними випробуваннями, визначенням органолептичних характеристик, фізичних характеристик, таких як щільність, pH, показник заломлення тощо. Заявник у кожному конкретному випадку вказує на норми і граничнодопустимі значення для кожної з цих характеристик.
Докладно описуються умови проведення випробувань, обладнання/прилади, які використовуються, та норми, якщо вони не наведені в монографіях ДФУ або Європейської фармакопеї і якщо описані у фармакопеях методи не застосовуються.
Крім того, тверді лікарські форми, призначені для приймання внутрішньо, підлягають дослідженню in vitro для визначення ступеня вивільнення та розчинення діючої речовини або речовин; такі дослідження здійснюються також у тих випадках, коли введення лікарського засобу відбувається в інший спосіб, якщо Центр вважатиме це за необхідне.
2.7.3. Ідентифікація та кількісне визначення діючої(их) речовини(н):
Ідентифікація та кількісне визначення діючої(их) речовини(н) проводяться або на усередненій репрезентативній пробі, відібраній з виробленої серії, або на певній кількості одиниць дозування, які аналізуються окремо.
Максимально припустиме відхилення вмісту діючої речовини в готовому лікарському засобі на дату його виробництва не повинно перевищувати ±5%, за винятком відповідним чином обґрунтованих випадків.
На основі проведених випробувань стабільності виробник повинен запропонувати та обґрунтувати граничнодопустиму кількість діючої речовини в готовому лікарському засобі аж до закінчення зазначеного терміну зберігання.
В окремих випадках, якщо лікарський засіб має особливо складний склад і кількісне визначення діючих речовин, яких багато або які присутні в дуже малих кількостях, потребує проведення складних досліджень, що важко виконати для кожної виробленої серії, то кількісне визначення однієї або декількох діючих речовин, які містяться у готовому лікарському засобі, можна не проводити за умови, що такі кількісні визначення проводять на проміжних стадіях технологічного процесу. Це положення може не розповсюджуватися на визначення характеристик цих речовин. Така спрощена методика повинна бути доповнена методом кількісної експертизи, яка дає змогу встановити відповідність лікарського засобу, що надійшов у продаж, його специфікаціям.
Якщо фізико-хімічні методи не можуть надати необхідну інформацію про якість лікарського засобу, обов’язково проводиться кількісне визначення біологічними методами in vivo та in vitro. При таких дослідженнях використовуються стандартні речовини і статистичний аналіз, який дає змогу визначити довірчі границі відповідного показника. Якщо такі випробування неможливо виконати щодо готового лікарського засобу, їх проводять на проміжній стадії якомога ближче до закінчення виробничого процесу.
Якщо відповідно до докладних відомостей, наведених у розділі ІІB, показано, що при виробництві лікарського засобу використовують значний надлишок АФІ або діючої речовини, то опис контрольних випробувань готового лікарського засобу повинен включати хімічне, а за необхідності й фармако-токсикологічне, вивчення змін, яким була піддана ця АФІ або діюча речовина, а також, можливо, визначення характеристик та/або кількісне визначення продуктів розпаду.
2.7.4. Ідентифікація та кількісне визначення допоміжних речовин:
Допоміжні речовини за необхідності піддаються тестам на тотожність.
Пропоновані методики визначення тотожності барвників повинні дати змогу встановити їх відповідність переліку барвників, наведеному у наказі Міністерства охорони здоров’я України від 19 червня 2007 року № 339 «Про затвердження Переліків назв допоміжних речовин та барвників, що входять до складу лікарських засобів» та у додатку до Директиви Ради 78/25/ЄЕС від 12 грудня 1977 року «Про зближення правил країн ЄС щодо барвників, дозволених для застосування у лікарських засобах».
Для консервантів є обов’язковими випробування щодо визначення верхньої та нижньої меж вмісту. Для інших допоміжних речовин, здатних негативно впливати на фізіологічні функції, обов’язковим є визначення верхньої межі. Якщо допоміжна речовина може вплинути на біодоступність діючої речовини, то обов’язковим є визначення верхньої та нижньої меж її вмісту, за винятком випадків, коли необхідна біодоступність гарантується іншими відповідними випробуваннями.
2.7.5. Випробування безпеки:
Крім фармако-токсикологічних випробувань, які надаються у матеріалах реєстраційного досьє, в аналітичних документах наводяться докладні відомості про випробування безпеки, такі як визначення стерильності, бактеріальних ендотоксинів, пірогенності та місцевої переносності на тваринах, мікробіологічної чистоти у тих випадках, коли випробування необхідно виконувати регулярно для підтвердження якості лікарського засобу.
Для радіофармацевтичних лікарських засобів описуються чистота радіонуклідів, радіохімічна чистота та специфічна активність. Відхилення від значення радіоактивності, указаного на етикетці, не повинно перевищувати ±10%. Для джерел потрібні докладні відомості про випробування первинних та вторинних радіонуклідів. Для генераторів-елюатів повинні бути передбачені випробування первинних радіонуклідів та інших компонентів системи генератора.
Для наборів специфікації на готовий лікарський засіб включають випробування характеристик лікарського засобу після введення радіоактивної мітки.
Повинен бути передбачений відповідний контроль радіохімічної чистоти та чистоти радіонуклідів міченої сполуки. Необхідно проводити якісне та кількісне визначення будь-якої речовини, яка має суттєве значення для введення радіоактивної мітки.
2.7.6. Вимоги до упаковки:
Повинен бути наведений опис системи упаковка/укупорка, уключаючи зазначення матеріалів, з яких виготовлено кожний компонент первинної упаковки, а також зазначена специфікація для контролю їх якості.
Для нефункціональних компонентів вторинної упаковки (наприклад, для тих, що не забезпечують додаткового захисту) достатньо наводити лише скорочений опис. Для функціональних компонентів вторинної упаковки повинна бути наведена докладна інформація.
Для пристроїв для введення лікарських засобів та дозуючих пристроїв необхідно надати СЕ-сертифікат (підтвердження відповідності Директиві 93/42/ЕЕС «Медичні прилади. Медичне обладнання. Медичні пристрої»), за наявності.
2.8. Стабільність (ІІ.F.):
У матеріалах реєстраційного досьє наводиться нижчезазначена інформація. Наводяться опис досліджень, на підставі яких заявник визначив пропонований термін зберігання, рекомендовані умови зберігання та специфікації на дату закінчення терміну зберігання.
Якщо в готовому лікарському засобі можливе утворення продуктів розпаду, то заявник повинен указати ці продукти розпаду та навести методи визначення їх характеристик та методики випробувань.
Висновки містять результати аналізів, які обґрунтовують пропонований термін зберігання при рекомендованих умовах зберігання і специфікації на готовий лікарський засіб на час закінчення терміну зберігання при цих рекомендованих умовах зберігання.
Указується граничнодопустимий рівень продуктів розпаду на дату закінчення терміну зберігання.
Якщо можливий ризик взаємодії лікарського засобу з первинною упаковкою, особливо в разі ін’єкційних лікарських засобів або аерозолів для системного застосування, то надаються результати досліджень з цього питання.
Для радіофармацевтичних лікарських засобів необхідно наводити інформацію щодо стабільності джерел радіонуклідів, радіонуклідних наборів та лікарських засобів, які містять радіоактивну мітку. Слід задокументовувати стабільність під час використання радіофармацевтичних лікарських засобів у багатодозових флаконах.
2.9. Біодоступність/біоеківалентність (II.G.):
Наводяться дані щодо біодоступності/біоеквівалентності або даються посилання на відповідні розділи частини IV матеріалів реєстраційного досьє (за потреби). У цьому разі відповідні розділи частини IV мають бути включені до складу матеріалів реєстраційного досьє.
У разі застосування процедури біовейвер у частині ІV необхідно надати звіт про проведення досліджень in vitro. Оцінка та проведення досліджень біоеквівалентності або обґрунтування щодо її непроведення мають бути надані відповідно до вимог розділу ХХ цього Порядку та керівних настанов.
3. Вимоги до частини IІІ «Фармакологічна та токсикологічна документація» матеріалів реєстраційного досьє
3.1. Матеріали реєстраційного досьє подаються відповідно до наведених нижче вимог.
Відповідність досліджень безпеки лікарських засобів забезпечується виконанням положень належної лабораторної практики.
Токсикологічні та фармакологічні випробування повинні визначити:
а) потенційну токсичність лікарського засобу та будь-які небезпечні або побічні токсичні ефекти, які можуть спостерігатися у людини при додержанні рекомендованих умов застосування лікарського засобу; ця експертиза повинна бути проведена з урахуванням відповідних патологічних станів;
б) фармакологічні властивості лікарського засобу з урахуванням взаємозв’язку його якісних та кількісних характеристик з рекомендованими умовами застосування у людини. Усі результати повинні бути достовірними та загально прийнятними. При плануванні експериментальних досліджень, а також при експертизі одержаних даних використовуються відповідні методи математичної та статистичної обробки результатів.
Крім того, необхідно надати клініцистам інформацію про потенційний терапевтичний ефект лікарського засобу.
3.2. Якщо лікарський засіб призначений для місцевого застосування, матеріали реєстраційного досьє щодо доклінічних досліджень мають містити дані щодо досліджень системної абсорбції, з урахуванням можливості застосування лікарського засобу на ушкоджених ділянках шкіри та його всмоктування через інші відповідні поверхні. Тільки в тому випадку, коли доведено, що системна абсорбція в даних умовах незначна, можна не досліджувати системну токсичність у разі використання повторних доз та фетотоксичність, а також вплив на репродуктивну функцію.
Однак, якщо в ході досліджень терапевтичної ефективності відзначається системне всмоктування лікарського засобу, матеріали реєстраційного досьє щодо доклінічних досліджень мають містити дані щодо проведених досліджень токсичності на тваринах, а, за необхідності, фетотоксичності.
У всіх випадках необхідно з особливою увагою проводити дослідження місцевої переносності після застосування повторних доз лікарського засобу, включаючи гістологічні дослідження; досліджувати ймовірність сенсибілізації та наявність можливої канцерогенної дії.
3.3. Для біологічних лікарських засобів, таких як лікарські засоби, одержані з крові або плазми крові людини, заявник повинен обґрунтувати виконану програму випробувань лікарського засобу.
При плануванні програми випробувань необхідно також взяти до уваги таке:
усі випробування, які потребують повторного введення лікарського засобу, необхідно планувати з урахуванням імовірної стимуляції утворення антитіл та впливу антитіл на організм;
слід розглянути питання про проведення досліджень репродуктивної функції, ембріональної/фетальної та перинатальної токсичності, а також можливої мутагенної та канцерогенної дії. Якщо причиною токсичності є неактивні речовини, то можна не проводити дослідження, якщо доведено, що ці компоненти видалені з лікарського засобу.
3.4. Для радіофармацевтичних лікарських засобів вважається, що токсичність може бути пов’язана з дозою радіоактивного випромінення. При діагностиці це є наслідком застосування даних лікарських засобів, при терапії — необхідною властивістю. При експертизі безпеки та ефективності радіофармацевтичних лікарських засобів ураховуються їх застосування та дані дозиметрії. Повинен бути задокументований вплив випромінення на органи та тканини. Визначення дози випромінення, що поглинається, проводиться відповідно до прийнятої міжнародної системи залежно від особливостей способу введення цих лікарських засобів.
3.5. Якщо допоміжна речовина використовується у фармацевтичній практиці вперше, необхідно провести її токсикологічні та фармакокінетичні дослідження.
3.6. Якщо існує імовірність значного розпаду лікарського засобу в процесі його зберігання, то слід розглянути питання про проведення токсикологічного дослідження продуктів розпаду.
3.7. Токсичність при однократному введенні та введенні повторних доз (ІІІ.А.):
3.7.1. Дослідження токсичності при однократному введенні (ІІІ.А.1.):
Ці дослідження призначені для якісного та кількісного вивчення гострих токсичних реакцій, які можуть виникнути після однократного введення АФІ або діючої речовини або речовин, які містяться в лікарському засобі, у тих пропорціях та фізико-хімічному стані, в яких вони присутні в готовому лікарському засобі.
Матеріали реєстраційного досьє щодо доклінічних досліджень мають містити інформацію про дослідження гострої токсичності, проведені на двох або більше видах ссавців, які належать до вивчених ліній, а іноді й на одному виді тварин за умови обґрунтування причин для прийняття такого рішення; способів введення (зазвичай — не менше двох різних способів, один з яких ідентичний або подібний до того, який рекомендується для введення лікарського засобу людині, а інший забезпечує системну дію речовини).
Матеріали мають містити інформацію щодо вивчень різних ознак токсичності, уключаючи місцеві реакції. Під час досліджень на тваринах має бути отримана максимальна кількість інформації.
При дослідженні токсичності при однократному введенні мають бути встановлені ознаки гострої токсичності та з’ясована причина загибелі тварини, проведена кількісна експертиза приблизної величини летальної дози у тварин відповідних видів та одержана інформація про залежність ефекту від дози, однак при цьому високого ступеня точності не потрібно.
Ці дослідження можуть дати уявлення про ймовірні прояви гострого передозування у людини та бути корисними для планування досліджень токсичності при введенні тваринам відповідних видів повторних доз.
У разі комбінації діючих речовин матеріали реєстраційного досьє мають містити інформацію щодо проведеного дослідження з визначення наявності або відсутності ефекту підсилення токсичності, а також встановлення нових токсичних ефектів.
3.7.2. Дослідження токсичності при повторних введеннях (ІІІ.А.2.):
Дослідження токсичності при введенні повторних доз призначені для виявлення фізіологічних та/або патолого-анатомічних змін, спричинених повторним введенням досліджуваної діючої речовини або комбінації діючих речовин, та для визначення залежності цих змін від дози.
Матеріали реєстраційного досьє щодо доклінічних досліджень мають містити інформацію щодо проведених двох видів (бажано) випробувань: одне короткострокове (протягом 2-4 тижнів), інше — тривале, тривалість якого залежить від умов клінічного застосування, мета якого — експериментальне визначення нетоксичного діапазону доз лікарського засобу, а звичайна тривалість — 3-6 місяців. Якщо лікарський засіб призначений лише для однократного введення пацієнту, то можлива наявність у матеріалах реєстраційного досьє даних лише одного випробування тривалістю від 2 до 4 тижнів. Якщо тривалість дослідження скорочена або збільшена дослідником, таке його рішення має бути обґрунтоване у матеріалах реєстраційного досьє.
Має бути надано обґрунтування доз.
Матеріали реєстраційного досьє щодо доклінічних досліджень мають містити інформацію щодо досліджень токсичності при введенні повторних доз, проведених на двох видах ссавців, один з яких не повинен належати до гризунів.
Вибір способу введення лікарського засобу залежить від рекомендованого терапевтичного застосування та можливості системної абсорбції лікарського засобу. Слід чітко обумовити метод та частоту введення доз.
Максимальна доза має бути підібрана таким чином, аби можна було виявити шкідливий вплив лікарського засобу. Тоді нижчі дози допоможуть визначити переносність лікарського засобу для тварин.
При проведенні досліджень на дрібних гризунах експеримент та методики контролю мають відповідати масштабам проблеми, що вивчалася, та дати змогу визначити довірчі межі.
Експертиза матеріалів доклінічних вивчень щодо токсичних ефектів повинна базуватися на вивченні поведінки тварин та їх зросту, гематологічних та біохімічних дослідженнях, особливо тих, що мають відношення до механізмів виведення лікарського засобу, а також на даних розтину та супутніх гістологічних дослідженнях.
Якщо лікарський засіб представлений новою комбінацією відомих діючих речовин, досліджених відповідно до вимог, матеріали реєстраційного досьє щодо доклінічних досліджень можуть містити інформацію щодо внесення дослідником необхідних змін у методику проведення тривалих досліджень, обґрунтувавши такі зміни. Виняток становлять випадки, коли при дослідженні гострої або підгострої токсичності виявлено нові токсичні ефекти або їх потенціювання.
3.8. Репродуктивна функція (фертильність та загальна характеристика репродуктивної функції) (ІІІ.В.):
Якщо внаслідок проведення інших випробувань одержано дані, які вказують на те, що лікарський засіб може спричиняти шкідливий вплив на потомство або порушувати чоловічу або жіночу репродуктивну функцію, слід провести відповідні випробування.
3.9. Дані щодо ембріотоксичності та тератогенності (ІІІ.С.):
Ці дослідження спрямовані на виявлення токсичного та особливо тератогенного ефектів, які спостерігаються на стадії розвитку організму після запліднення, внаслідок введення досліджуваного лікарського засобу самкам у період вагітності.
Хоча до теперішнього часу такі випробування мали лише обмежене прогностичне значення щодо прийнятності даних результатів для людини, вважається, що вони дають важливу інформацію при виявленні токсичних ефектів, таких як резорбція та інші аномалії.
Відмова від проведення таких випробувань у зв’язку з тим, що лікарський засіб не призначений для жінок дітородного віку або з якихось інших причин, повинна бути відповідним чином обґрунтована.
Матеріали реєстраційного досьє щодо доклінічного вивчення ембріональної/фетальної токсичності мають містити інформацію щодо досліджень на двох видах ссавців, один з яких не повинен належати до гризунів; у пери- та постнатальний періоди — як мінімум на одному виді тварин. Бажано, аби один з використовуваних видів тварин відповідав тому виду, на якому проводились дослідження токсичності при введенні повторних доз лікарського засобу.
Матеріали реєстраційного досьє щодо доклінічного вивчення мають містити інформацію щодо особливостей випробувань (кількість тварин, кількість лікарського засобу, що вводиться, вибір часу введення та критерії експертизи результатів) та залежать від рівня сучасних наукових знань на момент подання заяви та ступеня статистичної достовірності результатів.
3.10. Мутагенний потенціал (ІІІ.D.):
Метою вивчення мутагенної активності є виявлення змін, які досліджувана речовина може спричинити в генетичному матеріалі індивідів або в клітинах та які можуть призвести до постійної та спадкової відмінності нащадків від своїх пращурів. Проведення таких досліджень обов’язкове при вивченні будь-якої нової речовини.
Об’єм і характер результатів, а також критерії їх експертизи залежать від рівня сучасних наукових знань на момент подання заяви.
3.11. Канцерогенний потенціал (ІІІ.Е.):
Проведення випробувань з метою виявлення канцерогенної дії, як правило, потрібне:
а) для речовин, які подібні за хімічною будовою з відомими канцерогенами або канцерогенними сполуками;
б) для речовин, які при проведенні тривалих токсикологічних випробувань спричинили підозрілі зміни;
в) для речовин, у яких під час вивчення мутагенної активності або при проведенні короткострокових випробувань на канцерогенність були одержані підозрілі результати.
Проведення таких випробувань може знадобитися також для речовин, які входять до складу лікарського засобу, який передбачається регулярно приймати протягом тривалого періоду життя пацієнта.
Об’єм і характер результатів, а також критерії їх експертизи залежать від рівня сучасних наукових знань на момент подання заяви.
3.12. Фармакодинаміка (III.F.):
Фармакодинаміка вивчає зміни функцій фізіологічних систем, які спричиняє лікарський засіб, незалежно від того, чи є ці функції нормальними або модифікованими з метою експерименту.
Таке вивчення повинно включати два різних підходи.
По-перше, необхідно адекватно описати всі види дії лікарського засобу, на підставі яких рекомендується його терапевтичне застосування. Результати повинні бути надані в кількісному вираженні (наприклад, за допомогою кривих доза-ефект, час-ефект тощо) та за можливості зіставлені з даними, одержаними при вивченні речовини з відомою активністю. Якщо в заяві стверджується, що терапевтична ефективність досліджуваної речовини вище, то слід продемонструвати статистично достовірну різницю між ефектами.
По-друге, має бути надана загальна фармакологічна характеристика речовини та посилання на побічні реакції. Повинні бути досліджені основні функції фізіологічних систем. Чим ближчі дози, що спричиняють побічні явища, до доз, які спричиняють основний терапевтичний ефект, у зв’язку з яким речовину планується застосовувати, тим більшою повинна бути глибина такого дослідження.
Експериментальні методики (за винятком стандартних методик) повинні бути описані так докладно, аби їх можна було відтворити. Необхідно зрозуміло викласти експериментальні результати та довести їх статистичну достовірність.
Слід вивчити всі кількісні зміни реакцій організму, які виникають при повторному введенні діючої речовини, за винятком окремих випадків, які переконливо обґрунтовані.
Вивчення комбінацій діючих речовин повинно ґрунтуватися або на фармакологічних передумовах, або на характері терапевтичної дії лікарського засобу.
У першому випадку дослідження з вивчення фармакодинаміки демонструють ті взаємодії, які роблять таку комбінацію значущою для терапевтичного застосування.
У другому випадку, коли наукове обґрунтування такої комбінації речовин базується на експериментальній терапії, дослідження встановлюють, чи можливо довести в експериментах на тваринах терапевтичну ефективність такої комбінації речовин. Крім того, ще й значущість будь-якого відміченого побічного явища.
Нова діюча речовина комбінованого лікарського засобу має бути ретельно вивчена.
3.13. Фармакокінетика (ІІІ.G.):
Фармакокінетичні дослідження мають на увазі вивчення усіх процесів, які відбуваються з діючою речовиною в організмі, та охоплюють вивчення її всмоктування, розподіл, біотрансформацію та виведення.
Дослідження цих різних етапів може виконуватися як за допомогою фізичних, хімічних або біологічних методів, так і за допомогою вивчення фактичної фармакодинамічної активності діючої речовини.
Інформація щодо розподілу речовини та її елімінації (тобто щодо біотрансформації та виведення) потрібна в тих випадках, коли ці дані необхідні при визначенні дози лікарського засобу для людини. Вона повинна бути одержана і для хіміотерапевтичних речовин (антибіотиків тощо), і для речовин, застосування яких залежить від їх нефармакодинамічних ефектів (наприклад, різних діагностичних засобів тощо).
Дослідження фармакокінетики фармакологічно діючих речовин є обов’язковим.
Можна не проводити дослідження фармакокінетики нової комбінації відомих речовин, кожна з яких вже вивчена відповідно до положень цього Порядку, якщо таке рішення обґрунтоване результатами випробувань токсичності та експериментальними терапевтичними дослідженнями.
3.14. Місцева переносність (ІІІ.Н.):
Дослідження місцевої переносності проводиться з метою вивчення впливу лікарського засобу (як діючих, так і допоміжних речовин) на тканини організму в тих ділянках, які можуть контактувати з лікарським засобом унаслідок його введення при клінічному застосуванні. Дослідження плануються таким чином, щоб можна було відрізнити будь-який механічний вплив введення або фізико-хімічний вплив лікарського засобу від токсичного або фармакодинамічного ефекту.
4. Вимоги до частини IV «Клінічна документація» матеріалів реєстраційного досьє
4.1. Матеріали реєстраційного досьє подаються відповідно до зазначених нижче вимог:
Матеріали реєстраційного досьє щодо клінічних випробувань містять дані щодо науково-дослідницької роботи, метою якої є дослідження за участю людини як суб’єкта дослідження, призначеного для виявлення або підтвердження клінічних, фармакологічних та/або інших фармакодинамічних ефектів одного або декількох досліджуваних лікарських засобів, та/або виявлення побічних реакцій на один або декілька досліджуваних лікарських засобів, та/або для вивчення усмоктування, розподілу, метаболізму та виведення одного або кількох лікарських засобів з метою підтвердження його (їх) безпеки та/або ефективності.
Експертиза матеріалів реєстраційного досьє щодо клінічних випробувань базується на даних клінічних випробувань, у тому числі клінічних фармакологічних досліджень, спланованих з метою визначення ефективності та безпеки лікарського засобу за звичайних умов його застосування з урахуванням терапевтичних показань для застосування у людини. Терапевтична користь лікарського засобу повинна переважати потенційний ризик.
Документація щодо клінічних випробувань повинна дати змогу скласти достатньо обґрунтований та науково достовірний висновок щодо ефективності та безпеки лікарського засобу. До матеріалів реєстраційного досьє мають бути надані результати всіх клінічних випробувань як позитивного, так і негативного характеру.
4.2. Клінічний досвід (IV.В.):
Матеріали реєстраційного досьє щодо клінічних випробувань мають містити інформацію щодо планування та проведення усіх фаз клінічних випробувань, у тому числі досліджень з біодоступності та біоеквівалентності, а також звітність, які повинні відповідати нормам належної клінічної практики.
Усі клінічні випробування, інформація про які міститься у матеріалах реєстраційного досьє, повинні бути проведені відповідно до етичних принципів, установлених в останній редакції Гельсінської декларації.
4.3. Надання результатів (IV.С.):
4.3.1. Матеріали реєстраційного досьє мають містити докладні звіти про кожне клінічне випробування з детальними відомостями, достатніми для складання об’єктивної думки про нього:
протокол випробування, який містить обґрунтування, завдання, методи статистичної обробки та методологію випробування, умови проведення та контролю випробування, а також особливості досліджуваного лікарського засобу;
сертифікат(и) аудиту, інспекційної перевірки (якщо такі є);
список дослідників із зазначенням їх прізвищ, місць їх проживання, посад, кваліфікації та клінічних обов’язків, зазначенням країни, у якій проводилося випробування, а також зібраної інформації про кожного окремого пацієнта, у тому числі індивідуальних реєстраційних форм;
заключний звіт, підписаний дослідником, а при багатоцентрових випробуваннях — усіма дослідниками або дослідником-координатором (відповідальним дослідником).
4.3.2. Матеріали реєстраційного досьє щодо клінічних випробувань мають містити клінічні спостереження, резюмовані для кожного випробування із зазначенням:
а) кількості хворих, які отримали лікування, та їх статі;
б) критеріїв відбору до груп та вікового розподілу в групах досліджуваних пацієнтів та контрольних групах;
в) кількості пацієнтів, передчасно виключених з випробувань, із зазначенням причин, з яких їх участь у випробуванні було перервано;
г) при проведенні контрольованих випробувань за вищеперелічених умов — переліку осіб, які складали контрольну групу:
тих, що не отримували лікування;
отримували плацебо;
отримували інший лікарський засіб з відомим терапевтичним ефектом;
тих, що отримували інше, немедикаментозне лікування;
ґ) частоти побічних реакцій, що спостерігалися;
д) особливостей щодо пацієнтів, які можуть належати до груп ризику, наприклад людей похилого віку, дітей, жінок у період вагітності або менструації, пацієнтів, фізіологічний або патологічний стан яких потребує особливого підходу;
е) параметрів та критеріїв експертизи ефективності та результатів з урахуванням цих параметрів або критеріїв;
є) статистичної експертизи результатів, якщо цього вимагає дизайн випробування та наявність перемінних чинників.
4.3.3. У матеріалах реєстраційного досьє щодо клінічних випробувань дослідник у висновках за експериментальними даними повинен висловити свою думку про безпеку лікарського засобу в умовах його звичайного застосування; про його переносність, ефективність; указати корисну інформацію, яка стосується показань та протипоказань, дозування та середньої тривалості лікування; особливі заходи безпеки, яких необхідно дотримуватись при лікуванні; клінічні симптоми передозування. При наданні результатів багатоцентрових досліджень у матеріалах реєстраційного досьє мають міститись висновки відповідального дослідника від імені всіх центрів, які проводили дослідження, щодо безпеки та ефективності досліджуваного лікарського засобу.
4.3.4. Крім того, у матеріалах реєстраційного досьє щодо клінічних випробувань мають міститись спостереження дослідника щодо:
а) будь-яких ознак звикання, залежності або виникнення синдрому відміни лікарського засобу;
б) будь-яких лікарських взаємодій, які спостерігались при одночасному введенні лікарського засобу з іншими лікарськими засобами;
в) критеріїв, які слугували підставою для виключення окремих пацієнтів з випробування;
г) будь-яких випадків смерті, які були відмічені під час проведення випробування або в період наступного нагляду.
4.3.5. Докладні звіти про нову комбінацію лікарських діючих речовин повинні бути аналогічними документації, яка необхідна при вивченні нових лікарських засобів, також у них повинні бути обґрунтовані безпека та ефективність цієї комбінації.
4.3.6. Повну або часткову відсутність даних клінічних випробувань необхідно обґрунтувати. При одержанні непередбачених результатів під час випробувань слід провести додаткові токсикологічні та фармакологічні випробування та дати їм оцінку.
Якщо лікарський засіб призначено для тривалого застосування, необхідно надати докладні звіти щодо будь-яких змін фармакологічної дії після призначення повторних доз лікарського засобу, а також відносно встановленої тривалості курсу приймання лікарського засобу.
4.4. Клінічна фармакологія (IV.D.):
4.4.1. Фармакодинаміка:
У матеріалах реєстраційного досьє має бути наведений наявний взаємозв’язок фармакодинамічної дії та ефективності, у тому числі:
залежність «доза-ефект» та її зміни в часі;
обґрунтування доз та способу введення;
механізм дії (якщо це можливо).
Необхідно описати фармакодинамічну дію, яка не має відношення до ефективності.
Сам по собі доказ наявності фармакодинамічних ефектів лікарського засобу в людини не може бути достатньою підставою для того, щоб зробити висновки відносно якого-небудь конкретного ймовірного терапевтичного ефекту.
4.4.2. Фармакокінетика:
У матеріалах реєстраційного досьє має бути наведений опис таких фармакокінетичних характеристик:
усмоктування (швидкість та ступінь);
розподіл;
метаболізм;
виведення.
Описуються клінічно значущі особливості, у тому числі значення одержаних даних щодо кінетики лікарського засобу для визначення режиму його дозування, особливо щодо пацієнтів, які входять до групи ризику, а також відмінності фармакокінетики в людини від кінетики лікарського засобу, що спостерігалася в доклінічних випробуваннях на тваринах.
4.4.3. Лікарські взаємодії:
Якщо лікарський засіб при рекомендованому застосуванні буде вводитися водночас з іншими лікарськими засобами, до матеріалів реєстраційного досьє надаються докладні звіти про випробування, проведені з метою виявлення можливої зміни фармакологічної дії при їх сумісному введенні.
Якщо виявлені фармакодинамічні/фармакокінетичні взаємодії діючої речовини з іншими лікарськими засобами або іншими речовинами, наприклад, алкоголем, кофеїном, тютюном або нікотином, які можуть вживатися водночас, або якщо такі взаємодії вірогідні, то їх слід описати, особливо з точки зору клінічної значущості і у зв’язку із заявленими взаємодіями у тексті короткої характеристики лікарського засобу.
4.5. Біодоступність/біоеквівалентність (IV.E.):
Експертизу матеріалів реєстраційного досьє щодо біодоступності слід проводити в усіх необхідних випадках, наприклад, якщо терапевтична доза близька до токсичної або якщо в ході попередніх випробувань виявлено відхилення, які можуть бути обумовлені фармакокінетичними властивостями, наприклад, варіабельність усмоктування.
Крім того, вивчення біодоступності проводиться, якщо необхідно довести біоеквівалентність для лікарських засобів.
У разі застосування процедури біовейвер до матеріалів реєстраційного досьє необхідно надати звіт про проведення досліджень in vitro. Оцінка та проведення досліджень біоеквівалентності або обґрунтування щодо його непроведення мають бути надані відповідно до вимог розділів ХІХ та ХХ цього Порядку та керівних настанов.
4.6. Клінічна ефективність та безпека (IV.F.):
4.6.1. У матеріалах реєстраційного досьє щодо клінічних випробувань має міститись інформація щодо контрольованих клінічних випробувань та за можливості рандомізованих; будь-яка інша схема проведення клінічних випробувань повинна бути обґрунтована; щодо характеру лікування в контрольних групах, яке може варіювати від випадку до випадку та залежить також від етичних норм; тому в окремих випадках порівнюють ефективність нового лікарського засобу не з дією плацебо, а з терапевтичною ефективністю уже відомого лікарського засобу.
За будь-якої можливості й особливо у тих випадках, коли при проведенні випробувань дію лікарського засобу не можна об’єктивно виміряти, необхідно надати інформацію щодо вжитих заходів для уникнення систематичної помилки при проведенні випробувань, у тому числі використання методів сліпих досліджень та рандомізації.
4.6.2. У протоколі клінічних випробувань, що міститься у матеріалах реєстраційного досьє, мають бути наведені ретельно описані статистичні методи, які використовуватимуться, та кількість пацієнтів, критерії їх включення у випробування (у тому числі розрахунки статистичної потужності випробувань), рівень значущості та опис статистичної одиниці (одиниці відбору), а також заходи, ужиті для запобігання систематичній помилці, особливо методи рандомізації. Інформація про включення у клінічне випробування великої кількості досліджуваних осіб не є адекватною заміною контрольованого випробування, проведеного відповідно до вимог належної клінічної практики.
4.6.3. Якщо клінічні звіти з ефективності та безпеки лікарського засобу за умови додержання рекомендацій стосовно його застосування недостатньо обґрунтовані з наукової точки зору, вони не є підставою для надання висновку щодо ефективності та безпеки лікарського засобу.
4.6.4. Значущість даних щодо ефективності та безпеки лікарського засобу за умови додержання рекомендацій щодо його застосування суттєво зросте, якщо вони одержані декількома незалежними компетентними дослідниками.
4.6.5. Експертний звіт, що міститься у матеріалах реєстраційного досьє, має містити інформацію з оцінки безпеки, а також значущості методів її експертизи для різних випробувань.
4.6.6. Усі побічні явища, у тому числі патологічні відхилення показників лабораторних досліджень, повинні бути надані у матеріалах реєстраційного досьє окремо, особливо щодо:
загальної кількості випадків виникнення побічних реакцій;
характеру, тяжкості та первинної обумовленості побічного явища.
4.6.7. Експертиза відносної безпеки застосування лікарського засобу з урахуванням побічних реакцій повинна бути проведена щодо:
захворювання, при якому лікарський засіб призначається;
інших терапевтичних підходів;
конкретних характеристик у різних групах пацієнтів;
даних доклінічних досліджень з токсикології та фармакології.
4.6.8. У матеріалах реєстраційного досьє щодо клінічних випробувань мають бути надані рекомендації щодо умов, за яких лікарський засіб повинен застосовуватись для зниження частоти розвитку побічних реакцій.
4.7. Матеріали реєстраційного досьє, які супроводжують заяву у виняткових випадках (IV.G.):
Якщо заявник може довести, що для лікарського засобу неможливо надати повні дані про ефективність і безпечність за нормальних умов використання, оскільки:
показання, для яких застосовується лікарський засіб, зустрічаються вкрай рідко; у такий спосіб заявник не може із цих причин надати достатньо даних або
наявні наукові дані не дають змоги надати повну інформацію, або
збір такої інформації буде суперечити загальноприйнятим принципам медичної етики,
то реєстраційне посвідчення може видаватися з урахуванням певних зобов’язань.
Ці зобов’язання можуть включати таке:
заявник повинен здійснити певну програму досліджень у період часу, зазначений МОЗ, результати якої мають бути основою для повторної оцінки співвідношення користь/ризик;
лікарський засіб, що подається на реєстрацію, має бути рецептурним та в деяких випадках застосовуватися тільки під медичним наглядом, можливо в лікарні, а у випадку радіофармацевтичних препаратів — кваліфікованим спеціалістом;
інструкція для медичного застосування має звертати увагу лікаря на той факт, що наявний опис даного лікарського засобу є певною мірою неповним.
4.8. Післяреєстраційний досвід (IV.Н.):
Якщо лікарський засіб уже зареєстрований в інших країнах, то у матеріалах реєстраційного досьє надається інформація про побічні реакції (якщо можна, з указанням їх частоти) на цей лікарський засіб та лікарські засоби, які містять ту саму діючу речовину (речовини). Також надаються дані з досліджень безпеки даного лікарського засобу, які проводяться у світі.
VIІІ. Спеціальні вимоги до матеріалів реєстраційного досьє (у форматі чотирьох частин) за різними типами заяв
1. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів з добре вивченим медичним застосуванням
Для лікарських засобів, діючу(і) речовину(и) яких добре вивчено в медичному застосуванні з підтвердженою ефективністю та прийнятним рівнем безпеки, застосовуються такі спеціальні вимоги до матеріалів реєстраційного досьє.
Заявник повинен подавати частину І матеріалів реєстраційного досьє за вимогами, наведеними у главі 1 розділу VIІ цього Порядку, та частину ІІ матеріалів реєстраційного досьє за вимогами, наведеними у главі 2 розділу VIІ цього Порядку.
Для матеріалів реєстраційного досьє частин ІІІ та ІV у докладній науковій бібліографії необхідно вказувати доклінічні та клінічні характеристики лікарського засобу.
Для підтвердження добре вивченого медичного застосування мають застосовуватися спеціальні вимоги:
а) фактори, які необхідно враховувати при визначенні добре вивченого медичного застосування компонентів лікарських засобів:
час, протягом якого використовується діюча речовина;
кількісні аспекти використання діючої речовини;
ступінь наукового інтересу у використанні діючої речовини (з посиланням на опубліковані наукові джерела);
послідовність наукових оцінок.
Таким чином, для визначення добре вивченого застосування різних діючих речовин може знадобитися оцінка за різні періоди часу. Однак у будь-якому разі період часу, необхідний для визначення добре вивченого медичного застосування діючої речовини, має бути не меншим за 10 років з дати першого систематичного та документованого використання цієї діючої речовини як лікарського засобу;
б) матеріали реєстраційного досьє, надані заявником, повинні включати всі аспекти оцінки безпеки та ефективності, містити або давати посилання на огляд відповідної літератури з урахуванням перед- та післяреєстраційних досліджень та опублікованої наукової літератури стосовно результатів епідеміологічних досліджень і особливо порівняльних епідеміологічних досліджень; всю документацію, як позитивну, так і негативну. Щодо аспектів про «добре вивчене медичне застосування» необхідно уточнити, що «бібліографічне посилання» на інші джерела доказів (післяреєстраційні дослідження, епідеміологічні дослідження тощо), а не тільки наявні дані, що стосуються методів контролю та випробувань, можуть бути вагомим доказом безпеки та ефективності лікарського засобу за умови, що в матеріалах реєстраційного досьє чітко пояснено та обґрунтовано використання цих джерел інформації;
в) необхідно обґрунтувати, чому може вважатися доведеним прийнятний рівень безпеки та/або ефективності, незважаючи на відсутність деяких досліджень;
г) у доклінічних та/або клінічних оглядах необхідно пояснити значимість будь-яких поданих даних, що стосуються вже зареєстрованого лікарського засобу, відмінного від того, який пропонується до перереєстрації. Необхідно надати обґрунтування з приводу того, чи можна заявлений лікарський засіб уважати подібним до вже зареєстрованого лікарського засобу, незважаючи на існуючі розбіжності;
ґ) післяреєстраційний досвід використання інших лікарських засобів, що містять ті самі компоненти, є особливо важливим, і заявники під час надання матеріалів реєстраційного досьє мають приділити цьому питанню належну увагу.
2. Спеціальні вимоги до матеріалів реєстраційного досьє для генеричних лікарських засобів
2.1. Матеріали реєстраційного досьє на генеричні лікарські засоби повинні містити дані, описані у частині І матеріалів реєстраційного досьє за вимогами, наведеними у главі 1 розділу VIІ цього Порядку, та частині ІІ матеріалів реєстраційного досьє за вимогами, наведеними у главі 2 розділу VIІ цього Порядку, за умови, що заявник має дозвіл власника реєстраційного посвідчення оригінального лікарського засобу щодо посилання на частини ІІІ та ІV матеріалів його реєстраційного досьє.
2.2. У випадку, якщо заявник не має дозволу власника реєстраційного посвідчення оригінального лікарського засобу щодо посилання на частини ІІІ та ІV матеріалів його реєстраційного досьє, він надає дані, описані у частині І матеріалів реєстраційного досьє за вимогами, наведеними у главі 1 розділу VIІ цього Порядку, та частині ІІ матеріалів реєстраційного досьє за вимогами, наведеними у главі 2 розділу VIІ цього Порядку, а також дані, що доводять біодоступність і біоеквівалентність з оригінальним лікарським засобом, за умови, що він не є подібним біологічним лікарським засобом.
Для цих лікарських засобів матеріали реєстраційного досьє щодо доклінічних/клінічних оглядів/резюме (у разі їх проведення) мають включати такі елементи:
обґрунтування того, що лікарський засіб є по суті аналогічним;
короткий опис домішок, наявних у серіях діючої(их) речовини(н), а також у готовому лікарському засобі, який реєструється (у відповідних випадках про продукти розпаду, що виникають при зберіганні), разом з оцінкою цих домішок;
оцінка досліджень біоеквівалентності або обґрунтування того, чому дослідження не проводилися відповідно до вимог розділів ХІХ та ХХ цього Порядку та керівних настанов;
остання опублікована література, що є актуальною для АФІ або діючої речовини та даного реєстраційного досьє. Для цього можна використовувати анотації статей наукових журналів;
кожне твердження в короткій характеристиці лікарського засобу, яке до цього не було відомо, або не витікає, виходячи з властивостей лікарського засобу та/або його терапевтичної групи, необхідно розглянути в доклінічних/клінічних оглядах/резюме та підтвердити опублікованою літературою та/або додатковими дослідженнями. При цьому інструкція для медичного застосування генеричного лікарського засобу має містити інформацію, ідентичну до тієї, яка наведена в інструкції для медичного застосування референтного лікарського засобу. Будь-які відмінності інструкції для медичного застосування генеричного лікарського засобу мають обґрунтовуватись результатами, отриманими при багатоцентрових клінічних дослідженнях;
якщо до складу генеричного лікарського засобу входять солі, ефіри або похідні зареєстрованої АФІ або діючої речовини, відмінні від тих, що входять до складу референтного лікарського засобу, заявник повинен надати додаткові дані з метою доведення еквівалентності характеристик безпеки та ефективності;
якщо діюча речовина генеричного лікарського засобу має таку саму терапевтичну дію, що й оригінальний зареєстрований лікарський засіб, але ця дія пов’язана з іншою сіллю/ефіром/комплексом/похідним, необхідно підтвердити, що при цьому не змінюється фармакокінетика, фармакодинаміка та/або токсичність тієї частини речовини, яка є відповідальною за дані властивості, і може змінити профіль безпеки/ефективності. Якщо таких доказів не існує, то дану речовину можна вважати новою діючою речовиною;
якщо лікарський засіб призначено для іншого терапевтичного використання, або його представлено в іншій лікарській формі, або він має застосовуватися іншими шляхами введення, або в інших дозах, або за іншої позології, необхідно надати результати відповідних токсикологічних і фармакологічних досліджень та/або клінічних випробувань.
3. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів з фіксованою комбінацією
Матеріали реєстраційного досьє повинні стосуватися нових лікарських засобів, вироблених з використанням принаймні двох відомих АФІ або діючих речовин, які раніше не застосовувалися у такій комбінації з терапевтичною метою.
Для таких лікарських засобів необхідно надати матеріали повного реєстраційного досьє (частина І матеріалів реєстраційного досьє за вимогами, наведеними у главі 1 розділу VIІ цього Порядку, частина ІІ матеріалів реєстраційного досьє за вимогами, наведеними у главі 2 розділу VIІ цього Порядку, частина ІІІ матеріалів реєстраційного досьє за вимогами, наведеними у главі 3 розділу VIІ цього Порядку, частина ІV матеріалів реєстраційного досьє за вимогами, наведеними у главі 4 розділу VIІ цього Порядку).
За можливості треба надати інформацію щодо дільниць виробництва, допоміжних речовин та оцінки безпеки.
4. Спеціальні вимоги до матеріалів реєстраційного досьє за гібридною заявою про реєстрацію лікарського засобу
У такому випадку заявник має подавати частину І матеріалів реєстраційного досьє за вимогами, наведеними у главі 1 розділу VIІ цього Порядку, частину ІІ матеріалів реєстраційного досьє за вимогами, наведеними у главі 2 розділу VIІ цього Порядку.
Матеріали реєстраційного досьє частин ІІІ та ІV мають містити звіти про обмежені доклінічні дослідження та/або клінічні випробування, що проводилися заявником (залежно від певної ситуації щодо відмінностей запропонованого лікарського засобу та референтного препарату (пункт 6.5 розділу VI цього Порядку)), та докладні бібліографічні дані щодо решти необхідної інформації для підтвердження безпеки та/або ефективності запропонованого лікарського засобу. Необхідно надати обґрунтування з приводу того, чи можна запропонований лікарський засіб уважати подібним до вже зареєстрованого лікарського засобу, незважаючи на наявні розбіжності.
5. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів з продукції in bulk
Для готових лікарських засобів, які виготовлені за такими стадіями виробничого процесу, як кінцеве пакування та/або маркування, застосовуються особливі правила при поданні матеріалів реєстраційного досьє.
Заявник повинен подавати частину І матеріалів реєстраційного досьє за вимогами, наведеними у главі 1 розділу VIІ цього Порядку.
Для частин ІІ, ІІІ та ІV фармацевтичні, доклінічні та клінічні характеристики лікарського засобу необхідно вказувати у повному обсязі на підставі матеріалів реєстраційного досьє виробника продукції in bulk.
Відповідні розділи частини ІІ також мають містити додаткові дані та документацію заявника (власника реєстраційного посвідчення на такий лікарський засіб) щодо стадій виробничого процесу, які ним виконуються.
Крім того, у відповідних розділах матеріалів реєстраційного досьє необхідно чітко вказувати найменування та місцезнаходження виробничих потужностей виробника продукції in bulk (який відповідає за контроль якості) та виробничих потужностей виробника, який відповідає за випуск серії. Інформація щодо виробників має бути наведена у тексті маркування.
Необхідно надати підтвердження реєстрації заявленого лікарського засобу у споживчій упаковці в країні виробника продукції in bulk.
6. Спеціальні вимоги до матеріалів реєстраційного досьє для гомеопатичних лікарських засобів
Даний розділ стосується особливих вимог до оформлення частин ІІ та ІІІ матеріалів реєстраційного досьє на гомеопатичні лікарські засоби.
Спеціальні вимоги до частини ІІ матеріалів реєстраційного досьє для гомеопатичних лікарських засобів:
а) термінологія
Латинська назва гомеопатичної сировини, описаної в матеріалах реєстраційного досьє, повинна відповідати латинській назві, зазначеній в Європейській фармакопеї, або в разі відсутності — Німецькій гомеопатичній фармакопеї (GHP), Гомеопатичній фармакопеї США (HPUS), Британській гомеопатичній фармакопеї (BHP), Гомеопатичній фармакопеї Швабе. У відповідних випадках необхідно надати традиційно використовувану назву;
б) контроль вихідних матеріалів
Опис та дані щодо вихідних матеріалів, тобто щодо всіх використаних матеріалів, уключаючи сировину та проміжні матеріали, до кінцевого розведення у готовому лікарському засобі повинні бути підкріплені додатковими даними про гомеопатичну сировину.
Загальні вимоги до якості стосуються всіх вихідних матеріалів і сировини, а також проміжних етапів виробничого процесу до кінцевого розведення у готовому лікарському засобі. Якщо можна, потрібно провести дослідження за наявності токсичних компонентів, а також тоді, коли якість неможливо контролювати при кінцевому розведенні через високий ступінь розведення. Необхідно надати повний опис кожної стадії виробничого процесу, починаючи від вихідних матеріалів до кінцевого розведення у готовому лікарському засобі.
Якщо застосовуються декілька розведень, ці стадії розведення необхідно проводити відповідно до методів виробництва гомеопатичних засобів, які викладено у відповідній монографії Європейської фармакопеї, або за її відсутності в іншій національній фармакопеї;
в) методи контролю готового лікарського засобу
До готових гомеопатичних лікарських засобів застосовуються загальні вимоги до якості. Будь-які винятки вимагають ретельного обґрунтування заявником.
Необхідно ідентифікувати та проаналізувати всі компоненти, які мають потенційну токсичність. Якщо неможливо ідентифікувати та проаналізувати всі компоненти, які можуть виявитися токсичними, наприклад, через їх високий ступінь розведення в готовому лікарському засобі, потрібно обґрунтувати якість лікарського засобу за допомогою повної валідації виробничого процесу та процесу розведення;
г) випробування на стабільність
Необхідно підтвердити стабільність готового лікарського засобу. Дані щодо стабільності гомеопатичного лікарського засобу, як правило, дійсні як для розчинів, так і для порошків, з яких був вироблений даний лікарський засіб. Якщо ідентифікація або аналіз діючої речовини неможливі через високий ступінь розведення, можуть розглядатися дані щодо стабільності лікарського засобу.
Спеціальні вимоги до частини ІІІ матеріалів реєстраційного досьє для гомеопатичних лікарських засобів: необхідно обґрунтувати відсутність будь-якої інформації, наприклад, має надаватися обґрунтування, яким чином може доводитися прийнятний рівень безпеки, незважаючи на те, що відсутні деякі дослідження.
7. Спеціальні вимоги до матеріалів реєстраційного досьє для рослинних лікарських засобів
До заяви на перереєстрацію рослинних лікарських засобів додаються матеріали повного реєстраційного досьє, до якого повинні бути включені такі специфічні дані.
Положення частини ІІ, включаючи відповідність монографії Європейської фармакопеї, стосуються готових рослинних лікарських засобів. При цьому береться до уваги рівень наукових знань на момент подання заяви.
Стосовно рослинних лікарських засобів розглядаються такі специфічні аспекти:
а) рослинні субстанції та рослинні препарати
Щодо номенклатури рослинної субстанції необхідно вказати ботанічні характеристики рослини (рід, вид, різновид та автора), хемотип (у відповідних випадках) частин рослини, інші назви (синоніми, зазначені в інших фармакопеях) та лабораторний код.
Щодо номенклатури рослинного препарату необхідно вказати ботанічні характеристики рослини (рід, вид, різновид та автора), хемотип (у відповідних випадках) частин рослини, визначення препарату, співвідношення рослинної субстанції до рослинного препарату, розчинник(и), інші назви (синоніми, зазначені в інших фармакопеях) та лабораторний код.
При підготовці документів щодо структури рослинної субстанції та рослинного препарату (у відповідних випадках) необхідно надати інформацію про фізичну форму, опис компонентів з відомою терапевтичною дією або маркерів (молекулярну формулу, відносну молекулярну масу, структурну формулу, уключаючи відносну та абсолютну стереохімію), а також інформацію про інші компоненти.
При підготовці документів щодо виробництва рослинної субстанції у відповідних випадках необхідно надати інформацію про кожного постачальника сировини, включаючи підрядників, а також інформацію про кожну передбачену виробничу дільницю або устаткування, задіяне при виробництві/заготовці та контролі рослинної субстанції.
При підготовці документів щодо виробництва рослинного препарату у відповідних випадках необхідно вказати інформацію про кожного постачальника сировини, уключаючи підрядників, а також інформацію про кожну передбачену виробничу дільницю або устаткування, задіяне при виробництві та контролі рослинного засобу.
Щодо опису виробничого процесу рослинної субстанції необхідно надати інформацію, у якій буде відповідним чином описано виробництво субстанції та заготівлю рослинної сировини, уключаючи географічне місце вирощування та культивування лікарської рослини, умови її збору, висушування та зберігання.
Щодо опису виробничого процесу та процедури контролю рослинного препарату необхідно надати інформацію, у якій буде відповідним чином описане виробництво засобу, включаючи опис обробки сировини, розчинників і реагентів, стадії очищення та стандартизації.
Щодо розробки виробничого процесу необхідно надати (у відповідних випадках) короткий опис розробки рослинної субстанції та рослинного препарату з урахуванням передбачуваного шляху застосування та показань. У відповідних випадках має надаватися обговорення результатів порівняння фітохімічної будови рослинної субстанції та рослинного препарату, що використовувались на підтримку бібліографічних даних, та рослинної субстанції і рослинного препарату, що є діючою речовиною готового рослинного лікарського засобу, на який подано заяву про державну перереєстрацію.
При описі структури та інших характеристик рослинної субстанції за необхідності має надаватися інформація про ботанічні, макро- та мікроскопічні, фітохімічні характеристики та біологічну активність.
При описі структури та інших характеристик рослинного препарату за необхідності має надаватися інформація про фітохімічні і фізико-хімічні характеристики та біологічну активність.
За необхідності мають надаватися специфікації для рослинної субстанції та рослинного препарату.
У відповідних випадках потрібно надати опис аналітичних методів, що використовувалися при випробуванні рослинної субстанції та рослинного препарату.
При описі валідації аналітичних методик у відповідних випадках потрібно надати інформацію про валідацію методик, включаючи експериментальні дані для методів аналізу, які використовуються при випробуваннях рослинної субстанції та рослинного препарату.
При описі аналізу серій у відповідних випадках потрібно надати опис та результати аналізу серій рослинних субстанцій та рослинних препаратів у такому вигляді, як монографії фармакопей.
У відповідних випадках потрібно надати обґрунтування специфікацій рослинних субстанцій і рослинних препаратів.
У відповідних випадках потрібно надати інформацію про стандартні зразки або стандартні препарати, що використовуються при випробуваннях рослинної субстанції та рослинного препарату;
б) готові рослинні лікарські засоби
Короткий опис розробки складу лікарського засобу повинен уключати інформацію про розробку готового рослинного лікарського засобу з увагою на передбачуваний спосіб застосування та показання. У відповідних випадках потрібно надати відомості про результати порівняння фітохімічної будови лікарського засобу, який використовується на підтримку бібліографічних даних, та лікарського засобу, на який подано заяву.
ІХ. Вимоги до матеріалів реєстраційного досьє
(у форматі загального технічного документа)
1. Вимоги до матеріалів реєстраційного досьє, наведених у Модулі 1 «Адміністративна інформація»
1.1. Зміст:
Необхідно надати повний зміст модулів 1 — 5 реєстраційного досьє, яке подається разом із заявою про державну реєстрацію лікарського засобу.
1.2. Форма заяви:
У заяві мають бути зазначені назва лікарського засобу, який подається на державну реєстрацію, назва діючої(их) речовини(н), лікарська форма, шлях введення, сила дії (дозування) та форма випуску готового лікарського засобу, включаючи упаковку.
Зазначаються найменування/П.І.Б. та місцезнаходження/місце проживання заявника разом з найменуванням/П.І.Б. виробників та місцезнаходженням виробничих потужностей, задіяних на різних стадіях виробництва (включаючи виробника готового лікарського засобу та виробника(ів) діючої(их) речовини(н)), а також за потреби інформацію щодо імпортера.
Заявник має визначити тип заяви та вказати, які зразки (якщо вони додаються) надано.
До адміністративних даних додають:
копію ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника (є необхідними як для повного, так і неповного виробництва, а також для різних процесів фасування, пакування і маркування);
засвідчену копію документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист від заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи (крім АФІ, діючих речовин);
перелік країн, де даний лікарський засіб зареєстрований/перереєстрований;
копію короткої характеристики лікарського засобу/інструкції для медичного застосування, розробленої та затвердженої відповідно до нормативних вимог країни заявника/виробника, відповідно до встановлених вимог;
перелік країн, у яких було подано заяви на реєстрацію.
У заяві заявник надає докладну інформацію про лікарський засіб, про пропонованого власника реєстраційного посвідчення та виробника(ів), інформацію про статус препарату обмеженого застосування (препарату-сироти), наукові консультації та програми розробок у педіатрії та іншу інформацію, передбачену додатком 1.
1.3. Коротка характеристика лікарського засобу, маркування та інструкція для медичного застосування:
1.3.1. Копія короткої характеристики лікарського засобу/інструкції для медичного застосування, затвердженої відповідно до нормативних вимог країни заявника/виробника або інших країн, регуляторні органи яких керуються високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ.
1.3.2. Маркування:
Заявник має надати пропонований текст маркування для первинної та вторинної упаковок, складений згідно з вимогами розділу ХVIІІ цього Порядку.
1.3.3. Інструкція для медичного застосування:
Заявник має надати пропонований текст інструкції для медичного застосування (на паперовому та електронному носіях), складений згідно з вимогами розділу ХVI цього Порядку.
1.3.4. Коротка характеристика лікарського засобу:
Заявник пропонує проект короткої характеристики лікарського засобу, складений згідно з вимогами розділу ХVIІ цього Порядку.
1.4. Інформація про незалежних експертів:
Експерти мають надавати резюме із зауваженнями, зробленими при розгляді документів та матеріалів реєстраційного досьє, зокрема до Модулів 3, 4 і 5 (хімічна, фармацевтична та біологічна документація, доклінічна документація та клінічна документація відповідно). Резюме незалежного експерта має висвітлювати критичні моменти, пов’язані з якістю лікарського засобу, доклінічними дослідженнями та клінічними випробуваннями, і містити всі дані, необхідні для оцінювання.
Цих вимог необхідно дотримуватися при складанні загального резюме з якості, доклінічних досліджень та клінічних випробувань, які включено до Модуля 2 реєстраційного досьє на лікарський засіб. У Модулі 1 має бути надана інформація, підписана незалежними експертами, про їх освіту, спеціалізацію та професійний досвід. В експертів має бути відповідна кваліфікація.
1.5. Спеціальні вимоги до різних типів заяв:
Спеціальні вимоги до різних типів заяв наведено в розділі Х цього Порядку.
Додаток до Модуля 1. Оцінка небезпеки для довкілля.
Заява про державну реєстрацію лікарських засобів за необхідності має містити резюме оцінки ризику, у якому оцінюють можливі ризики для довкілля через використання та/або утилізацію лікарського засобу, а також наводять пропозиції щодо відповідної інформації в маркуванні. Необхідно розглянути ризик для довкілля, пов’язаний з вивільненням лікарських засобів, що містять генетично модифіковані організми (далі — ГМО), відповідно до вимог Закону України «Про державну систему біобезпеки при створенні, випробуванні, транспортуванні та використанні генетично модифікованих організмів».
Інформація щодо ризику для довкілля надається у вигляді додатка до Модуля 1.
Інформація включає:
вступ;
копію письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою;
дані, включаючи методи виявлення та ідентифікації, а також унікальний код ГМО і будь-яку додаткову інформацію про ГМО або лікарський засіб, які мають значення при оцінці ризику для довкілля;
звіт про оцінку ризику для довкілля (далі — ОРД), підготовлений на підставі наявної інформації;
на основі вищевикладеної інформації та ОРД — висновок стосовно відповідної стратегії управління ризиками щодо ОРД і досліджуваного лікарського засобу, план моніторингу в післяреєстраційний період та визначення будь-якої спеціальної інформації, яка повинна надаватися в короткій характеристиці лікарського засобу, маркуванні та інструкції для медичного застосування;
відповідні заходи щодо інформування населення.
Наведена інформація має бути засвідчена підписом автора із зазначенням дати, даних щодо освіти, стажування та професійного досвіду. Необхідно вказати професійні стосунки між автором і заявником.
2. Вимоги до матеріалів реєстраційного досьє, наведених у Модулі 2 «Резюме Загального технічного документа»
У цьому модулі наводять резюме хімічної, фармацевтичної та біологічної документації, доклінічних та клінічних даних, наданих у Модулях 3, 4 і 5 реєстраційного досьє на лікарський засіб, а також у резюме незалежних експертів.
Необхідно визначити та проаналізувати важливі питання. Слід передбачити узагальнені фактичні дані, включаючи матеріали у вигляді таблиць. У цих звітах передбачають перехресні посилання на таблиці або на інформацію, що міститься в основній документації, наданій в Модулі 3 (Якість. Хімічна, фармацевтична та біологічна інформація про лікарські засоби, що містять хімічні та/або біологічні діючі речовини), Модулі 4 (Звіти про доклінічні дослідження) і Модулі 5 (Звіти про клінічні випробування).
Огляди і резюме повинні відповідати основним принципам і вимогам, викладеним нижче:
2.1. Загальний зміст:
У Модулі 2 надають зміст наукової документації, наведеної у Модулях 2-5.
2.2. Вступ:
Має бути надана інформація про фармакологічну групу, механізм дії та запропоноване клінічне застосування лікарського засобу.
2.3. Загальне резюме з якості:
У загальному резюме з якості слід надавати огляд інформації, пов’язаної з хімічними, фармацевтичними та біологічними даними.
Необхідно звернути особливу увагу на основні критичні параметри та питання, пов’язані з аспектами якості, а також надати обґрунтування у тих випадках, коли не дотримано відповідних вимог та настанов. Цей документ повинен охоплювати питання і описувати відповідні дані, які докладно висвітлено в Модулі 3.
2.4. Огляд доклінічних даних:
Необхідно наводити узагальнену та критичну оцінку доклінічних досліджень лікарського засобу на тваринах/in vitro, а також обговорення та обґрунтування стратегії випробування і за необхідності відхилення від відповідних вимог.
Необхідно включити оцінку домішок і продуктів розпаду лікарського засобу разом з їх потенційними фармакологічними та токсикологічними ефектами, за винятком лікарських засобів біологічного походження. Слід розглянути будь-які розходження у хіральності, хімічній формі та чистоті сполук, які використовувались у доклінічних дослідженнях і у лікарському засобі, що буде вироблятися.
Для лікарських засобів біологічного походження необхідно оцінити порівнянність матеріалу, використаного у доклінічних дослідженнях, клінічних випробуваннях і лікарському засобі, який буде реєструватися.
Будь-яка нова допоміжна речовина має бути предметом окремої оцінки з безпеки.
Необхідно визначити властивості лікарського засобу, доведені у доклінічних дослідженнях, а також представити значення результатів з безпеки лікарського засобу для планованого клінічного застосування за участю людини.
2.5. Огляд клінічних даних:
Огляд клінічних даних має містити критичний аналіз клінічних даних, які включено у резюме та Модуль 5. Необхідно надати спосіб клінічної розробки лікарського засобу, включаючи дизайн випробування, рішення, прийняті стосовно дослідження, а також проведення досліджень.
Необхідно надати короткий огляд даних клінічних досліджень, включаючи важливі обмежувальні фактори, а також оцінку ризику/користі, яка базується на висновках клінічних досліджень, обґрунтувати запропоновану дозу та показання для застосування, виходячи з отриманих клінічних даних щодо ефективності та безпеки, а також оцінити, як за допомогою короткої характеристики лікарського засобу та інших підходів можна оптимізувати користь та обмежити ризики.
Необхідно пояснити всі питання щодо ефективності та безпеки, які виникають у процесі розробки.
2.6. Резюме доклінічних даних:
Резюме доклінічних даних потрібно надавати на основі фактичних результатів фармакологічних, фармакокінетичних і токсикологічних досліджень, проведених на тваринах/in vitro, у текстовому форматі та у вигляді таблиць у такому порядку:
вступ;
резюме фармакологічних даних у текстовому форматі;
резюме фармакологічних даних у вигляді таблиць;
резюме фармакокінетичних даних у текстовому форматі;
резюме фармакокінетичних даних у вигляді таблиць;
резюме токсикологічних даних у текстовому форматі;
резюме токсикологічних даних у вигляді таблиць.
2.7. Резюме клінічних даних:
Необхідно надати докладне із наведенням фактичних даних резюме клінічної інформації з вивчення лікарського засобу, включеної в Модуль 5. Резюме має включати результати всіх біофармацевтичних досліджень, досліджень з клінічної фармакології, а також досліджень з клінічної ефективності та безпеки. Необхідно надати короткий огляд індивідуальних досліджень.
Клінічна інформація у вигляді резюме повинна надаватися в такому порядку:
резюме біофармацевтичних досліджень та пов’язаних з ними аналітичних методів;
резюме досліджень з клінічної фармакології;
резюме з клінічної ефективності;
резюме з клінічної безпеки;
короткі огляди індивідуальних досліджень.
3. Вимоги до матеріалів реєстраційного досьє, наведених у Модулі 3: «Якість. Хімічна, фармацевтична та біологічна інформація про лікарські засоби, що містять хімічні та/або біологічні діючі речовини»
3.1. Формат та подання.
Загальна схема Модуля 3:
зміст;
основні дані;
діюча речовина.
Загальна інформація:
назва;
структура;
загальні властивості.
Процес виробництва діючої(их) речовини(н):
виробник(и);
опис виробничого процесу та контролю у процесі виробництва;
контроль матеріалів;
контроль критичних стадій і проміжної продукції;
валідація процесу та/або його оцінка;
розробка виробничого процесу.
Опис характеристик діючої(их) речовини(н):
доказ структури та інші характеристики;
домішки.
Контроль діючої(их) речовини(н):
специфікація;
аналітичні методики;
валідація аналітичних методик;
аналізи серій;
обґрунтування специфікації.
Стандартні зразки та матеріали.
Система упаковки/укупорки.
Стабільність:
резюме щодо стабільності та висновки;
протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності;
дані про стабільність.
Готовий лікарський засіб.
Опис і склад лікарського засобу.
Фармацевтична розробка:
компоненти лікарського засобу;
діюча(і) речовина(и);
допоміжні речовини;
лікарський засіб;
розробка складу;
надлишки;
фізико-хімічні та біологічні властивості;
розробка виробничого процесу;
система упаковки/укупорки;
мікробіологічні характеристики;
сумісність.
Виробництво:
виробник(и);
склад на серію;
опис виробничого процесу та контролю у процесі виробництва;
контроль критичних стадій і проміжної продукції;
валідація процесу та/або його оцінка.
Контроль допоміжних речовин:
специфікації;
аналітичні методики;
валідація аналітичних методик;
обґрунтування специфікацій;
допоміжні речовини людського або тваринного походження;
нові допоміжні речовини.
Контроль готового лікарського засобу:
специфікація(ї);
аналітичні методики;
валідація аналітичних методик;
аналізи серій;
характеристика домішок;
обґрунтування специфікації(й).
Стандартні зразки та матеріали.
Система упаковки/укупорки.
Стабільність:
резюме щодо стабільності та висновки;
протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності;
дані про стабільність.
Додатки:
приміщення та обладнання (тільки для біологічних лікарських засобів);
оцінка безпеки щодо сторонніх агентів;
допоміжні речовини.
Додаткова інформація:
схема валідації виробничого процесу лікарського засобу;
пристрій для введення лікарського засобу;
сертифікат(и) відповідності;
для лікарських засобів, які містять або в процесі виробництва яких використовують матеріали тваринного та/або людського походження, сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії;
посилання на джерела літератури.
3.2. Зміст: основні принципи та вимоги:
а) хімічні, фармацевтичні та біологічні дані, які необхідно надавати, включають для діючої(их) речовини(н) і готового лікарського засобу всю інформацію про розробку, виробничий процес, характеристики та властивості, процедуру та вимоги контролю за якістю, стабільність, а також опис складу та оформлення готового лікарського засобу;
б) необхідно надати дві основні складові інформації про діючу(і) речовину(и) та готовий лікарський засіб відповідно;
в) у цьому модулі додатково має бути надана докладна інформація про вихідні матеріали та сировину, які використовуються при виробництві діючої(их) речовини(н) і допоміжних речовин, що входять до складу готового лікарського засобу;
г) усі методики та методи випробувань, використані при виробництві та контролі діючої(х) речовини(н) та готового лікарського засобу, мають бути викладені чітко і детально, щоб можна було відтворити їх при проведенні контрольних випробувань, на вимогу вповноваженого органу. Усі методи випробувань повинні відповідати сучасному науковому рівню, а також бути валідованими. Слід надавати результати досліджень з валідації. Якщо використовуються методи випробувань, включені до ДФУ або Європейської фармакопеї, то цей виклад необхідно замінити відповідним посиланням на монографію(ї) та загальний(і) розділ(и);
ґ) для всіх речовин, препаратів і лікарських форм, вказаних у монографіях ДФУ або Європейської фармакопеї, необхідно робити посилання на ці фармакопеї. Стосовно речовин, які не вказано в цих фармакопеях, мають надаватися посилання на інші національні фармакопеї.
Однак, якщо матеріал, зазначений в ДФУ, або Європейській фармакопеї, або в інших національних фармакопеях, одержують способом, при якому можуть виникати домішки, що не контролюються за монографією фармакопеї, то необхідно вказати ці домішки та їх допустимі межі, а також надати методику їхнього визначення. У разі якщо специфікація, включена до монографії ДФУ, або Європейської фармакопеї, або іншої національної фармакопеї, є недостатньою для забезпечення якості речовини, може знадобитися докладніша специфікація. Центр може надати інформацію вповноваженим органам, відповідальним за відповідну фармакопею. Заявник повинен надати вповноваженим органам цієї фармакопеї докладні відомості про виявлені недоліки та про додаткові специфікації, що застосовуються.
Якщо методи аналізу включено до ДФУ або Європейської фармакопеї, то немає необхідності наводити їх повний виклад, достатньо в кожному розділі, в якому планувався виклад цього методу, робити відповідне посилання на монографію(ї) та загальний(і) розділ(и);
д) якщо вихідні матеріали та сировина, діючі або допоміжні речовини не описано ані в ДФУ, ані в Європейській фармакопеї, ані в інших національних фармакопеях, то може бути прийнятним посилання на монографію фармакопеї іншої країни. У таких випадках заявник повинен надати копію монографії разом з валідацією аналітичних методик, описаних у монографії, а також за необхідності переклад;
е) якщо діючу та/або допоміжну(і) речовину(и) і вихідний матеріал описано в монографії Європейської фармакопеї, заявник може надати сертифікат відповідності Європейській фармакопеї у відповідному пункті цього модуля. Вважається, що сертифікати відповідності монографії Європейської фармакопеї заміняють суттєві дані у відповідних розділах, зазначених у цьому модулі. Виробник речовини повинен підтвердити заявнику письмово, що виробничий процес не змінювався з часу видачі цього сертифіката відповідності;
є) для добре вивчених діючих речовин виробник діючої речовини або заявник може підготувати матеріали з:
докладного опису виробничого процесу,
контролю за якістю в процесі виробництва та валідації процесу,
у вигляді окремого документа, а виробник може подати їх у вигляді мастер-файла на діючу речовину (матеріали реєстраційного досьє (Drug Master File)).
Однак у такому випадку виробник діючої речовини має надати заявнику готового лікарського засобу всі дані, які можуть знадобитися йому. Виробник повинен надати заявнику письмове підтвердження, що він гарантує відповідність між серіями (партіями), а також, що він не буде вносити зміни у виробничий процес або специфікації, не поінформувавши про це заявника. Документи та докладну інформацію, що супроводжують заяву на внесення такої зміни, необхідно подавати в уповноважені органи; ці документи та дані також надаються заявнику в тих розділах, де вони стосуються відкритої частини мастер-файла на діючу речовину;
ж) необхідно описати особливі запобіжні заходи щодо передачі губчатої енцефалопатії тварин (сировина, отримана від жуйних тварин): на кожній стадії виробничого процесу заявник повинен продемонструвати відповідність використаних матеріалів, посилаючись, зокрема, на Керівництво з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами. Підтвердити відповідність можна, посилаючись на вищевказаний документ, або надавши сертифікат відповідності конкретній монографії Європейської фармакопеї, або шляхом надання наукових даних для обґрунтування цієї відповідності;
з) слід надати інформацію про оцінку ризику потенційного зараження сторонніми агентами (незалежно від того, мають вони вірусне походження чи ні) згідно з вимогами, викладеними в спеціальних керівництвах, а також у загальних монографіях та загальних розділах ДФУ або Європейської фармакопеї;
и) необхідно описати будь-які спеціальні прилади та обладнання, що може застосовуватися на будь-якій стадії виробничого процесу та етапі контролю за лікарським засобом;
і) для пристроїв з введення лікарських засобів необхідно надати СЕ-сертифікат (підтвердження відповідності Директиві 93/42/ЕЕС «Медичні прилади. Медичне обладнання. Медичні пристрої») за наявності.
3.3. Особливу увагу приділяють таким підрозділам Модуля 3:
3.3.1. Діюча(і) речовина(и) (3.2.S.):
Загальна інформація щодо вихідних матеріалів та сировини (3.2.S.1):
а) надається інформація про назву діючої речовини, включаючи рекомендовану МНН, за наявності — фармакопейну назву, зазначену в ДФУ, Європейській фармакопеї, та хімічну назву.
Надається структурна формула, включаючи відносну та абсолютну стереохімію, молекулярна формула та відносна молекулярна маса. Для біотехнологічних лікарських засобів за потреби необхідно надати схематичну послідовність амінокислот і відносну молекулярну масу.
Для біологічних лікарських засобів необхідно надати перелік фізико-хімічних та інших важливих властивостей діючої речовини, включаючи її біологічну активність;
б) у контексті цього пункту вихідними матеріалами вважають всі матеріали, з яких виробляються або екстрагуються діючі речовини.
Для лікарських засобів біологічного походження за вихідні матеріали вважають будь-яку речовину біологічного походження, таку як мікроорганізми, органи та тканини рослинного або тваринного походження, клітини або рідини (включаючи кров або плазму) людського або тваринного походження, біотехнологічні клітинні компоненти (рекомбінантні або нерекомбінантні субстрати клітини, включаючи первинні клітини).
У контексті цього пункту лікарським засобом біологічного походження вважають усі лікарські засоби, діюча речовина яких є біологічною речовиною. Так, біологічна речовина — це речовина, що виробляється або екстрагується з біологічного джерела, для опису та визначення якості якої необхідно надавати комбінацію фізичних, хімічних та біологічних методів аналізу разом із описом процесу виробництва та контролю за ним. Біологічними лікарськими засобами є імунологічні лікарські засоби та лікарські засоби, що є похідними крові та плазми людини; лікарські засоби, що отримані за допомогою біотехнологічних методів (наприклад, технології рекомбінантної ДНК; контрольованої експресії генів, що кодують біологічно активні білки прокаріотів і еукаріотів, у тому числі трансформованих клітин тварин; методів отримання гібридом і моноклональних антитіл тощо), а також препарати прогресивної терапії.
Будь-які інші речовини, які використовують для виробництва або екстрагування діючої(их) речовини(н), але з яких цю діючу речовину безпосередньо не одержують, а саме: реагенти, живильні середовища, сироватка зародка ембріона, добавки та буфери, що застосовуються у препаративній хроматографії тощо, вважають вихідними матеріалами.
3.3.2. Процес виробництва діючої(их) речовини(н) (3.2.S.2):
а) заявник зобов’язаний надати опис виробничого процесу діючої речовини. Для адекватного опису процесу виробництва та контролю за ним потрібну інформацію необхідно викласти у відповідності до вимог, встановлених цим Порядком;
б) усі матеріали, необхідні для виробництва діючої(их) речовини(н), потрібно перераховувати із зазначенням стадії виробництва, на якій використовується кожний матеріал. Необхідно надати інформацію про якість і контроль цих матеріалів, а також інформацію, яка доводить, що всі матеріали відповідають вимогам щодо їхнього запланованого використання.
Необхідно перерахувати вихідні матеріали (сировину), а також навести показники їх якості та методи контролю.
Необхідно надати найменування/П.І.Б. та місцезнаходження виробничих потужностей та вказати обов’язки кожного виробника, включаючи контрактні виробництва, а також інформацію про кожну із запланованих виробничих дільниць або лабораторій;
в) для біологічних лікарських засобів встановлено такі додаткові вимоги:
необхідно надати опис та документальне підтвердження походження та історії вихідних матеріалів.
Стосовно особливих заходів щодо запобігання передачі губчатої енцефалопатії заявник повинен підтвердити, що діюча речовина відповідає, зокрема, Керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами.
При використанні банків клітин надаються докази того, що характеристики клітин залишилися незмінними при тій кількості пасажів, які використовуються для виробництва, а також протягом наступного періоду.
Посівні матеріали, банки клітин, пули сироваток або плазми та інші матеріали біологічного походження, а також за можливості вихідні речовини, з яких вони отримані, випробовуються на наявність сторонніх агентів.
Якщо присутності потенційно патогенних сторонніх агентів уникнути неможливо, то вихідні речовини необхідно використовувати тільки в тому випадку, коли при подальшій обробці буде забезпечуватися видалення та/або інактивація даних сторонніх агентів, і це має бути доведено валідацією.
Там, де це можливо, виробництво вакцин повинно ґрунтуватись на системі посівних партій та відомих банків клітин. При виробництві бактеріальних та вірусних вакцин характеристики збудника інфекції повинні бути продемонстровані на посівному матеріалі. Крім того, по живих вакцинах стабільність характеристики атенуації повинна бути продемонстрована на посівному матеріалі; якщо докази будуть недостатніми, характеристики атенуації також повинні бути продемонстровані на стадії виробництва.
Для лікарських засобів, отриманих з крові або плазми людини, відповідно до положень, викладених у розділі Х цього Порядку, необхідно описати та документально підтвердити походження, критерії та методики відбору, транспортування та зберігання вихідних матеріалів.
Необхідно описати виробничі приміщення та обладнання;
г) у відповідному порядку необхідно надати інформацію про методи контролю та критерії прийнятності на кожній критичній стадії, інформацію про якість і контроль проміжних продуктів, а також про процес валідації та/або його аналіз;
ґ) якщо присутності потенційно патогенних сторонніх агентів уникнути неможливо, то вихідні речовини необхідно використовувати тільки в тих випадках, коли наступна обробка забезпечує їхнє видалення та/або інактивацію, що має доводитись у відповідному модулі, який стосується оцінки вірусної безпеки;
д) необхідно передбачити для діючої речовини опис і обговорення суттєвих змін, внесених у виробничий процес під час розробки, та/або заміни виробничої дільниці.
3.3.3. Опис характеристик діючої(их) речовини(н) (3.2.S.3):
Необхідно надати дані щодо структури та інших характеристик діючої(их) речовини(н).
Слід підтвердити структуру діючої(их) речовини(н), ґрунтуючись на сучасних фізико-хімічних та/або імунохімічних та/або біологічних методах, а також надати інформацію про домішки.
3.3.4. Контроль діючої(их) речовини(н) (3.2.S.4):
Необхідно надати докладну інформацію про специфікації, які використовуються для посерійного контролю діючої(их) речовини(н), обґрунтування вибору цих специфікацій, методів аналізу та їх валідації.
Необхідно надати результати контролю окремих серій, виготовлених на етапі розробки.
3.3.5. Стандартні зразки або матеріали (3.2.S.5):
Необхідно визначити та докладно описати стандартні матеріали та стандартні зразки. За можливості необхідно застосовувати хімічні стандартні зразки та біологічні стандартні матеріали, описані в ДФУ або Європейській фармакопеї.
3.3.6. Система упаковки/укупорки (3.2.S.6):
Необхідно надати опис контейнера та системи упаковки/укупорки і специфікації її компонентів. За можливості необхідно використовувати пакувальні засоби, які відповідають вимогам ДФУ або Європейської фармакопеї.
3.3.7. Стабільність (3.2.S.7):
а) необхідно надати короткі відомості про тип проведених досліджень, використані протоколи і отримані під час досліджень результати;
б) необхідно надати оформлені у відповідному форматі докладні результати дослідження стабільності, включаючи відомості про аналітичні методики, які використовуються для одержання даних, і валідацію цих методик;
в) необхідно надати протокол про дослідження стабільності в післяреєстраційний період та гарантії заявника щодо стабільності.
3.4. Готовий лікарський засіб (3.2.P.):
3.4.1. Опис і склад лікарського засобу (3.2.P.1):
Необхідно надати опис готового лікарського засобу і його склад. Інформація повинна охоплювати опис лікарської форми та складу з переліком усіх компонентів готового лікарського засобу, їх кількостей в перерахунку на одиницю дози, функції в лікарському засобі:
діюча(і) речовина(и);
допоміжна(і) речовина(и) незалежно від її (їх) походження або кількості, включаючи барвники, консерванти, модифікатори, стабілізатори, згущувачі, емульгатори, смакові та ароматичні речовини тощо;
складові компоненти лікарської форми, зовнішніх оболонок лікарських засобів, що потрапляють в організм пацієнта при застосуванні внутрішньо або будь-яким іншим шляхом введення (тверді капсули, м’які капсули, капсули ректальні, таблетки, вкриті оболонкою, таблетки, вкриті плівковою оболонкою тощо);
ці відомості необхідно доповнити будь-якими суттєвими даними, що стосуються типу контейнера та за потреби способу його укупорювання, разом з докладною інформацією про пристрої, за допомогою яких буде використовуватися або вводитися лікарський засіб і які поставлятимуться разом з лікарським засобом.
Вираз «прийнята термінологія», який використовується при описі компонентів лікарських засобів, незалежно від застосування інших положень, означає таке:
для речовин, які наводяться в ДФУ, або Європейській фармакопеї, або ж в інших національних фармакопеях, — основна назва, наведена у заголовку відповідної монографії, та посилання на конкретну фармакопею;
для інших речовин — МНН, рекомендована ВООЗ, або за відсутності такої точна наукова назва; для речовин, що не мають МНН або точної наукової назви;
дані про те, яким чином та із чого вони вироблені та які добавки у них вводилися, супроводжуючи за потреби цю інформацію відповідними деталями;
для барвників — відповідний код «Е», визначений Директивою Ради 78/25/ЄЕС від 12 грудня 1977 року «Про зближення правил країн ЄС щодо барвників, дозволених для застосування у лікарських засобах» та/або Директивою Європейського Парламенту та Ради 94/36/ЄС від 30 червня 1994 року «Про барвники, що застосовуються у харчових продуктах».
Для того, щоб надати «кількісний склад» діючої(их) речовини(н) у готових лікарських засобах, необхідно залежно від розглянутої лікарської форми вказати масу або кількість одиниць біологічної активності в розрахунку або на одиницю дози, або на одиницю маси, або на одиницю вмісту для кожної діючої речовини.
Якщо діючі речовини представлено у вигляді сполук або похідних, то необхідно надати їх кількісне вираження, указавши їхню загальну масу, а за потреби — масу активної частини молекули.
Для лікарських засобів, що містять діючу речовину, яку вперше заявлено у складі лікарського засобу, кількісну характеристику діючої речовини, що є сіллю або гідратом, необхідно завжди зазначати в перерахунку на масу активної частини молекули. Ця кількісна характеристика має бути однаковою у всіх матеріалах реєстраційного досьє незалежно від країни подання.
Для АФІ або діючої речовини, які не можна визначати хімічним шляхом, указують одиниці біологічної активності або, якщо такі є, міжнародні одиниці біологічної активності, установлені ВООЗ. Якщо міжнародні одиниці ВООЗ не встановлено, то одиниці біологічної активності потрібно виражати таким чином, аби надати точну інформацію про активність АФІ або діючої речовини, використовуючи, де необхідно, одиниці Європейської фармакопеї. За можливості має бути вказана біологічна активність на одиницю маси.
3.4.2. Фармацевтична розробка (3.2.P.2):
У цьому розділі надається інформація про дослідження з розробки, проведені з метою доведення того, що лікарська форма, склад, виробничий процес, система упаковки/укупорки контейнера, мікробіологічні властивості та інструкції для медичного застосування відповідають планованому застосуванню, зазначеному в матеріалах реєстраційного досьє заявника.
Дослідження, описані в цьому розділі, мають відрізнятися від посерійних контрольних випробувань, які проводяться відповідно до специфікацій. Необхідно визначити та описати критичні параметри складу та характеристики процесу, які можуть впливати на відтворюваність серій, дію та якість лікарського засобу. За потреби при наданні додаткових підтверджувальних даних необхідно посилатися на відповідні пункти Модуля 4 (звіти про доклінічні дослідження) та Модуля 5 (звіти про клінічні випробування) матеріалів реєстраційного досьє:
а) необхідно обґрунтувати сумісність діючої речовини з допоміжними речовинами, а також основні фізико-хімічні властивості діючої речовини, які можуть вплинути на дію готового лікарського засобу, або сумісність різних діючих речовин одна з одною у випадку лікарських засобів у вигляді комбінацій;
б) необхідно обґрунтувати вибір допоміжних речовин, особливо щодо їх відповідних функцій і концентрацій;
в) необхідно надати опис розробки готового лікарського засобу, беручи до уваги пропонований шлях введення та спосіб застосування;
г) наявність будь-яких надлишків у складі має бути обґрунтованою;
ґ) необхідно вказати та обґрунтувати будь-які фізико-хімічні і біологічні властивості та будь-які параметри, що стосуються дії готового лікарського засобу;
д) необхідно надати інформацію про вибір і оптимізацію виробничого процесу, а також про розбіжності між виробничим процесом, який використовували при виготовлені серій, задіяних у фазах клінічних випробувань, і планованим серійним процесом виробництва готового лікарського засобу;
е) необхідно обґрунтувати придатність контейнера та системи укупорки, яка використовується для зберігання, перевезення та застосування готового лікарського засобу. При цьому може знадобитися опис потенційної взаємодії між лікарським засобом і матеріалом контейнера;
є) як для нестерильних, так і для стерильних лікарських засобів необхідно надати та задокументувати мікробіологічні властивості лікарської форми відповідно до вимог ДФУ або Європейської фармакопеї;
ж) з метою підтвердження відповідної додаткової інформації, яка міститься у маркуванні щодо застосування розчинника(ів) чи дозатора, необхідно обґрунтувати сумісність готового лікарського засобу з розчинником(ами), призначеним(и) для розведення перед застосуванням, або дозатором.
3.4.3. Процес виробництва лікарського засобу (3.2.P.3):
а) опис способу виробництва, зазначеного в заяві про державну реєстрацію лікарського засобу (додаток 1 до цього Порядку), викладається таким чином, щоб надати адекватне коротке резюме характеру виконуваних операцій.
З цією метою він має включати, як мінімум:
опис різних стадій виробництва, включаючи контроль у процесі виробництва та відповідні критерії прийнятності для оцінки того, чи можуть процеси, що використовуються при виробництві, спричинити будь-які небажані зміни компонентів лікарської форми;
у випадку безперервного виробничого процесу — докладний опис запобіжних заходів, необхідних для забезпечення однорідності готового лікарського засобу;
експериментальні дослідження з валідації виробничого процесу при використанні нестандартних методів виробництва або у випадку, коли процес критичний для лікарського засобу;
для стерильних лікарських засобів — докладний опис існуючих процесів стерилізації та/або асептичних процесів;
детальну виробничу рецептуру (склад на серію).
Необхідно надати найменування/П.І.Б. та місцезнаходження виробничих потужностей та вказати обов’язки кожного виробника, включаючи контрактні виробництва, а також інформацію про кожну із запланованих виробничих дільниць або лабораторій;
б) необхідно навести опис аналітичних методик для контролю якості лікарського засобу, які можуть застосовуватися на проміжних стадіях технологічного процесу, з метою забезпечення стандартності виробничого процесу.
Ці методики є важливими з точки зору перевірки відповідності лікарського засобу виробничій рецептурі, особливо у тих випадках, коли заявник пропонує аналітичний метод контролю готового лікарського засобу, який не включає кількісного визначення усіх діючих речовин (або всіх допоміжних речовин, які повинні відповідати таким самим вимогам, що й діючі).
Це стосується й випадків, коли контроль за якістю готового лікарського засобу залежить від контрольних випробувань у процесі виробництва, особливо в тих випадках, коли метод виготовлення лікарського засобу суттєво впливає на його якість;
в) необхідно надати опис, документацію та результати досліджень з валідації для критичних стадій виробництва або критичних методів кількісного визначення, які використовуються у виробничому процесі.
3.4.4. Контроль допоміжних речовин (3.2.P.4):
а) слід надати перелік усіх вихідних матеріалів, що використовуються для виробництва допоміжних речовин, із зазначенням того, на якому етапі процесу застосовується кожний із них. Має надаватися інформація про якість і контроль цих вихідних матеріалів, а також інформація, яка свідчить про те, що матеріали відповідають стандартам з точки зору їх передбачуваного застосування.
Пропоновані методики визначення тотожності барвників повинні дати змогу встановити їх відповідність переліку барвників, наведеному у наказі Міністерства охорони здоров’я України від 19 червня 2007 року № 339 «Про затвердження Переліків назв допоміжних речовин та барвників, що входять до складу лікарських засобів» та Директиві Європейського Парламенту 78/25/ЄЕС від 12 грудня 1977 року «Про зближення правил країн ЄС щодо барвників, дозволених для застосування у лікарських засобах» та/або Директиві Європейського Парламенту та Ради 94/36/ЄС від 30 червня 1994 року «Про барвники, що застосовуються у харчових продуктах». Крім того, барвник має відповідати критеріям чистоти, наведеним у Директиві Європейського Парламенту та Ради 95/45/ЄС від 26 липня 1995 року «Про встановлення критеріїв чистоти до барвників, що застосовуються у харчових продуктах» зі змінами;
б) для кожної допоміжної речовини мають бути надані специфікації та їхнє обґрунтування. Необхідно описати та належним чином валідувати аналітичні методики, що використовуються для контролю їх якості;
в) особливу увагу необхідно приділити допоміжним речовинам людського або тваринного походження.
З метою дотримання особливих заходів щодо запобігання передачі збудників губчатої енцефалопатії тварин заявник також повинен підтвердити і для допоміжних речовин, що лікарський засіб виробляється відповідно до, зокрема, Керівництва з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами.
Відповідність вимогам вищезгаданого керівництва можна підтвердити, надавши сертифікат відповідності конкретній монографії Європейської фармакопеї щодо збудників губчатої енцефалопатії або наукові дані, які обґрунтовують цю відповідність;
г) нові допоміжні речовини:
Для допоміжних речовин, що використовуються уперше в лікарському засобі або застосовуються новим шляхом введення, необхідно надавати повний опис виробництва, властивостей і контролю, з посиланням на підтверджені доклінічні та клінічні дані щодо безпеки. Ця інформація має бути оформлена так, як зазначено вище для діючої речовини.
Необхідно надати документ, що містить докладну хімічну, фармацевтичну та біологічну інформацію. Ця інформація має бути оформлена так, як зазначено в Модулі 3 стосовно діючої речовини.
Інформацію про нову допоміжну речовину може бути надано у вигляді окремого документа.
Якщо заявник і виробник нової допоміжної речовини не є однією й тією самою особою, такий окремий документ має надаватися виробником заявнику для подачі до Центру.
Додаткова інформація про результати дослідження токсичності нової допоміжної речовини повинна надаватися в Модулі 4 матеріалів реєстраційного досьє.
Результати клінічних досліджень для нової допоміжної речовини слід описувати в Модулі 5.
3.4.5. Контроль лікарського засобу (3.2.P.5):
До серії готового лікарського засобу, що підлягає контролю, належать усі одиниці лікарської форми, які виробляли з однієї кількості сировини та піддавали однаковій послідовності технологічних операцій та/або стерилізації, або в разі безперервного виробничого процесу усі одиниці готової продукції, які виробляли за певний проміжок часу.
Максимально припустиме відхилення вмісту діючої речовини в готовому лікарському засобі на дату його виробництва не повинно перевищувати ±5%, за винятком відповідним чином обґрунтованих випадків.
Необхідно надати докладну інформацію про специфікації (при випуску й протягом терміну придатності) з обґрунтуванням їхнього вибору, методів аналізу та валідації.
3.4.6. Стандартні зразки та матеріали (3.2.P.6):
Необхідно визначити та докладно описати стандартні матеріали та стандартні зразки, які використовуються під час контролю готового лікарського засобу, якщо про них не вказано в розділі, що стосується діючої речовини.
3.4.7. Система упаковки/укупорки (3.2.P.7):
Необхідно надати опис контейнера та укупорювальної системи, включаючи матеріали, з яких вироблено кожний компонент первинної упаковки, а також їх специфікації. Специфікації повинні включати опис і методи контролю. За потреби має надаватись інформація про нефармакопейні методи (включаючи валідацію).
Для нефункціональних зовнішніх пакувальних матеріалів надається тільки короткий опис. Для функціональних компонентів вторинної упаковки надається додаткова інформація.
Для пристроїв для введення лікарських засобів необхідно надати СЕ-сертифікат (підтвердження відповідності Директиві 93/42/ЕЕС «Медичні прилади. Медичне обладнання. Медичні пристрої») за наявності.
3.4.8. Стабільність (3.2.P.8):
а) необхідно надати короткі резюме про види проведених досліджень, використані протоколи і отримані під час досліджень результати;
б) необхідно надати оформлені у відповідному форматі докладні результати дослідження стабільності, включаючи відомості про аналітичні методики, які використовуються для одержання даних, і валідацію цих методик.
Для вакцин потрібно надати інформацію про кумулятивну стабільність за необхідності;
в) необхідно надати протокол з вивчення стабільності у післяреєстраційний період і гарантії заявника щодо стабільності.
4. Вимоги до матеріалів реєстраційного досьє, наведених у Модулі 4 «Звіти про доклінічне дослідження»
4.1. Загальний план Модуля 4:
зміст;
звіти про дослідження.
Фармакологія:
первинна фармакодинаміка;
вторинна фармакодинаміка;
фармакологія безпеки;
фармакодинамічні взаємодії.
Фармакокінетика:
аналітичні методики та звіти щодо їх валідації;
всмоктування;
розподіл;
метаболізм;
виведення;
фармакокінетичні взаємодії (доклінічні);
інші фармакокінетичні дослідження.
Токсикологія:
токсичність при одноразовому введенні;
токсичність при повторних введеннях;
генотоксичність;
канцерогенність;
репродуктивна токсичність та токсичний вплив на розвиток потомства;
місцева переносимість.
Додаткові дослідження токсичності.
Посилання на джерела літератури.
4.2. Звіти про дослідження.
Особливу увагу необхідно звернути на таке:
у матеріалах реєстраційного досьє щодо фармакологічних та токсикологічних випробувань повинні визначати:
а) потенційну токсичність лікарського засобу та будь-які небезпечні або небажані токсичні реакції, що можуть спостерігатися за запропонованих умов його застосування людиною; має бути наведена їх оцінка з урахуванням відповідних патологічних станів;
б) фармакологічні властивості лікарського засобу за якісними і кількісними показниками стосовно запропонованого клінічного застосування. Усі результати повинні бути достовірними і мати загальне застосування. При плануванні експериментальних досліджень і оцінці отриманих даних необхідно використовувати методи математичної та статистичної обробки результатів.
Крім того, у матеріалах реєстраційного досьє необхідно надати інформацію про терапевтичний і токсикологічний потенціал лікарського засобу.
У матеріалах реєстраційного досьє щодо програми дослідження необхідно враховувати, що:
усі дослідження, що вимагають повторного введення лікарського засобу, мають плануватися з огляду на ймовірну стимуляцію утворення антитіл і впливу антитіл на організм;
також необхідно розглянути питання доцільності проведення досліджень репродуктивної функції, ембріональної/фетальної і перинатальної токсичності і можливої мутагенної та канцерогенної дії. Якщо причиною токсичності є не діюча речовина, а інші речовини, то можна не проводити дослідження, за умови, коли існує достовірне підтвердження вилучення цих компонентів з лікарського засобу.
Якщо допоміжна речовина використовується у фармацевтичній практиці вперше, необхідно провести її токсикологічні та фармакокінетичні дослідження.
Якщо існує ймовірність значного розпаду лікарського засобу під час його зберігання, необхідно розглянути питання про проведення токсикологічного дослідження продуктів розпаду.
4.2.1. Фармакологія:
У матеріалах реєстраційного досьє щодо фармакологічних досліджень необхідно висвітлити два різних підходи:
по-перше, фармакодинамічна активність лікарського засобу, що пропонується до терапевтичного застосування, має бути відповідним чином дослідженою і описаною. По змозі повинні використовуватися визнані й валідовані методики дослідження як in vivo, так і in vitro. Опис нових експериментальних методик повинен бути достатньо докладним, щоб забезпечити їхнє відтворення. Результати потрібно надавати за кількісними показниками, наприклад, кривими доза-ефект та/або час-ефект тощо. Результати повинні зіставлятися з даними, що характеризують речовину або речовини з аналогічною терапевтичною дією. Відсутність порівняльних досліджень має бути обґрунтованою;
по-друге, дані про основні фармакологічні властивості діючої речовини, із зазначенням її побічної дії на основні функції фізіологічних систем організму. Якщо дози лікарського засобу, що викликають негативні побічні реакції, є близькими до доз, рекомендованих для медичного застосування, ці дослідження мають бути поглибленими.
Якщо експериментальні методи не є стандартними, вони мають бути достатньо детально описаними, аби мати можливість їх відтворення і підтвердження їх достовірності. Результати експерименту мають бути чітко викладеними і доведено їх статистичну достовірність. Будь-які кількісні зміни реакцій, що виникли у відповідь на повторне введення діючої речовини, мають бути дослідженими.
Матеріали реєстраційного досьє щодо вивчень фіксованих комбінацій діючих речовин стосовно їх фармакодинамічної взаємодії повинні ґрунтуватися або на фармакологічних передумовах, або на показаннях для їх застосування. У першому випадку фармакодинамічне дослідження повинне підтвердити взаємодії, що роблять таку комбінацію значимою для терапевтичного застосування. У другому випадку, коли наукове обґрунтування такої комбінації речовин базується на експериментальній терапії, дослідження має встановити можливість підтвердження на тваринах дії, що очікується від такої комбінації речовин, і, принаймні, значимість будь-яких виявлених побічних реакцій.
4.2.2. Фармакокінетика:
Матеріали реєстраційного досьє щодо фармакокінетичних досліджень включають аналіз всіх процесів, що відбуваються з діючою речовиною і її метаболітами в організмі, та охоплюють вивчення всмоктування, розподілу, біотрансформації та виведення цих речовин.
Дослідження кожного з цих етапів може виконуватися як за допомогою фізичних, хімічних або біологічних методів, так і шляхом вивчення фактичної фармакодинамічної активності самої діючої речовини.
Інформація про розподіл і виведення з організму є необхідною у всіх випадках, коли такі дані є обов’язковими для визначення доз для людини, та стосовно хіміотерапевтичних лікарських засобів (антибіотиків тощо) і речовин, використання яких залежить від їх нефармакодинамічних ефектів (наприклад, численні діагностичні засоби тощо).
Можна також провести дослідження in vitro, перевагою яких є використання тест-систем людського походження та порівняння з тест-системами тваринного походження (тобто зв’язування з білками, метаболізм, взаємодія між лікарськими засобами).
У матеріалах реєстраційного досьє необхідно обов’язково наводити інформацію щодо фармакокінетичних досліджень фармакологічно активних речовин. При використанні нових фіксованих комбінацій відомих діючих речовин, що вже були дослідженими, відповідно до положень цього Порядку інформація щодо фармакокінетичних досліджень може не наводитись, якщо таке рішення обґрунтовано результатами дослідження токсичності та експериментальних терапевтичних випробувань.
Дизайн фармакокінетичних досліджень має забезпечувати порівняння даних для тварин і людини та екстраполяцію на людину результатів, отриманих для тварин.
4.2.3. Токсикологія:
а) токсичність при одноразовому введенні
Матеріали реєстраційного досьє щодо дослідження токсичності при одноразовому введенні включають якісний і кількісний аналіз токсичних проявів, що можуть виникнути внаслідок одноразового введення діючої речовини або речовин, які містяться в лікарському засобі у таких пропорціях і фізико-хімічному стані, як і в готовому лікарському засобі;
б) токсичність при повторних (багаторазових) введеннях
Матеріали реєстраційного досьє щодо дослідження токсичності при повторному (багаторазовому) введенні мають бути спрямованим на виявлення будь-яких фізіологічних та/або паталогоанатомічних змін, що виникли внаслідок багаторазового введення діючої речовини або комбінації діючих речовин, та визначення того, як ці зміни залежать від дози.
У матеріалах реєстраційного досьє бажано наводити інформацію щодо двох досліджень — короткострокового, тривалістю 2-4 тижні та довгострокового. Тривалість останнього залежить від тривалості клінічного застосування лікарського засобу. Його метою є експериментальне визначення і опис потенційних побічних реакцій, що мають бути врахованими при проведенні клінічних випробувань;
в) генотоксичність
У матеріалах реєстраційного досьє наводиться інформація щодо мутагенного та кластогенного потенціалу, метою якої є виявлення порушень, які може спричинити діюча речовина в генетичному матеріалі окремого організму або в клітинах. Мутагенні речовини є небезпечними для здоров’я людини, оскільки дія мутагену спричиняє мутації в статевих клітинах і виникнення спадкових порушень, а також у соматичних клітинах, що може призводити до розвитку злоякісних новоутворень. Ці дослідження є обов’язковими для всіх нових діючих речовин;
г) канцерогенність
У матеріалах реєстраційного досьє наводиться інформація щодо досліджень канцерогенного потенціалу, які зазвичай проводяться, якщо:
лікарський засіб призначено для тривалого безперервного або періодичного (з перервами) застосування протягом усього життя хворого;
при проведенні токсикологічних досліджень токсичності при повторному (багаторазовому) введенні лікарського засобу виявлено зміни, що викликають занепокоєння з приводу їхнього канцерогенного потенціалу;
діюча речовина належить до хімічного класу або є близькою за структурою до відомих канцерогенів або коканцерогенів.
Немає необхідності проводити такі дослідження з безумовно генотоксичними сполуками, оскільки вважається, що вони є трансвидовими канцерогенами і становлять небезпеку для людини;
ґ) репродуктивна токсичність та токсичний вплив на розвиток потомства
У матеріалах реєстраційного досьє наводиться інформація щодо дослідження можливих порушень репродуктивної функції у чоловіків та жінок, а також негативного впливу на потомство, які здійснюються за допомогою відповідних випробувань. Вони включають дослідження впливу на репродуктивну функцію статевозрілих самців і самиць, дослідження токсичного та тератогенного впливу на потомство на всіх стадіях розвитку від зачаття до статевої зрілості, а також латентних ефектів, коли досліджуваний лікарський засіб застосовувався для лікування вагітних самиць.
Відсутність подібних досліджень у матеріалах реєстраційного досьє має бути відповідним чином обґрунтовано.
У матеріалах реєстраційного досьє наводиться інформація залежно від показань для застосування лікарського засобу щодо проведення додаткових досліджень (розвитку потомства) (за необхідності), наприклад, за наявності обґрунтування введення лікарського засобу статевонезрілим тваринам.
Матеріали реєстраційного досьє щодо доклінічних досліджень мають містити інформацію щодо дослідження ембріотоксичності, які проводяться, як правило, на двох видах ссавців, одним із яких не є гризуни. Дослідження пери- та постнатальної токсичності має проводитися принаймні на одному виді тварин. Якщо відомо, що метаболізм лікарського засобу для певного виду тварин є аналогічним метаболізму у людини, при проведенні досліджень доцільно використовувати саме цей вид. Бажано також, щоб один з видів тварин був тим самим, який використовувався при проведенні досліджень токсичності при повторному (багаторазовому) введенні;
д) місцева переносимість
Матеріали реєстраційного досьє щодо доклінічних досліджень мають містити інформацію щодо місцевої переносимості, метою якої є вивчення та визначення місцевої дії лікарського засобу (діючих і допоміжних речовин) на тканини організму в тих ділянках, що можуть контактувати з лікарським засобом, внаслідок його введення при клінічному застосуванні.
Стратегія дослідження має бути спрямованою на те, щоб відрізнити будь-який механічний вплив введення або дію, зумовлену фізико-хімічними властивостями лікарського засобу, від токсичного або фармакодинамічного ефекту.
У матеріалах реєстраційного досьє має бути доведено, що дослідження місцевої переносимості здійснювалися з використанням лікарського засобу, розробленого для застосування людиною, під час якого тваринам контрольної групи вводиться наповнювач/розчинник для введення досліджуваного лікарського засобу та/або допоміжні речовини. За необхідності слід надати інформацію щодо додаткового включення групи позитивного контролю або речовини порівняння.
Інформація щодо дизайну дослідження місцевої переносимості (вибір видів тварин, тривалість, частота, спосіб введення, дози) повинна містити завдання дослідження і рекомендовані умови клінічного застосування лікарського засобу. За необхідності наводять інформацію щодо проведених досліджень зворотності місцевих ушкоджень.
Інформацію щодо досліджень на тваринах можна замінити інформацією щодо випробувань з використанням валідованих методів in vitro, якщо результати досліджень дозволяють визначити співвідношення користь/ризик.
Для хімічних речовин, що застосовуються місцево (наприклад, дермальні, ректальні, вагінальні), має наводитись оцінка їх сенсибілізуючого потенціалу з використанням щонайменше однієї тест-системи (дослідження на морських свинках або місцевих лімфатичних вузлах).
5. Вимоги до матеріалів реєстраційного досьє, наведених у Модулі 5 «Звіти про клінічні випробування»
5.1. Модуль 5 має таку загальну схему:
зміст;
перелік усіх клінічних випробувань у вигляді таблиць;
звіти про клінічні випробування;
звіти про біофармацевтичні дослідження;
звіти, що стосуються дослідження фармакокінетики при використанні біоматеріалів людського походження;
звіти про фармакокінетичні дослідження у людини;
звіти про фармакодинамічні дослідження у людини;
звіти про дослідження ефективності та безпеки;
звіти про контрольовані клінічні дослідження щодо підтвердження заявлених показань для застосування;
звіти про неконтрольовані клінічні дослідження, звіти про аналізи даних за кількома дослідженнями і звіти про інші клінічні дослідження;
звіти про післяреєстраційний досвід застосування;
зразки індивідуальних реєстраційних форм та індивідуальні списки пацієнтів;
посилання на джерела літератури.
5.2. Перелік усіх клінічних випробувань у вигляді таблиць:
Особливу увагу необхідно звернути на наявність у матеріалах реєстраційного досьє інформації щодо такого:
а) клінічна інформація, яку необхідно надати, повинна дозволити сформувати достатньо обґрунтовані й достовірні з наукової точки зору висновки щодо ефективності та безпеки лікарського засобу. Отже, важливою вимогою є те, що оприлюдненню підлягають результати всіх клінічних випробувань як сприятливі, так і несприятливі;
б) клінічним випробуванням завжди мають передувати відповідні фармакологічні та токсикологічні дослідження, проведені на тваринах, інформація щодо яких наведена у Модулі 4 матеріалів реєстраційного досьє. Дослідник повинен ознайомитися з висновками, зробленими в результаті фармакологічних і токсикологічних досліджень, і тому заявник повинен надати йому принаймні брошуру дослідника, у яку повинна ввійти вся відповідна інформація, відома на дату початку клінічних випробувань, включаючи хімічні, фармакологічні та біологічні дані, результати токсикологічних, фармакокінетичних і фармакодинамічних досліджень на тваринах, а також результати попередніх клінічних випробувань, що надають адекватні дані для обґрунтування характеру, масштабу та тривалості планованого дослідження; повні звіти про фармакологічні та токсикологічні дослідження повинні надаватися на вимогу. Відносно матеріалів людського або тваринного походження повинні бути задіяні всі наявні засоби щодо забезпечення безпеки у зв’язку з можливим поширенням збудників інфекції до початку дослідження;
в) власники реєстраційного посвідчення повинні забезпечити, щоб основна документація клінічного дослідження (у тому числі індивідуальні реєстраційні форми) зберігалася у власників отриманих результатів, за винятком медичних карт стаціонарних/амбулаторних хворих (пацієнтів).
Медичні карти стаціонарних/амбулаторних хворих (пацієнтів) повинні зберігатися у відповідних умовах та протягом строку, передбаченого чинним законодавством.
Інформація щодо будь-якої зміни права власності стосовно наявних даних повинна бути відповідно оформленою;
г) опис кожного клінічного дослідження повинен містити достатню кількість інформації, щоб скласти об’єктивний висновок про:
протокол, що містить обґрунтування, цілі, статистичний метод і методологію дослідження з умовами його проведення та організації, і докладну інформацію про досліджуваний лікарський засіб;
сертифікат про проходження аудиту (якщо такий проводився);
список дослідників;
кожний дослідник повинен повідомити своє прізвище, місце проживання, місце роботи, займану посаду, кваліфікаційні дані та обов’язки при проведенні клінічних досліджень;
вказати, де проводилося дослідження, надати інформацію щодо кожного окремого пацієнта, включаючи індивідуальні реєстраційні форми;
заключний звіт, підписаний дослідником, а при багатоцентровому дослідженні — всіма співдослідниками або відповідальним дослідником;
ґ) повна документація щодо опису клінічного дослідження, про яке йшлося вище, може надаватись на вимогу.
У матеріалах реєстраційного досьє має бути відображена думка дослідника на основі експериментальних доказів про безпеку лікарського засобу за звичайних умов його застосування, його переносимість, ефективність, про будь-яку корисну інформацію відносно показань для застосування та протипоказань, дозувань, тривалості терапії, а також щодо особливих запобіжних заходів, яких необхідно вжити під час лікування та при появі клінічних симптомів передозування. У звіті про результати багатоцентрового дослідження, який наводиться у матеріалах реєстраційного досьє, відповідальний дослідник у своїх висновках повинен висловити думку про безпеку та ефективність досліджуваного лікарського засобу під час багатоцентрового дослідження;
д) клінічні спостереження щодо кожного дослідження повинні бути узагальненими із зазначенням:
кількості та статі пацієнтів, які отримали лікування;
відбору і розподілу за віком пацієнтів у досліджуваних та контрольних групах;
кількості пацієнтів, які достроково вибули з дослідження, і причин, з яких це відбулося;
якщо контрольовані дослідження проводилися за зазначених вище умов, указати, що відбувалося з контрольною групою:
не одержувала лікування,
одержувала плацебо,
одержувала інший лікарський засіб з відомою дією,
одержувала інший вид лікування без застосування лікарських засобів;
частоти побічних реакцій, що спостерігалися;
подробиць щодо пацієнтів, що входять до груп підвищеного ризику, наприклад, люди літнього віку, діти, вагітні або жінки репродуктивного віку, або хворі, фізіологічний або патологічний стан яких вимагає особливої уваги;
параметрів або критеріїв оцінки ефективності та отриманих результатів;
статистичної оцінки результатів, якщо цього вимагає дизайн дослідження, і використаних змінних факторів.
У матеріалах реєстраційного досьє повинна відображатись інформація щодо спостереження дослідника про:
будь-які ознаки звикання, залежності або труднощів у пацієнтів, що виникають при відміні лікарського засобу;
будь-які взаємодії, що мали місце при одночасному введенні з іншими лікарськими засобами;
критерії, що визначають необхідність виключення деяких пацієнтів із дослідження;
фатальні випадки під час дослідження або в період подальшого спостереження.
Інформація про нову комбінацію діючих речовин повинна бути ідентичною даним про новий лікарський засіб і до неї необхідно включати обґрунтування безпеки та ефективності комбінації.
У разі повної або часткової відсутності вищенаведених даних у матеріалах реєстраційного досьє необхідно надати пояснення. За необхідності повинна надаватись інформація про огляд та аналіз додаткових доклінічних токсикологічних та фармакологічних досліджень у разі отримання при проведенні клінічних досліджень несподіваних результатів.
Якщо лікарський засіб призначено для тривалого застосування, у матеріалах реєстраційного досьє необхідно надати опис будь-яких змін фармакологічної дії в результаті багаторазового застосування лікарського засобу, а також необхідно обґрунтувати вибір дозувань для тривалого застосування.
5.3. Звіти про клінічні випробування:
5.3.1. Звіти про біофармацевтичні дослідження:
До матеріалів реєстраційного досьє необхідно надати звіти про дослідження біодоступності, порівняльної біодоступності, біоеквівалентності, кореляції in vitro — in vivo і опис біоаналітичних та аналітичних методик.
Крім того, за потреби демонстрації біоеквівалентності лікарських засобів має бути наведена інформація про проведену оцінку їх біодоступності.
У разі застосування процедури біовейвер до матеріалів реєстраційного досьє необхідно надати звіт про проведення досліджень in vitro. Оцінка та проведення досліджень біоеквівалентності або обґрунтування щодо його непроведення має бути надана відповідно до вимог розділів ХІХ та ХХ цього Порядку та керівних настанов.
5.3.2. Звіти, що стосуються дослідження фармакокінетики при використанні біоматеріалів людського походження:
До біоматеріалів людського походження відносять білки, клітини, тканини й пов’язані з ними матеріали, отримані від людини, які використовуються при проведенні досліджень in vitro або ex vivo з метою оцінки фармакокінетичних властивостей діючих речовин.
У матеріалах реєстраційного досьє необхідно надати звіти про дослідження зв’язування з білками плазми, метаболізму в печінці та взаємодії діючої речовини, а також дослідження з використанням інших біоматеріалів людського походження.
5.3.3. Звіти про фармакокінетичні дослідження у людини:
а) у матеріалах реєстраційного досьє необхідно описати такі фармакокінетичні характеристики:
абсорбція (швидкість і ступінь);
розподіл;
метаболізм;
виведення.
Необхідно надати інформацію щодо опису клінічно важливих характеристик, включаючи значення кінетичних даних при визначенні схеми прийому лікарського засобу для пацієнтів із груп ризику, і відмінностей між людиною та видами тварин, використаних при проведенні доклінічних досліджень.
У матеріалах реєстраційного досьє крім інформації щодо стандартних фармакокінетичних досліджень з використанням значної кількості зразків при фармакокінетичних аналізах у популяції, що базуються на розрідженому відборі проб під час клінічних досліджень, також може наводитись інформація щодо впливу внутрішніх і зовнішніх факторів на варіабельність взаємозв’язку між дозою і фармакокінетичним відгуком. Необхідно надати звіти про дослідження фармакокінетики та перші дослідження переносимості лікарського засобу здоровими добровольцями та пацієнтами, звіти про вивчення фармакокінетики з метою оцінки впливу внутрішніх і зовнішніх факторів, а також звіти про фармакокінетичні дослідження в популяції;
б) якщо лікарський засіб зазвичай застосовується разом з іншими лікарськими засобами, у матеріалах реєстраційного досьє має бути наданий опис дослідження їх одночасного застосування, що проводилося для демонстрації можливої зміни фармакологічної дії.
Необхідно надати інформацію щодо вивчення фармакокінетичної взаємодії діючої речовини з іншими лікарськими засобами або речовинами.
5.3.4. Звіти про фармакодинамічні дослідження у людини:
а) у матеріалах реєстраційного досьє необхідно підтвердити кореляцію фармакодинамічної дії та ефективності, включаючи:
взаємозв’язок доза — відгук та її розвиток у часі;
обґрунтування дозувань і способів введення;
механізм дії, якщо можливо.
Необхідно надати опис фармакодинамічної дії, не пов’язаний з ефективністю.
Інформація щодо демонстрації фармакодинамічної дії в людини сама по собі є недостатньою для того, щоб зробити висновки стосовно будь-якої конкретної потенційної терапевтичної дії;
б) якщо лікарський засіб, зазвичай, застосовується разом з іншими лікарськими продуктами, повинен бути наданий опис дослідження їх одночасного застосування, що проводилося для демонстрації можливої зміни фармакологічної дії.
Необхідно зазначити інформацію щодо вивчення фармакодинамічної взаємодії діючої речовини з іншими лікарськими засобами або речовинами.
5.3.5. Звіти про дослідження ефективності та безпеки:
5.3.5.1. Звіти про контрольовані клінічні дослідження щодо підтвердження заявлених показань для застосування:
Інформація повинна бути надана щодо проведених клінічних досліджень, які за можливості є рандомізованими «контрольованими клінічними дослідженнями», де досліджуваний лікарський засіб порівнюється із плацебо та відомим лікарським засобом відомої терапевтичної ефективності; використання будь-якого іншого дизайну дослідження необхідно обґрунтувати. Інформація щодо лікування контрольних груп буде різною у кожному конкретному випадку та буде залежати від етичних норм й галузі терапії, тому в окремих випадках, можливо, буде наводитися інформація щодо порівняння ефективності нового лікарського засобу з ефективністю відомого лікарського засобу відомої терапевтичної ефективності, ніж з дією плацебо.
Під час надання оцінки необхідно застосовувати заходи, що дозволяють уникнути необ’єктивності, включаючи методи рандомізації та сліпого контролю.
Протокол дослідження, що міститься у матеріалах реєстраційного досьє, повинен включати докладний опис використаних статистичних методів, число пацієнтів й причини для їхнього включення (включаючи розрахунки статистичної потужності досліджень), застосований рівень значимості та опис статистичної одиниці. Заходи, застосовані для уникнення необ’єктивної оцінки, особливо методи рандомізації, мають бути відповідно обґрунтовані.
Інформація щодо включення великої кількості пацієнтів для участі в дослідженні не повинна вважатися рівноцінною заміною відповідному контрольованому дослідженню.
При наведенні інформації щодо аналізу даних з безпеки необхідно приділити увагу обставинам, що призвели до зміни дози або необхідності супутнього застосування іншого лікарського засобу, серйозних побічних реакцій, реакцій, які стали причиною виключення з участі в дослідженні та смерті. Необхідно ідентифікувати пацієнтів або групи пацієнтів дослідження з підвищеним ступенем ризику й звернути особливу увагу на потенційно вразливі групи, кількість яких може бути невеликою, наприклад, діти, вагітні, люди літнього віку зі слабким здоров’ям, люди зі значними порушеннями обміну речовин або екскреції та ін. Необхідно приділити увагу значенню оцінки безпеки для можливих видів застосування лікарського засобу.
5.3.5.2. Звіти про неконтрольовані клінічні дослідження, звіти про аналізи даних за кількома дослідженнями і звіти про інші клінічні дослідження:
Ці звіти необхідно надати до матеріалів реєстраційного досьє.
5.3.6. Звіти про післяреєстраційний досвід застосування:
Якщо лікарський засіб уже зареєстровано в інших країнах, у матеріалах реєстраційного досьє необхідно надати інформацію про побічні реакції на розглянутий лікарський засіб та лікарські засоби з тією(ими) самою(ими) діючою(ими) речовиною(ами), за можливості, порівняно з показниками їхнього застосування.
5.3.7. Зразки індивідуальних реєстраційних форм та індивідуальні списки пацієнтів:
До матеріалів реєстраційного досьє додаються зразки індивідуальних реєстраційних форм та списки пацієнтів зі збереженням конфіденційності персональних даних пацієнтів даного дослідження.
Х. Спеціальні вимоги до матеріалів реєстраційного досьє (у форматі загального технічного документа) за різними типами заяв
1. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів з добре вивченим медичним застосуванням
Для лікарських засобів, АФІ або діючу(і) речовину(и) яких добре вивчено в медичному застосуванні з підтвердженою ефективністю та прийнятним рівнем безпеки, застосовуються такі спеціальні вимоги до матеріалів реєстраційного досьє.
Заявник повинен подавати Модулі 1, 2 і 3, як зазначено у розділі ІХ цього Порядку.
Для матеріалів реєстраційного досьє Модулів 4 і 5 у докладній науковій бібліографії необхідно вказувати доклінічні та клінічні характеристики лікарського засобу.
Для підтвердження добре вивченого медичного застосування мають використовуватися спеціальні вимоги:
а) фактори, які необхідно враховувати при визначенні добре вивченого медичного застосування компонентів лікарських засобів:
час, протягом якого використовується АФІ або діюча речовина,
кількісні аспекти використання АФІ або діючої речовини,
ступінь наукового інтересу у використанні АФІ або діючої речовини (з посиланням на опубліковані наукові джерела),
послідовність наукових оцінок.
Таким чином, для визначення добре вивченого застосування різних АФІ або діючих речовин може знадобитися оцінка за різні періоди часу. Однак у будь-якому разі період часу, необхідний для визначення добре вивченого медичного застосування АФІ або діючої речовини, має бути не меншим за 10 років з часу першого систематичного та документованого використання цієї АФІ або діючої речовини як лікарського засобу;
б) матеріали реєстраційного досьє, надані заявником, повинні включати всі аспекти оцінки безпеки та ефективності, містити або давати посилання на огляд відповідної літератури, з урахуванням перед- та післяреєстраційних досліджень, та опублікованої наукової літератури стосовно результатів епідеміологічних досліджень і, особливо, порівняльних епідеміологічних досліджень; всю документацію, як позитивну, так і негативну. Щодо аспектів про «добре вивчене медичне застосування» необхідно уточнити, що «бібліографічне посилання» на інші джерела доказів (післяреєстраційні дослідження, епідеміологічні дослідження тощо), а не тільки наявні дані, що стосуються методів контролю та випробувань, можуть бути вагомим доказом безпеки та ефективності лікарського засобу за умови, що в матеріалах реєстраційного досьє чітко пояснено та обґрунтовано використання цих джерел інформації;
в) особливу увагу необхідно приділити інформації, якої не вистачає, та необхідно обґрунтувати, чому може вважатися доведеним прийнятний рівень безпеки та/або ефективності, незважаючи на відсутність деяких досліджень;
г) у доклінічних та/або клінічних оглядах необхідно пояснити значимість будь-яких поданих даних, що стосуються вже зареєстрованого лікарського засобу, відмінного від того, який пропонується до перереєстрації. Необхідно надати обґрунтування з приводу того, чи можна заявлений лікарський засіб уважати подібним до вже зареєстрованого лікарського засобу, незважаючи на існуючі розбіжності;
ґ) післяреєстраційний досвід використання інших лікарських засобів, що містять ті самі компоненти, є особливо важливим, і заявники під час надання матеріалів реєстраційного досьє мають приділити цьому питанню належну увагу.
2. Спеціальні вимоги до матеріалів реєстраційного досьє для генеричних лікарських засобів
а) Матеріали реєстраційного досьє на генеричні лікарські засоби повинні містити дані, описані в Модулях 1, 2 і 3 розділу ІХ цього Порядку за умови, що заявник має дозвіл власника реєстраційного посвідчення оригінального/референтного лікарського засобу щодо посилання на Модулі 4 і 5 матеріалів його реєстраційного досьє;
б) Матеріали реєстраційного досьє повинні містити дані, описані в Модулях 1, 2 і 3 розділу ІХ цього Порядку, а також дані, що доводять біодоступність і біоеквівалентність з оригінальним лікарським засобом, за умови, що він не є біологічним лікарським засобом.
Для цих лікарських засобів матеріали реєстраційного досьє щодо доклінічних/клінічних оглядів/резюме (у разі їх проведення) мають включати такі елементи:
обґрунтування того, що лікарський засіб є по суті аналогічним;
короткий опис домішок, наявних у серіях АФІ або діючої(их) речовини(н), а також у готовому лікарському засобі, який реєструється (у відповідних випадках про продукти розпаду, що виникають при зберіганні), разом з оцінкою цих домішок;
оцінка досліджень біоеквівалентності або обґрунтування того, чому дослідження не проводилися відповідно до розділів ХІХ та ХХ цього Порядку та керівних настанов;
остання опублікована література, що є актуальною для АФІ або діючої речовини та цього реєстраційного досьє. Для цього можна використовувати анотації статей наукових журналів;
кожне твердження в короткій характеристиці лікарського засобу, яке до цього не було відомо, або не витікає, виходячи з властивостей лікарського засобу та/або його терапевтичної групи, необхідно розглянути в доклінічних/клінічних оглядах/резюме та підтвердити опублікованою літературою та/або додатковими дослідженнями. При цьому інструкція для медичного застосування генеричного лікарського засобу має містити інформацію, ідентичну тій, яка наведена в інструкції для медичного застосування референтного лікарського засобу. Будь-які відмінності інструкції для медичного застосування генеричного лікарського засобу мають обґрунтовуватись результатами, отриманими при багатоцентрових клінічних дослідженнях;
якщо до складу генеричного лікарського засобу входять солі, ефіри або похідні зареєстрованої АФІ або діючої речовини, відмінні від тих, що входять до складу референтного лікарського засобу, заявник повинен надати додаткові дані з метою доведення еквівалентності характеристик безпеки та ефективності;
якщо АФІ або діюча речовина генеричного лікарського засобу має таку саму терапевтичну дію, що й оригінальний зареєстрований лікарський засіб, але ця дія пов’язана з іншою сіллю/ефіром/комплексом/похідним, необхідно підтвердити, що при цьому не змінюється фармакокінетика, фармакодинаміка та/або токсичність тієї частини речовини, яка є відповідальною за ці властивості, і може змінити профіль безпеки/ефективності. Якщо таких доказів не існує, то дану речовину можна вважати новою діючою речовиною;
якщо лікарський засіб призначено для іншого терапевтичного використання або представлено в іншій лікарській формі, або він має застосовуватися іншими шляхами введення, або в інших дозах, або за іншої позології, необхідно надати результати відповідних токсикологічних і фармакологічних досліджень та/або клінічних випробувань.
3. Спеціальні вимоги до матеріалів реєстраційного досьє для подібних біологічних лікарських засобів (біосимілярів)
У випадку реєстрації подібних біологічних лікарських засобів, які не можуть вважатись генеричними лікарськими засобами через специфічність процесу виробництва, застосовуваної сировини, особливостей молекулярної будови та терапевтичної дії, до матеріалів реєстраційного досьє необхідно надати додаткові дані, що підтверджують належний рівень безпеки (токсикологічні чи інші доклінічні) та ефективності (клінічні).
При поданні заяви на реєстрацію подібного біологічного лікарського засобу після закінчення патентного захисту даних щодо зареєстрованого раніше оригінального біологічного лікарського засобу необхідно надавати такі матеріали:
інформація, що подається, не повинна обмежуватися Модулями 1, 2 і 3 (фармацевтичні, хімічні та біологічні дані), доповненими даними з біоеквівалентності та біодоступності. Вид і кількість додаткових даних (тобто токсикологічних, інших доклінічних та відповідних клінічних даних) мають визначатися у кожному окремому випадку згідно з відповідними вимогами та керівництвами;
у зв’язку з різноманітністю біологічних лікарських засобів може знадобитися надання інформації щодо проведення ідентифікувальних досліджень, передбачених у Модулях 4 і 5 реєстраційного досьє, враховуючи спеціальну характеристику кожного конкретного лікарського засобу.
У випадку, коли зареєстрований оригінальний біологічний лікарський засіб має більше одного показання до застосування, ефективність і безпека подібного біологічного лікарського засобу повинні підтверджуватися або за необхідності доводитися окремо для кожного із заявлених показань.
4. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів з фіксованою комбінацією
Матеріали реєстраційного досьє повинні стосуватися нових лікарських засобів, вироблених з використанням, принаймні, двох відомих АФІ або діючих речовин, які раніше не застосовувалися у такій комбінації з терапевтичною метою.
Для таких лікарських засобів необхідно надати матеріали повного реєстраційного досьє (Модулі 1-5).
За можливості треба надати інформацію щодо дільниць виробництва, допоміжних речовин та оцінки безпеки.
5. Спеціальні вимоги до матеріалів реєстраційного досьє у виняткових випадках
Якщо заявник може довести, що для лікарського засобу неможливо надати повні дані про ефективність і безпечність за нормальних умов використання, оскільки:
показання, для яких застосовується лікарський засіб, зустрічаються вкрай рідко; у такий спосіб заявник не може зі зрозумілих причин надати достатньо даних, або
існуючі наукові дані не дають змоги надати повну інформацію, або
збір такої інформації буде суперечити загальноприйнятим принципам медичної етики,
то реєстраційне посвідчення може видаватися з урахуванням певних зобов’язань.
Ці зобов’язання можуть включати таке:
заявник повинен здійснити певну програму досліджень у період часу, зазначений МОЗ, результати якої мають бути основою для повторної оцінки співвідношення користь/ризик,
лікарський засіб, що подається на реєстрацію, має бути рецептурним та в деяких випадках застосовуватися тільки під медичним наглядом, можливо в лікарні, а у випадку радіофармацевтичних препаратів — кваліфікованим спеціалістом,
інструкція для медичного застосування та будь-яка медична інформація мають звертати увагу лікаря на той факт, що наявний опис даного лікарського засобу є певною мірою неповним.
6. Спеціальні вимоги до матеріалів реєстраційного досьє за гібридною заявою про реєстрацію лікарського засобу
У такому випадку заявник має подавати Модулі 1, 2 та 3 як зазначено в розділі IХ цього Порядку.
Матеріали реєстраційного досьє для Модулів 4 та/або 5 мають містити звіти про обмежені доклінічні дослідження та/або клінічні випробування, що проводилися заявником (залежно від певної ситуації щодо відмінностей запропонованого лікарського засобу та референтного препарату (пункт 6.5 розділу VI цього Порядку), та докладні бібліографічні дані щодо решти необхідної інформації для підтвердження безпеки та/або ефективності запропонованого лікарського засобу. Необхідно надати обґрунтування з приводу того, чи можна запропонований лікарський засіб уважати подібним до вже зареєстрованого лікарського засобу, незважаючи на існуючі розбіжності.
7. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів з продукції in bulk
Для готових лікарських засобів, які виготовлені за такими стадіями виробничого процесу як кінцеве пакування та/або маркування, застосовуються особливі правила при поданні матеріалів реєстраційного досьє.
Заявник повинен подавати Модулі 1 та 2 матеріалів реєстраційного досьє за вимогами, наведеними у розділі ІХ цього Порядку.
Для Модулів 3, 4 і 5 фармацевтичні, доклінічні та клінічні характеристики лікарського засобу необхідно вказувати у повному обсязі на підставі матеріалів реєстраційного досьє виробника продукції in bulk. Відповідні розділи Модуля 3 також мають містити додаткові дані та документацію заявника (власника реєстраційного посвідчення на такий лікарський засіб) щодо стадій виробничого процесу, які ним виконуються.
Крім того, у відповідних розділах матеріалів реєстраційного досьє необхідно чітко вказувати найменування та місцезнаходження виробничих потужностей виробника продукції in bulk (який відповідає за контроль якості) та виробничих потужностей виробника, який відповідає за випуск серії. Інформація щодо виробників має бути наведена у тексті маркування.
Необхідно надати підтвердження реєстрації заявленого лікарського засобу у споживчій упаковці в країні виробника продукції in bulk.
8. Спеціальні вимоги до матеріалів реєстраційного досьє для біологічних лікарських засобів
8.1. Лікарські засоби, отримані з крові або плазми людини:
Положення Модуля 3 можуть частково не застосовуватися до лікарських засобів, отриманих з крові або плазми людини, для яких матеріали реєстраційного досьє, оформлені згідно з вимогами, викладеними в пунктах 3.2 та 3.3 глави 3 розділу IХ цього Порядку, для вихідних матеріалів, отриманих з крові або плазми людини, можуть бути замінені мастер-файлом на плазму, оформленим відповідно до цієї частини.
Додатково до матеріалів реєстраційного досьє може надаватись:
мастер-файл на плазму, що є окремим документом, який не входить до матеріалів реєстраційного досьє, та містить всю відповідну докладну інформацію про характеристики цільної плазми людини, що використовується як вихідний матеріал та/або сировина для виробництва субфракцій/проміжних фракцій, компонентів допоміжних та діючих речовин, які є частиною лікарського засобу;
інформація щодо всіх закладів охорони здоров’я/установ, які займаються фракціонуванням/обробкою плазми людини, має підготувати та поповнювати свіжими даними набір відповідної докладної інформації, про яку йдеться у мастер-файлі на плазму;
якщо заявник не є власником мастер-файла на плазму, то власник повинен надати свій мастер-файл заявнику для його подання. У будь-якому випадку заявник є відповідальним за цей лікарський засіб;
у будь-якому реєстраційному досьє на лікарський засіб, що містить компоненти, отримані з плазми людини, повинне бути посилання на мастер-файл на плазму, який відповідає саме тій плазмі, що використовувалася як вихідний матеріал/сировина.
8.1.1. У мастер-файлі на плазму повинна міститися інформація про плазму, що використовувалася як вихідний матеріал/сировина, а саме:
а) про походження плазми:
інформація про заклади охорони здоров’я/установи, у яких проводиться відбір крові/плазми, уключаючи дані про проведені інспекції та отриману акредитацію, а також епідеміологічні дані про інфекції, які передаються через кров;
інформація про заклади охорони здоров’я/установи, у яких проводиться дослідження зразків донорської крові і пулів плазми, включаючи дані про проведені інспекції та отриману акредитацію;
критерії включення/виключення донорів крові/плазми;
прийнята система, що дозволяє відстежити шлях кожного донорського зразка від забору крові/плазми до готового лікарського засобу та навпаки;
б) про якість та безпеку плазми:
відповідність монографії Європейської фармакопеї або у разі її відсутності іншій визнаній національній фармакопеї;
аналіз зразків донорської крові/плазми та пулів на наявність збудників інфекцій, уключаючи опис методів аналізу, та у випадку пулів плазми дані з валідації використаних методів аналізу;
технічні характеристики контейнерів для відбору крові та плазми, уключаючи інформацію про використані розчини антикоагулянтів;
умови зберігання та транспортування плазми;
процедура з підтримки обладнання та/або період карантину;
характеристика пулу плазми.
8.1.2. У мастер-файлі на плазму повинна міститися інформація про прийняту систему взаємодії між виробником одержаного з плазми лікарського засобу, що здійснює фракціонування/обробку плазми, з одного боку, та/або закладами охорони здоров’я/установами, які займаються відбором та аналізом крові/плазми, з іншого боку, яка визначає умови їхньої взаємодії та погодження специфікацій.
8.1.3. Мастер-файл на плазму повинен також містити перелік лікарських засобів, отриманих з даної плазми, незалежно від того, зареєстровані ці лікарські засоби чи ще перебувають у процесі реєстрації, включаючи лікарські засоби, виготовлені для клінічних випробувань.
8.1.4. У мастер-файлі на плазму повинна міститися інформація про оцінку та сертифікацію:
Для ще не зареєстрованих лікарських засобів заявник повинен надати матеріали повного реєстраційного досьє та окремо мастер-файл на плазму, якщо даний мастер-файл не проходив оцінку раніше.
Мастер-файл на плазму щорічно поповнюється свіжими даними та повторно сертифікується.
Мастер-файл на плазму містить інформацію щодо сертифікації, повторної сертифікації або зміни до мастер-файла на плазму, який відноситься до лікарського засобу.
8.2. Вакцини:
Стосовно вакцин для людини, за умови застосування системи загального файла на вакцинний антиген (Vaccine Antigen Master File) (далі — ВАЗФ), положення Модуля 3 можуть частково не застосовуватись при оформленні реєстраційного досьє згідно з вимогами, викладеними у розділах 3.2 та 3.3 частини ІХ цього Порядку для вихідних матеріалів.
Матеріали реєстраційного досьє для вакцин, за винятком вакцин проти грипу людини, повинні включати ВАЗФ на кожний антиген, який є активною речовиною цієї вакцини.
8.2.1. ВАЗФ є частиною матеріалів реєстраційного досьє, яка містить всю відповідну детальну інформацію про біологічний, фармацевтичний та хімічний характер кожної з активних речовин, що входять до складу цього лікарського засобу. Ця окрема частина може бути загальною для однієї та більше моновалентних та/або комбінованих вакцин, що представлені одним й тим самим заявником/власником реєстраційного посвідчення.
Вакцина може містити один або декілька різних антигенів, кожен з яких є активною речовиною.
Комбінована вакцина містить принаймні два різних антигени, призначені для профілактики одного або декількох інфекційних захворювань.
Моновалентна вакцина містить тільки один антиген, призначений для профілактики одного інфекційного захворювання.
8.2.2. ВАЗФ повинен містити інформацію щодо якості, про що йдеться у відповідному розділі (діюча речовина Модуля 3):
а) діюча речовина:
загальна інформація, включаючи відповідність монографіям ДФУ, Європейської фармакопеї;
інформація щодо виробництва діючої речовини, включаючи інформацію про процес виробництва, вихідні матеріали та сировину, особливі заходи з оцінки ризику передачі збудників та додаткові заходи оцінки безпеки споруд та обладнання;
характеристики діючої речовини;
контроль якості діючої речовини;
стандартні зразки речовин та матеріалів;
система упаковка/укупорка для діючої речовини;
стабільність діючої речовини;
б) оцінка та сертифікація.
Для нових вакцин, які містять новий вакцинний антиген, заявник повинен надати матеріали повного реєстраційного досьє, включаючи всі ВАЗФ, що належать до кожного окремого антигену, які є частиною нової вакцини, якщо загальний файл на кожний окремий антиген не пройшов оцінку раніше.
Зміни до ВАЗФ на вже зареєстровану вакцину підлягають науковій та технічній оцінці. При позитивній оцінці на кожний файл видається сертифікат відповідності, який супроводжується звітом про проведену оцінку.
Загальний файл вакцинного антигену має містити інформацію щодо сертифікації, повторної сертифікації або внесення змін до загального файла на вакцинний антиген, який має відношення до лікарського засобу.
9. Спеціальні вимоги до матеріалів реєстраційного досьє для радіофармацевтичних лікарських засобів та їх прекурсорів
9.1. Радіофармацевтичні лікарські засоби:
При реєстрації радіофармацевтичних лікарських засобів заяви потрібно подавати разом з повними матеріалами реєстраційного досьє, які повинні містити такі специфічні особливості:
9.1.1. У модулі 3:
а) у радіонуклідному наборі, який після постачання виробником має бути помічений радіоактивною міткою, діючою речовиною вважається та частина лікарської форми, яка зв’язується з радіонуклідом або є носієм радіонукліда. Опис методу виробництва радіонуклідних наборів повинен включати подробиці виробництва набору та подробиці його рекомендованої кінцевої обробки з метою створення радіофармацевтичного лікарського засобу. Необхідні специфікації радіонукліда повинні (у відповідних випадках) відповідати загальній монографії або спеціальним монографіям Європейської фармакопеї. Крім того, має надаватись опис усіх сполук для мічення радіоактивним ізотопом. Також необхідно надати опис структури сполуки з радіоактивною міткою.
Необхідно надавати інформацію про радіонукліди, які задіяні в ядерних реакціях.
Як первинні, так і вторинні радіонукліди вважаються активними речовинами генератора;
б) повинні бути надані докладні дані про характер радіонукліда, ідентичність ізотопу, можливі домішки, носій, використання та специфічну активність;
в) до вихідних матеріалів належать матеріали, що є метою опромінення;
г) необхідно розглянути хімічну/радіофармацевтичну чистоту лікарського засобу та її зв’язок з розподілом в організмі людини;
ґ) необхідно надати опис чистоти радіонукліда, радіохімічної чистоти та специфічної активності;
д) для радіонуклідних генераторів необхідно надати докладні дані щодо випробувань первинних і вторинних радіонуклідів. Для генераторів-елюатів необхідно надати методи випробування первинних радіонуклідів та інших компонентів генераторної системи;
е) вимога виражати вміст діючих речовин у перерахунку на масу активних компонентів відноситься тільки до радіонуклідних наборів. Для радіонуклідів радіоактивність необхідно виражати в бекерелях у конкретний день та за потреби у конкретний час з посиланням на часовий пояс. Має бути вказаний тип радіації;
є) для радіонуклідних наборів специфікації готового лікарського засобу повинні включати випробування з визначення дії лікарських засобів після мічення радіоактивним ізотопом. Мають бути включені відповідні засоби контролю радіохімічної та радіонуклідної чистоти сполуки з радіоактивною міткою. Потрібно ідентифікувати та проаналізувати будь-який матеріал, необхідний для мічення радіоактивним ізотопом;
ж) має бути надана інформація про стабільність радіонуклідних генераторів, радіонуклідних наборів і лікарських засобів з радіоактивною міткою. Необхідно документувати стабільність під час використання радіофармацевтичних лікарських засобів у флаконах, що містять декілька доз.
9.1.2. У Модулі 4:
Матеріали реєстраційного досьє щодо оцінки безпеки та ефективності радіофармацевтичних лікарських засобів повинні відповідати вимогам, що висуваються до лікарських засобів та радіаційної дозиметрії. Необхідно документувати рівень впливу опромінення на органи/тканини. Відповідно до міжнародної системи необхідно зробити попередній розрахунок поглиненої дози випромінювання при певному методі застосування.
9.1.3. У Модулі 5:
До матеріалів реєстраційного досьє у відповідних випадках необхідно надати результати клінічних випробувань, в інших — обґрунтувати показання до застосування за допомогою клінічних оглядів.
9.2. Прекурсори радіонуклідів, що використовуються при виробництві мічених радіоактивними ізотопами лікарських засобів:
В особливих випадках, коли розглядається прекурсор радіонукліда, який призначено тільки для мічення радіоактивним ізотопом, до матеріалів реєстраційного досьє необхідно надати інформацію, що стосується можливих наслідків низької ефективності мічення або дисоціації in vivo міченого радіоактивним ізотопом кон’югата, тобто питань, пов’язаних з впливом вільних радіонуклідів на хворого. Крім того, має бути надана відповідна інформація, пов’язана з професійною небезпекою, тобто впливом опромінення на медичний персонал та довкілля.
Зокрема, у відповідних випадках повинна бути надана така інформація:
9.2.1. У Модулі 3:
Як визначено у підпункті 9.1.1 пункту 9.1 цього розділу, у відповідних випадках положення Модуля 3 належать до реєстрації прекурсорів радіонуклідів.
9.2.2. У Модулі 4:
До матеріалів реєстраційного досьє мають бути надані результати досліджень щодо токсичності одноразової та багаторазової дози, проведених відповідно до принципів належної лабораторної практики, якщо інше обґрунтування відсутнє.
У цьому випадку надання інформації про дослідження мутагенності радіонукліда не вважається достатніми.
Повинна бути надана інформація про хімічну токсичність та розподіл відповідного «холодного» нукліда.
9.2.3. У Модулі 5:
До матеріалів реєстраційного досьє необхідно надати інформацію, що демонструє клінічне застосування прекурсору радіонукліда, приєднаного до відповідних молекул, оскільки у випадку, коли розглядається прекурсор радіонукліда, призначений тільки для мічення радіоактивним ізотопом досліджуваного лікарського засобу, клінічні дані, отримані в дослідженнях самого прекурсору, не мають значення.
10. Спеціальні вимоги до матеріалів реєстраційного досьє для гомеопатичних лікарських засобів
Ця глава стосується особливих вимог до оформлення Модулів 3 і 4 матеріалів реєстраційного досьє на гомеопатичні лікарські засоби, які визначені в абзацах сімнадцятому та вісімнадцятому розділу ІІ цього Порядку.
10.1. У Модулі 3:
Спеціальні вимоги до Модуля 3 матеріалів реєстраційного досьє для гомеопатичних лікарських засобів:
а) термінологія
Латинська назва гомеопатичної сировини, описаної у матеріалах реєстраційного досьє, повинна відповідати латинській назві, зазначеній в Європейській фармакопеї, або в разі відсутності — Німецькій гомеопатичній фармакопеї (GHP), Гомеопатичній фармакопеї США (HPUS), Британській гомеопатичній фармакопеї (BHP), Гомеопатичній фармакопеї Швабе). У відповідних випадках необхідно надати традиційно використовувану назву;
б) контроль вихідних матеріалів
Опис та дані щодо вихідних матеріалів, тобто щодо всіх використаних матеріалів, уключаючи сировину та проміжні матеріали, до кінцевого розведення у готовому лікарському засобі повинні бути підкріплені додатковими даними про гомеопатичну сировину.
Загальні вимоги до якості стосуються всіх вихідних матеріалів і сировини, а також проміжних етапів виробничого процесу до кінцевого розведення у готовому лікарському засобі. Якщо можна, потрібно провести дослідження у разі присутності токсичних компонентів, а також тоді, коли якість неможливо контролювати при кінцевому розведенні через високий ступінь розведення. Необхідно надати повний опис кожної стадії виробничого процесу, починаючи від вихідних матеріалів до кінцевого розведення у готовому лікарському засобі.
Якщо застосовуються декілька розведень, ці стадії розведення необхідно проводити відповідно до методів виробництва гомеопатичних засобів, які викладено у відповідній монографії Європейської фармакопеї, або, за її відсутності, в іншій фармакопеї;
в) методи контролю готового лікарського засобу
До готових гомеопатичних лікарських засобів застосовуються загальні вимоги до якості. Будь-які виключення вимагають ретельного обґрунтування заявником.
Необхідно ідентифікувати та проаналізувати всі компоненти, які мають потенційну токсичність. Якщо неможливо ідентифікувати та проаналізувати всі компоненти, які можуть виявитися токсичними, наприклад через їх високий ступінь розведення в готовому лікарському засобі, потрібно обґрунтувати якість лікарського засобу за допомогою повної валідації виробничого процесу та процесу розведення;
г) випробування на стабільність
Необхідно підтвердити стабільність готового лікарського засобу. Дані щодо стабільності гомеопатичного лікарського засобу, як правило, дійсні як для розчинів, так і для порошків, з яких був вироблений даний лікарський засіб. Якщо ідентифікація або аналіз АФІ або діючої речовини неможливі через високий ступінь розведення, можуть розглядатися дані щодо стабільності лікарського засобу.
10.2. У Модулі 4:
Спеціальні вимоги до Модуля 4 матеріалів реєстраційного досьє для гомеопатичних лікарських засобів:
Необхідно обґрунтувати відсутність будь-якої інформації, наприклад, має надаватися обґрунтування, яким чином може доводитися прийнятний рівень безпеки, незважаючи на те, що відсутні деякі дослідження.
11. Спеціальні вимоги до матеріалів реєстраційного досьє для рослинних лікарських засобів
До заяви на реєстрацію/перереєстрацію рослинних лікарських засобів додаються матеріали повного реєстраційного досьє, до якого повинні бути включені такі специфічні дані.
Спеціальні вимоги до Модуля 3 матеріалів реєстраційного досьє для рослинних лікарських засобів:
У Модулі 3:
Положення Модуля 3, включаючи відповідність монографії Європейської фармакопеї, стосуються реєстрації/перереєстрації готових рослинних лікарських засобів. При цьому береться до уваги рівень наукових знань на момент подання заяви.
Стосовно рослинних лікарських засобів розглядаються такі специфічні аспекти:
а) рослинні субстанції та рослинні препарати
Щодо номенклатури рослинної субстанції необхідно вказати ботанічні характеристики рослини (рід, вид, різновид та автора), хемотип (у відповідних випадках) частин рослини, інші назви (синоніми, зазначені в інших фармакопеях) та лабораторний код.
Щодо номенклатури рослинного препарату необхідно вказати ботанічні характеристики рослини (рід, вид, різновид та автора), хемотип (у відповідних випадках) частин рослини, визначення препарату, співвідношення рослинної субстанції до рослинного препарату, розчинник(и), інші назви (синоніми, зазначені в інших фармакопеях) та лабораторний код.
При підготовці документів щодо структури рослинної субстанції та рослинного препарату (у відповідних випадках) необхідно надати інформацію про фізичну форму, опис компонентів з відомою терапевтичною дією або маркерів (молекулярну формулу, відносну молекулярну масу, структурну формулу, уключаючи відносну та абсолютну стереохімію), а також інформацію про інші компоненти.
При підготовці документів щодо виробництва рослинної субстанції у відповідних випадках необхідно надати інформацію про кожного постачальника сировини, включаючи підрядників, а також інформацію про кожну передбачену виробничу дільницю або устаткування, задіяне при виробництві/заготовці та контролі рослинної субстанції.
При підготовці документів щодо виробництва рослинного препарату у відповідних випадках необхідно вказати інформацію про кожного постачальника сировини, уключаючи підрядників, а також інформацію про кожну передбачену виробничу дільницю або устаткування, задіяне при виробництві та контролі рослинного засобу.
Щодо опису виробничого процесу рослинної субстанції необхідно надати інформацію, у якій буде відповідним чином описано виробництво субстанції та заготівлю рослинної сировини, уключаючи географічне місце вирощування та культивування лікарської рослини, умови її збору, висушування та зберігання.
Щодо опису виробничого процесу та процедури контролю рослинного препарату необхідно надати інформацію, у якій буде відповідним чином описане виробництво засобу, включаючи опис обробки сировини, розчинників і реагентів, стадії очищення та стандартизації.
Щодо розробки виробничого процесу необхідно надати (у відповідних випадках) короткий опис розробки рослинної субстанції та рослинного препарату з урахуванням передбачуваного шляху застосування та показань. У відповідних випадках має надаватися обговорення результатів порівняння фітохімічної будови рослинної субстанції та рослинного препарату, що використовувались на підтримку бібліографічних даних, та рослинної субстанції і рослинного препарату, що є діючою речовиною готового рослинного лікарського засобу, на який подано заяву про державну перереєстрацію.
При описі структури та інших характеристик рослинної субстанції за необхідності має надаватися інформація про ботанічні, макро- та мікроскопічні, фітохімічні характеристики і біологічну активність.
При описі структури та інших характеристик рослинного препарату у разі необхідності має надаватися інформація про фітохімічні і фізико-хімічні характеристики та біологічну активність.
За необхідності мають надаватися специфікації для рослинної субстанції та рослинного препарату.
У відповідних випадках потрібно надати опис аналітичних методів, що використовувалися при випробуванні рослинної субстанції та рослинного препарату.
При описі валідації аналітичних методик у відповідних випадках потрібно надати інформацію про валідацію методик, включаючи експериментальні дані для методів аналізу, які використовуються при випробуваннях рослинної субстанції та рослинного препарату.
При описі аналізу серій у відповідних випадках потрібно надати опис та результати аналізу серій рослинних субстанцій та рослинних препаратів у такому вигляді, як монографії фармакопей.
У відповідних випадках потрібно надати обґрунтування специфікацій рослинних субстанцій і рослинних препаратів.
У відповідних випадках потрібно надати інформацію про стандартні зразки або стандартні препарати, що використовуються при випробуваннях рослинної субстанції та рослинного препарату;
б) готові рослинні лікарські засоби
Короткий опис розробки складу лікарського засобу повинен уключати інформацію про розробку готового рослинного лікарського засобу з увагою на передбачуваний спосіб застосування та показання. У відповідних випадках потрібно надати відомості про результати порівняння фітохімічної будови лікарського засобу, який використовується на підтримку бібліографічних даних, та лікарського засобу, на який подано заяву.
12. Спеціальні вимоги до матеріалів реєстраційного досьє для лікарських засобів прогресивної терапії (високотехнологічні препарати)
12.1. При поданні заяви на реєстрацію лікарських засобів прогресивної терапії (високотехнологічні препарати) необхідно дотримуватися вимог до матеріалів реєстраційного досьє (Модулі 1-5), як зазначено у розділі ІХ цього Порядку.
Для біологічних лікарських засобів необхідно застосовувати спеціальні вимоги до матеріалів реєстраційного досьє для Модулів 3, 4 та 5, як зазначено у главі 8 цього розділу. Додатково спеціальні вимоги до матеріалів реєстраційного досьє на лікарські засоби прогресивної терапії (високотехнологічні препарати) описані у пунктах 12.2, 12.3 та 12.4 цієї глави цього розділу. За необхідності, враховуючи особливості лікарських засобів прогресивної терапії (високотехнологічні препарати), до матеріалів реєстраційного досьє надається додаткова інформація.
Через специфічну природу лікарських засобів прогресивної терапії (високотехнологічних препаратів) можуть бути надані матеріали, що складені відповідно до наукових керівництв з ефективності, безпеки та якості високотехнологічних препаратів на основі оцінки ризику для визначення ступеня якості, доклінічні та клінічні дані, які будуть включені до матеріалів реєстраційного досьє.
Матеріали реєстраційного досьє повинні включати аналіз ризиків, що може охоплювати всю розробку. Інформація про фактори ризику, які можуть розглядатися, повинна відображати: походження клітин (аутологічні, галогенні, ксеногенні), здатність до розростання та/або видозмінювання та ініціювання імунної відповіді; маніпуляції на клітинному рівні; комбінація клітин з біоактивними молекулами або структурними матеріалами; природу лікарського засобу генної терапії; ступінь здатності до реплікації вірусів або мікроорганізмів, що використовуються in vivo; рівень інтеграції нуклеїнових кислот або генів у геном; тривала функціональність; ризик онкогенності та спосіб введення або застосування.
Матеріали реєстраційного досьє щодо аналізу ризиків можуть містити інформацію про відповідні доклінічні та клінічні дані або досвід застосування інших високотехнологічних препаратів.
Будь-яке відхилення від вимог цього пункту має бути науково обґрунтовано у Модулі 2. Матеріали реєстраційного досьє Модуля 2 також можуть включати інформацію щодо аналізу ризиків, що описаний вище, у разі застосування. У такому випадку має бути оцінено використовувану методику, природу певних ризиків та результати підходу на підставі оцінки ризику для програми з розробки та оцінки, а також має бути наданий опис будь-яких відхилень від вимог цього пункту внаслідок аналізу ризиків.
12.2. Спеціальні вимоги до Модуля 3:
12.2.1. Спеціальні вимоги для усіх лікарських засобів прогресивної терапії (високотехнологічні препарати):
У матеріалах реєстраційного досьє необхідно надати опис системи простежуваності, яку заявник буде встановлювати та підтримувати для гарантії того, що окремий продукт та його вихідні матеріали і сировина, включаючи усі речовини, що вступають у контакт з клітинами і тканинами, що можуть міститися у препараті, можуть бути простежені за допомогою встановлення походження, виробництва, упаковки, зберігання, транспортування та доставки у заклад охорони здоров’я/установу.
Система простежуваності щодо людських клітин та тканин, а також клітин крові має відповідати вимогам, встановленим у відповідних керівництвах.
12.2.2. Спеціальні вимоги для лікарських засобів генної терапії:
Лікарський засіб генної терапії — біологічний лікарський засіб, якому властиві такі характеристики:
діюча речовина такого лікарського засобу містить або складається з рекомбінантної нуклеїнової кислоти, що використовується або вводиться людині з метою впорядкування, відновлення, заміни, додавання або вилучення генетичної послідовності;
його терапевтична, профілактична або діагностична дія пов’язана безпосередньо з послідовністю рекомбінантної нуклеїнової кислоти, яку він містить, або з продуктом генетичної експресії цієї послідовності.
До лікарських засобів генної терапії не належать вакцини для профілактики інфекційних захворювань.
Лікарські засоби генної терапії включають:
чисту нуклеїнову кислоту;
складну нуклеїнову кислоту або невірусні вектори;
вірусні вектори;
генетично модифіковані клітини.
У матеріалах реєстраційного досьє для лікарських засобів генної терапії повинно бути відображено інформацію про три основні елементи виробничого процесу:
вихідні матеріали — матеріали, з яких виробляється діюча речовина, такі як, наприклад, ген, що становить інтерес, плазміди експресії, банки клітин та запаси вірусів або невірусний вектор;
АФІ або діюча речовина — рекомбінантний вектор, вірус, чисті або складні плазміди, клітини, що продукують вірус, генетично модифіковані in vitro клітини;
готовий лікарський засіб — діюча речовина в її кінцевій безпосередній упаковці для призначеного медичного застосування. Залежно від типу лікарського засобу прогресивної терапії (високотехнологічного препарату), способу та умов його застосування може бути потрібна обробка клітин пацієнта ex vivo.
До матеріалів реєстраційного досьє у Модулі 3 необхідно надати:
а) інформацію про відповідні характеристики лікарського засобу генної терапії, включаючи його експресію в популяції цільових клітин, а також інформацію щодо джерела, структури, характеристик та визначення справжності кодуючої послідовності гена, включаючи її цілісність і стабільність. Крім терапевтичного гена необхідно надати інформацію про повну послідовність інших генів, регуляторні елементи та скелет вектора;
б) інформацію щодо характеристик вектора, який використовувався для переносу та доставки гена. Вона повинна включати фізико-хімічні та/або біологічно-імунологічні характеристики.
Для лікарських засобів, в яких використано мікроорганізми (наприклад бактерії або віруси) для переносу гена (біологічний перенос гена), необхідно надати інформацію про патогенність вихідного штаму та про його тропність до певних тканин і видів клітин, а також про залежність взаємодії від клітинного циклу.
Для лікарських засобів, в яких використано небіологічні засоби переносу гена, необхідно надати інформацію про фізико-хімічні властивості окремих компонентів та всієї комбінації;
в) опис принципів створення або підтримки банку клітин та їх характеристик, які належать до лікарських засобів, що переносять ген;
г) інформацію про джерело клітин, що містять рекомбінантний вектор.
Якщо джерелом клітин є людина, то необхідно надати інформацію про її вік, стать, результати мікробіологічного та вірусного досліджень, критерії виключення та країну проживання.
Якщо джерелом клітин є тварина, то необхідно надавати докладну інформацію щодо:
джерела тварин;
утримання тварин та догляду за ними;
трансгенних тварин (методи створення, характеристика трансгенних клітин, характер введеного гена);
заходів щодо запобігання та контролю інфекцій у тваринних донорів;
тестування на наявність збудників інфекцій;
приміщень;
контролю вихідних матеріалів і сировини.
Необхідно надати опис методу збору клітин, включаючи місце, тип тканини, робочий процес, транспортування, зберігання та системи відстеження, а також процедури контролю під час збору клітин;
ґ) оцінку вірусної безпеки, а також системи відстеження лікарських засобів від донора до готового лікарського засобу, що є важливою частиною матеріалів реєстраційного досьє, що надається. Наприклад, має бути виключена присутність вірусу, що здатний до реплікації, у джерелах нереплікаційних компетентних вірусних векторів.
12.2.3. Спеціальні вимоги до лікарських засобів соматоклітинної терапії:
Лікарський засіб соматоклітинної терапії — біологічний лікарський засіб, якому властиві такі характеристики:
містить чи складається з клітин або тканин, біологічні характеристики, фізіологічні функції або структурні властивості яких є важливими для клінічного застосування, були змінені у результаті значних маніпуляцій або з клітин чи тканин, які не призначені для використання з тією самою метою у донора або реципієнта;
має властивості для, або використовується у, або вводиться людині з метою лікування, профілактики або діагностики захворювання шляхом фармакологічної, імунологічної або метаболічної дії його клітин або тканин.
Лікарські засоби соматоклітинної терапії включають:
клітини, які за допомогою маніпуляцій змінили якісні та кількісні параметри своїх імунологічних, метаболічних або інших функціональних властивостей;
клітини сортовані, відібрані, з якими проведено відповідні маніпуляції та які надалі будуть задіяні у виробничому процесі при отриманні готового лікарського засобу;
маніпульовані клітини та поєднані з неклітинними компонентами (наприклад, біологічними або неактивними матрицями або медичними засобами), які чинять основну призначену дію в готовому лікарському засобі;
похідні аутологічних клітин, експресованих in vitro за специфічних культуральних умов;
генетично модифіковані клітини або клітини, з якими проводилися інші маніпуляції, щоб експресувати раніше неекспресовані гомологічні або негомологічні функціональні властивості.
Весь виробничий процес, від збору клітин у пацієнта (аутологічна ситуація) до введення йому лікарського засобу, вважається одним втручанням.
У матеріалах реєстраційного досьє для лікарських засобів соматоклітинної терапії повинно бути відображено інформацію про три елементи виробничого процесу:
вихідні матеріали — матеріали, з яких виробляється діюча речовина, тобто органи, тканини, рідини організму або клітини;
АФІ або діюча речовина — маніпульовані клітини, клітинні лізати, проліферувальні клітини та клітини, що використовуються у сполученні з неактивними матрицями та медичними засобами;
готовий лікарський засіб — діюча речовина, підготовлена у її кінцевій безпосередній упаковці для призначеного медичного застосування.
У матеріалах реєстраційного досьє у Модулі 3 має бути представлена:
а) загальна інформація про діючу речовину
Діючі речовини лікарських засобів клітинної терапії складаються з клітин, які внаслідок обробки in vitro проявляють профілактичні, діагностичні або терапевтичні властивості, відмінні від їх первинних фізіологічних і біологічних властивостей.
Необхідно надати опис типу клітин та використаної культури. Має бути надана документація на тканини, органи або біологічні рідини, з яких отримано клітини, а також аутологічний, алогенний або ксеногенний характер донорських зразків та їх географічне походження. Необхідно надати детальний опис заготівлі клітин, зразків та умов їх зберігання до подальшої обробки. Для алогенних клітин особливу увагу необхідно приділити найпершій стадії процесу — відбору донорів. Має бути надана інформація про тип проведених маніпуляцій і фізіологічну функцію клітин, які використовуються як діюча речовина;
б) інформація щодо вихідних матеріалів для діючої речовини.
12.2.3.1. Соматичні клітини людини:
Лікарські засоби соматоклітинної терапії з використанням клітин людини виробляються з певної кількості (пулу) життєздатних клітин, які отримують у результаті виробничого процесу, що починається на рівні органів або тканин, отриманих від людини, або на рівні чітко визначеної системи банку клітин, де клітинний пул належить до безперервних клітинних ліній. У рамках цього розділу діючою речовиною вважається посіяний пул клітин людини, а готовим лікарським засобом вважається пул клітин людини, виготовлених для призначеного медичного застосування. У матеріалах реєстраційного досьє необхідно надати повну інформацію про вихідні матеріали та кожну стадію виробничого процесу, включаючи питання вірусної безпеки.
У матеріалах реєстраційного досьє необхідно надати інформацію про такі характеристики донора, як вік, стать, мікробіологічний статус, критерії виключення та країну проживання; опис відбору зразків, включаючи місце, тип, робочий процес створення пулу, перевезення; зберігання та моніторинг; методи контролю відбору зразків.
Окремі вимоги, викладені для соматичних клітин людини, стосуються інформації щодо підготовки та контролю якості систем створення банку клітин. Ці особливості належать до алогенних і ксеногенних клітин.
У матеріалах реєстраційного досьє необхідно надати інформацію про використання будь-якої сировини (наприклад, цитокинів, факторів росту або культурального середовища) або можливих допоміжних речовин і медичних засобів (наприклад, засоби для сортування клітин, біосумісні полімери, матриці, волокна, гранули) з точки зору їх біосумісності, функціональності, а також ризику передачі збудників інфекції.
12.2.3.2. Тваринні соматичні клітини (ксеногенні):
У матеріалах реєстраційного досьє необхідно надавати докладну інформацію щодо таких питань:
джерела тварин;
тримання та догляд за тваринами;
генетично модифіковані тварини (методи створення, характеристики трансгенних клітин, характер введеного або вилученого нокаутного гена);
заходи щодо запобігання та контролю інфекцій у тваринних донорів;
тестування на наявність збудників інфекції, включаючи мікроорганізми, що передаються вертикально (також ендогенні ретровіруси);
устаткування;
системи створення банку клітин;
контроль вихідних матеріалів і сировини.
У матеріалах реєстраційного досьє у Модулі 3 має бути представлена:
а) інформація про процес виробництва діючої речовини і готового лікарського засобу — необхідно надати інформацію про різні стадії виробничого процесу такі, як, наприклад, відділення органа/тканини, вибір популяції клітин, що становить інтерес, культивування клітин in vitro, трансформація клітин за допомогою фізико-хімічних агентів або шляхом переносу гена;
б) характеристика діючої речовини — необхідно надати всю інформацію, яка стосується цього пункту, про характеристики популяції клітин, що становлять інтерес, включаючи її ідентичність (оригінальні види, бендінгова цитогенетика (banding cytogenetics), морфологічний аналіз), чистоту (побічні мікробні агенти і клітинні забруднювачі), силу дії (певна біологічна активність) та придатність (аналіз каріотипу та онкогенності) для призначеного медичного застосування;
в) фармацевтична розробка готового лікарського засобу — крім інформації про специфічний спосіб введення (внутрішньовенна інфузія, локальна ін’єкція, хірургічна трансплантація) має бути також надана інформація про можливе використання допоміжних медичних засобів (біосумісних полімерів, матриць, волокон, гранул) з точки зору їх біосумісності та зносостійкості;
г) система простежування — необхідно надати докладну схему процесу, який забезпечує систему простежування препаратів від донора до готового лікарського засобу.
12.2.4. Спеціальні вимоги до лікарських засобів прогресивної терапії (високотехнологічні препарати), що містять пристрої для введення:
У матеріалах реєстраційного досьє необхідно надати опис фізичних характеристик та дії продукту, а також опис методів проектування продукту.
Необхідно описати взаємодію та сполучуваність генів, клітин та/або тканин і структурних компонентів пристрою.
Пристрій для введення або активний медичний виріб, що імплантується, можуть бути невід’ємною частиною діючої речовини. Якщо пристрій для введення або активний медичний виріб, що імплантується, комбінується з клітинами під час виробництва, застосування або введення готових продуктів, вони повинні розглядатися як невід’ємна частина готового лікарського засобу.
У матеріалах реєстраційного досьє необхідно надати інформацію, що стосується пристрою для введення або активного медичного виробу, що імплантується (що є невід’ємною частиною діючої речовини або готового лікарського засобу), та є необхідною для оцінки комбінованого високотехнологічного препарату. Така інформація має включати:
а) інформацію про вибір та заплановану функцію пристрою для введення та демонстрацію сумісності пристрою для введення з іншими складовими продукту;
б) докази відповідності частини пристрою для введення загальним вимогам, встановленим Директивою 93/42/ЄЄС «Медичні прилади. Медичне обладнання. Медичні пристрої» або іншим чинним нормативним актом, відповідності частини активного медичного виробу, що імплантується, загальним вимогам, встановленим Директивою 90/385/ЄЄС «Активні медичні пристрої, що імплантують»;
в) за необхідності доказ відповідності пристрою для введення або активного медичного виробу, що імплантується, посилаючись на Керівництво з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами (діюче видання, опубліковане Комісією в Офіційному бюлетені Європейського союзу);
г) за наявності/на вимогу Центру результати будь-якої оцінки компетентним органом, що проводив таку оцінку, частини пристрою для введення або частини активного медичного виробу, що імплантується, проведеної компетентним органом на відповідність вимогам Директиви 93/42/ЄЄС «Медичні прилади. Медичне обладнання. Медичні пристрої» або Директиви 90/385/ЄЄС «Активні медичні пристрої, що імплантують».
12.3. Спеціальні вимоги до Модуля 4:
Загальноприйняті вимоги до матеріалів реєстраційного досьє, викладених у Модулі 4 щодо доклінічного дослідження лікарських засобів, не завжди можуть застосовуватись щодо матеріалів реєстраційного досьє для лікарських засобів генної та соматоклітинної терапії у зв’язку з винятковими та різноманітними структурними та біологічними властивостями таких лікарських засобів, включаючи високий ступінь специфічності видів, специфічність суб’єкта, імунологічні бар’єри та відмінності у плейотропних реакціях.
У Модулі 2 необхідно надати відповідне обґрунтування доклінічної розробки та критеріїв, що використані для вибору відповідних видів і моделей.
За потреби необхідно ідентифікувати або розробити нові моделі на тваринах, щоб мати змогу екстраполювати на людину дані щодо специфічної активності та токсичності. У матеріалах реєстраційного досьє необхідно надати наукове обґрунтування використання цих моделей захворювання на тваринах для забезпечення безпеки та підтвердження ефективності лікарського засобу.
12.4. Спеціальні вимоги до Модуля 5:
У матеріалах реєстраційного досьє необхідно продемонструвати ефективність високотехнологічних лікарських засобів, як це описано у Модулі 5. Однак, може виявитися, що було неможливим проведення клінічних випробувань згідно із загальноприйнятими вимогами щодо певних лікарських засобів та певних терапевтичних показань. У матеріалах реєстраційного досьє Модуля 2 необхідно надати обґрунтування будь-якого відхилення від існуючих вимог.
У матеріалах реєстраційного досьє Модуля 5 повинно бути відображено ті особливості, що може мати клінічна розробка лікарських засобів прогресивної терапії через складний та лабільний характер діючих речовин. Це питання потребує додаткового розгляду через особливості життєздатності, проліферації, міграції та диференціації клітин (соматоклітинна терапія), через особливі клінічні обставини, за яких використовуються лікарські засоби, або через особливий механізм дії, що здійснюється через експресію гена (генна терапія).
У заяві про реєстрацію високотехнологічних лікарських засобів необхідно зазначити особливий ризик, що пов’язаний з такими лікарськими засобами та виникає через потенційне забруднення інфекційними збудниками. Особливий акцент необхідно зробити на ранніх стадіях розробки, включаючи вибір донорів, з одного боку, і терапевтичне втручання в цілому, уключаючи відповідне поводження з лікарським засобом, та його застосування, з іншого боку.
Більше того, у відповідних випадках матеріали реєстраційного досьє Модуля 5 повинні містити інформацію про заходи щодо перевірки та контролю функцій і розвитку живих клітин у реципієнта для запобігання передачі збудників інфекції реципієнту та мінімізації потенційного ризику для здоров’я інших людей, популяції.
12.4.1. Фармакологічні дослідження та дослідження ефективності у людини:
Матеріали реєстраційного досьє щодо фармакологічних досліджень у людини повинні надавати інформацію про очікуваний механізм дії, очікувану ефективність, виходячи з виправданих кінцевих цілей, розподілу в організмі, відповідного режиму дозування та способів введення або модальності застосування, необхідної для дослідження бажаної ефективності.
Проведення фармакокінетичних досліджень згідно із загальноприйнятими вимогами може виявитись неможливим для деяких високотехнологічних лікарських засобів. Іноді неможливо провести клінічні дослідження на здорових добровольцях, а визначення дози та кінетики лікарського засобу ускладнено. Однак у матеріалах реєстраційного досьє необхідно надати інформацію про вивчення розподілу та поведінки лікарського засобу in vivo, включаючи проліферацію клітин та їх довгострокову функцію, а також про ступінь розподілу генно-інженерних лікарських засобів та тривалості бажаної експресії гена, про використання та, за необхідності, розробку відповідних тестів для моніторингу клітинного препарату або клітини, що експресує необхідний ген в організмі людини, та для контролю функції клітин, які було введено або трансфектовано.
У матеріалах реєстраційного досьє має бути надана оцінка ефективності безпеки високотехнологічного лікарського засобу, що повинна включати докладний опис та оцінку терапевтичної процедури в цілому, включаючи особливі способи введення (такі як трансфекція клітин ex vivo, маніпуляції in vitro або використання методів інтервенції) і випробування можливих додаткових режимів лікування (включаючи імуносупресивне, антивірусне та цитотоксичне).
Усю процедуру необхідно апробувати при проведенні клінічних випробувань та викласти в інформації про лікарський засіб.
12.4.2. Надання даних щодо безпеки:
У матеріалах реєстраційного досьє необхідно відобразити питання безпеки, що виникають у зв’язку з імунною реакцією на лікарські засоби або на експресовані білки і пов’язані з імунним відторгненням, імунносупресією або відмовою імуноізоляційних структур.
Певні лікарські засоби генної та соматоклітинної терапії (наприклад, ксеногенні лікарські засоби та певні лікарські засоби генного переносу) можуть містити реплікаційно-компетентні частки та/або збудників інфекцій. Можливо на до- та післяреєстраційних стадіях необхідно буде контролювати стан пацієнта з точки зору розвитку можливих інфекцій та/або патологічних ускладнень; можливо, такий контроль необхідно розповсюдити на людей, з якими контактує пацієнт, включаючи медичний персонал.
При використанні деяких лікарських засобів соматоклітинної терапії та деяких лікарських засобів генного переносу неможливо повністю виключити ризик забруднення збудниками, передача яких потенційно можлива. Однак цей ризик можна мінімізувати за допомогою відповідних заходів, описаних у матеріалах реєстраційного досьє Модуля 3.
Заходи, що є частиною виробничого процесу, повинні доповнюватися супровідними методами аналізу, процедурами контролю якості та відповідними методами контролю, опис яких необхідно включити до Модуля 5.
Застосування деяких лікарських засобів соматоклітинної терапії може обмежуватися тимчасово або постійно закладами охорони здоров’я/установами, у яких є фахівці та обладнання для забезпечення специфічного нагляду за безпекою пацієнтів. Подібний підхід може відповідати певним лікарським засобам генної терапії, застосування яких пов’язане з потенційним ризиком наявності збудників інфекції, здатних до реплікації.
У відповідних випадках у матеріалах реєстраційного досьє необхідно передбачити та висвітлити питання тривалого нагляду за розвитком пізніх ускладнень.
У відповідних випадках заявнику необхідно надати детальний план процесу управління ризиками, що включає інформацію про клінічні та лабораторні дані пацієнта, епідеміологічні дані (які виявляються) та, якщо це має відношення, архівні дані про зразки тканин від донора та реципієнта. Така систематизована інформація необхідна для забезпечення моніторингу лікарського засобу та швидкої реакції при виявленні підозрілих ознак небажаних явищ.
ХІ. Вимоги до матеріалів реєстраційного досьє для афі або діючої речовини
1. Вимоги до заяви:
У заяві має бути вказана назва АФІ або діючої речовини, технологічна форма, включаючи упаковку.
Зазначаються найменування та місцезнаходження заявника разом з найменуванням та місцезнаходженням виробників і виробничих дільниць, задіяних на різних стадіях виробництва, а також, за потреби, дані щодо імпортера.
Сертифікати якості на три промислові серії АФІ або діючої речовини або сертифікат якості на одну промислову серію у супроводі зобов’язання надати сертифікати якості на дві інші серії, як тільки вони стануть доступними (усі сертифікати якості повинні подаватися за кожним заявленим місцем виробництва).
2. Маркування упаковки:
Заявник надає пропонований текст маркування упаковки (первинної, вторинної (за наявності)).
3. Оцінка небезпеки для довкілля:
У відповідних випадках до заяви про державну реєстрацію АФІ або діючої речовини додають резюме екологічної оцінки можливого ризику для довкілля через її використання та/або утилізацію у складі лікарського засобу, а також наводять пропозиції щодо відповідної інформації, яка має бути наведена у маркуванні. Необхідно розглянути ризик для довкілля, пов’язаний з вивільненням АФІ або діючих речовин, що містять ГМО, відповідно до вимог Закону України «Про державну систему біобезпеки при створенні, випробуванні, транспортуванні та використанні генетично модифікованих організмів».
Інформація повинна включати:
вступ;
копію письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою;
дані, включаючи методи виявлення та ідентифікації, а також унікальний код ГМО і будь-яку додаткову інформацію про ГМО або речовину, яка має значення при оцінці ризику для довкілля;
звіт про оцінку ризику для довкілля (ОРД), підготовлений на підставі наявної інформації;
відповідні заходи для інформування населення.
Наведена інформація має бути засвідчена підписом автора із зазначенням дати, даних щодо освіти, стажування та професійного досвіду. Необхідно вказати професійні стосунки між автором і заявником.
4. Основні дані:
4.1. Усі методики та методи, що використовуються при виробництві та контролі АФІ або діючої речовини, слід викладати чітко і детально, щоб можна було відтворити їх при проведенні контрольних випробувань, на вимогу уповноваженого органу. Усі методи випробувань повинні відповідати сучасному науковому рівню, а також бути валідованими. Слід надавати результати досліджень з валідації. Якщо використовуються методи випробувань, включені до ДФУ або Європейської фармакопеї, то цей виклад необхідно замінити відповідним посиланням на монографію(ї) та загальний(і) розділ(и).
4.2. Для всіх речовин, вказаних у монографіях ДФУ або Європейської фармакопеї, необхідно робити посилання на фармакопеї. Стосовно речовин, які не вказано в цих фармакопеях, мають надаватися посилання на інші фармакопеї, стандарти яких є не нижчими за стандарти ДФУ або Європейської фармакопеї.
Якщо методи аналізу включено до ДФУ або Європейської фармакопеї, то немає необхідності наводити їх повний виклад, достатньо в кожному розділі, в якому планувався виклад цього методу, робити відповідне посилання на монографію та загальний розділ.
4.3. Якщо вихідні матеріали та сировина, діючі або допоміжні речовини не описано ані в ДФУ, ані в Європейській фармакопеї, ані в інших фармакопеях, стандарти яких є не нижчими за стандарти ДФУ або Європейської фармакопеї, може бути прийнятним посилання на монографію фармакопеї іншої країни. У таких випадках заявник повинен надати результати досліджень з валідації аналітичних методик, описаних у монографії, а також, за необхідності, переклад.
4.4. Якщо АФІ або діюча речовина і вихідний матеріал описано в монографії Європейської фармакопеї, заявник може надати сертифікат відповідності Європейській фармакопеї, виданий Європейським директоратом з питань якості лікарських засобів. Виробник речовини повинен підтвердити, що виробничий процес не змінювався з часу видачі цього сертифіката відповідності.
4.5. Необхідно описати особливі запобіжні заходи щодо передачі губчатої енцефалопатії тварин (сировина, отримана від жуйних тварин): на кожній стадії виробничого процесу заявник повинен продемонструвати відповідність використаних матеріалів, посилаючись на відповідні керівництва з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами. Підтвердити це можна, або надавши сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії, виданий Європейським директоратом з питань якості лікарських засобів, або шляхом надання наукових даних для обґрунтування цієї відповідності.
4.6. Слід надати інформацію про оцінку ризику потенційного зараження сторонніми агентами (незалежно від того, мають вони вірусне походження чи ні) згідно з вимогами, викладеними в спеціальних керівництвах, а також у загальних монографіях та загальних розділах ДФУ або Європейської фармакопеї.
4.7. Особливу увагу приділяють викладенню таких розділів матеріалів реєстраційного досьє:
4.7.1. Загальна інформація:
Має бути надана інформація про назву АФІ або діючої речовини, включаючи рекомендовану МНН, за наявності — фармакопейну назву, зазначену в ДФУ, Європейській фармакопеї, та хімічну назву.
Має бути надана структурна формула, включаючи відносну та абсолютну стереохімію, молекулярна формула та відносна молекулярна маса. Для високотехнологічних (біотехнологічних) діючих речовин за потреби необхідно надати схематичну послідовність амінокислот і відносну молекулярну масу.
4.7.2. Виробництво:
а) заявник має надати опис виробничого процесу АФІ або діючої речовини;
б) необхідно перелічити усі матеріали, необхідні для виробництва, із зазначенням стадії виробництва, на якій використовується кожний матеріал. Необхідно надати інформацію про якість і контроль цих матеріалів, а також інформацію, яка доводить, що всі матеріали відповідають вимогам щодо їхнього запланованого використання.
Необхідно перелічити вихідні матеріали (сировину), а також надати документи щодо їх якості та методів контролю (за відсутності сертифіката відповідності Європейській фармакопеї, виданого Європейським директоратом з питань якості лікарських засобів);
в) необхідно представити відповідні методи контролю та критерії прийнятності на кожній критичній стадії виробництва, інформацію про якість і контроль проміжних продуктів, а також про процес валідації та/або його аналіз (за відсутності сертифіката відповідності Європейській фармакопеї, виданого Європейським директоратом з питань якості лікарських засобів);
г) якщо присутності потенційно патогенних сторонніх агентів уникнути неможливо, то вихідна речовина може використовуватися тільки в тих випадках, коли подальша обробка забезпечує їхнє видалення та/або інактивацію, що має доводитися в розділі, який стосується оцінки вірусної безпеки.
4.7.3. Опис характеристик:
Необхідно надати дані щодо структури та інших характеристик АФІ або діючої речовини.
Слід надати підтвердження структури АФІ або діючої речовини, ґрунтуючись на результатах фізико-хімічних, та/або імунохімічних, та/або біологічних методів, а також надати інформацію про домішки.
4.7.4. Контроль:
Необхідно надати докладну інформацію про специфікації, які використовуються для посерійного контролю діючої речовини, обґрунтування вибору цих специфікацій, методів аналізу та їх валідації (за відсутності сертифіката відповідності Європейській фармакопеї, виданого Європейським директоратом з питань якості лікарських засобів).
4.7.5. Система упаковка/укупорка:
Необхідно надати опис контейнера та системи упаковка/укупорка і специфікації її компонентів. За можливості необхідно використовувати пакувальні засоби, які відповідають вимогам ДФУ або Європейської фармакопеї.
4.7.6. Стабільність:
За необхідності слід надати короткі відомості про тип проведених досліджень, використані протоколи і отримані під час досліджень результати.
За необхідності слід надати оформлені у відповідному форматі докладні результати дослідження стабільності, включаючи відомості про аналітичні методики, які використовуються для одержання даних, і валідацію цих методик.
ХІІ. Вимоги до гомеопатичних лікарських засобів, матеріали реєстраційного досьє яких можуть не містити наукових клінічних даних, та обсяг даних, необхідних для підтвердження їхньої якості
1. Гомеопатичні лікарські засоби, матеріали реєстраційного досьє яких можуть не містити доказів їх терапевтичної ефективності, повинні відповідати таким умовам:
лікарські засоби призначені для перорального або зовнішнього застосування;
на етикетці лікарського засобу або в будь-якій інформації, що його стосується, не повинні наводитися конкретні показання для застосування;
ступінь розведення лікарського засобу є достатнім для того, аби гарантувати його безпеку; зокрема лікарський засіб не може містити більше однієї частини на 10000 маткового розчину або 1/100 від мінімальної дози, яка використовується в алопатії щодо субстанцій, наявність яких в алопатичному лікарському засобі потребує обов’язкового пред’явлення рецепта лікаря.
2. Заява може охоплювати одну серію лікарського засобу, одержаного з однієї й тієї самої гомеопатичної сировини або видів сировини. Для того, аби продемонструвати, зокрема, фармацевтичну якість лікарських засобів та їх однорідність від серії до серії, матеріали реєстраційного досьє мають включати:
наведену у фармакопеї (ДФУ, або Європейська фармакопея, або в разі відсутності — Німецька гомеопатична фармакопея (GHP), Гомеопатична фармакопея США (HPUS), Британська гомеопатична фармакопея (BHP), Гомеопатична фармакопея Швабе) наукову назву гомеопатичної сировини або видів сировини та дані про різні шляхи введення, лікарські форми та ступені розведення, які підлягають реєстрації;
досьє з описом способу одержання та контролю гомеопатичної сировини або видів сировини та обґрунтування його/їх гомеопатичної природи на основі відповідної бібліографії;
опис виробництва та контролю для кожної лікарської форми та опис способу розведення і підсилення дії лікарського засобу;
копію ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника;
засвідчену копію документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійного листа заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи;
копію реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розмішено лікарський засіб. Зазначити перелік країн, де цей лікарський засіб зареєстровано/перереєстровано;
пропозиції до маркування на упаковці;
інструкцію для медичного застосування у разі, якщо на зовнішній упаковці наведено неповну інформацію, передбачену розділом XVIII цього Порядку;
дані про стабільність лікарського засобу.
ХІІІ. Окремі вимоги до традиційних лікарських засобів рослинного походження та до їх матеріалів реєстраційного досьє
1. Допускається спрощена процедура реєстрації традиційних лікарських засобів рослинного походження, що відповідають таким умовам:
а) показання для застосування мають відповідати традиційним лікарським засобам рослинного походження, які завдяки своєму складу призначені для самостійного використання без контролю з боку лікаря;
б) лікарські засоби призначені виключно для застосовування відповідно до точно вказаного дозування та схеми застосування (у вказаних дозах та відповідно до наведеного режиму прийому);
в) лікарські засоби призначені для перорального, зовнішнього або інгаляційного застосування;
г) період традиційного застосування має становити понад 30 років у світі і понад 15 років в Україні;
ґ) є достатня кількість даних про традиційне використання лікарського засобу, зокрема доведена безпека застосування у звичайних умовах, фармакологічні ефекти або ефективність лікарського засобу доведені тривалим використанням і досвідом.
Наявність у складі лікарського засобу рослинного походження вітамінів та мінералів, безпеку яких підтверджено задокументованими доказами, не повинна бути перешкодою для реєстрації таких лікарських засобів, як традиційних лікарських засобів рослинного походження, за умови, що дія вітамінів або мінералів є допоміжною або здатна підсилювати дію активних інгредієнтів рослинного походження щодо заявлених показань.
2. Комплект реєстраційних документів на традиційні лікарські засоби, що подаються на державну реєстрацію, повинен містити докладні відомості, викладені у Модулях 1, 2 та 3 розділів ІХ і Х цього Порядку.
Крім того, необхідно надати:
а) копію реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розмішено лікарський засіб. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано;
б) бібліографічні або експертні дані (літературний огляд або експертний звіт) відносно ефектів даного або аналогічного лікарського засобу, що використовувався у медичній практиці протягом понад 30 років (з них понад 15 років в Україні).
Аналогічним лікарським засобом рослинного походження вважається лікарський засіб, який містить таку саму діючу речовину, незалежно від використовуваних допоміжних речовин, призначений для застосування за такими самими або подібними показаннями, з такою самою силою дії та способом застосування, таким самим або подібним шляхом введення, що й лікарський засіб, на який подається заява;
в) бібліографічний огляд, що описує безпеку застосування даного лікарського засобу та супроводжується експертним звітом, а на вимогу Центру — додаткові дані, необхідні для оцінки безпеки лікарського засобу;
г) копію ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника;
ґ) засвідчену копію документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійного листа заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи.
3. Вимога надання документа про медичне використання протягом 30 років вважається виконаною у випадку відсутності реєстрації лікарського засобу. Лікарський засіб також вважається таким, що відповідає даній вимозі, навіть, якщо кількість діючих речовин лікарського засобу була знижена протягом цього періоду.
4. У реєстрації традиційного лікарського засобу рослинного походження за спрощеною процедурою може бути відмовлено у разі, якщо лікарський засіб не відповідає вищенаведеному, та:
а) не наведено кількісний та якісний склад компонентів, або
б) показання до застосування не відповідають традиційним лікарським засобам рослинного походження, які завдяки своєму складу призначені для самостійного використання без контролю з боку лікаря, або
в) препарат може бути небезпечним за звичайних умов застосування, або
г) дані, що підтверджують традиційне застосування, недостатні особливо, якщо фармакологічні ефекти або ефективність не підтверджені тривалим досвідом застосування, або
ґ) не підтверджено фармацевтичну якість лікарського засобу.
ХІV. Окремі вимоги до лікарських засобів, що виробляються за прописами, та до їх матеріалів реєстраційного досьє
1. Спрощена процедура реєстрації може бути застосована до певних лікарських засобів, що виробляються за прописами, які відповідають такому:
а) склад лікарського засобу, технологія виробництва, специфікація та інструкція для медичного застосування повинні відповідати таким, що наведені у затверджених прописах;
б) діючі речовини, застосовані у лікарських засобах, мають відповідати вимогам, наведеним у затверджених прописах;
в) до матеріалів реєстраційного досьє допускається внесення тільки таких змін, які зазначені у цьому Порядку.
2. Комплект матеріалів реєстраційного досьє на лікарські засоби, що виробляються згідно із затвердженими прописами, які подаються на державну реєстрацію, повинен містити докладні відомості, викладені у Модулях 1 та 2 розділів ІХ та Х цього Порядку.
Крім того, необхідно надати:
а) методи контролю якості та інструкцію для медичного застосування;
б) дані щодо діючої речовини, які включають найменування та місцезнаходження виробника, інформацію щодо виробництва, стабільності, маркування, пакування, умов зберігання;
в) дані щодо готового лікарського засобу, які включають інформацію щодо об’єму серій, стабільності, маркування, пакування, умов та терміну зберігання;
г) копію реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розмішено лікарський засіб. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано;
ґ) копію ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника;
д) засвідчену копію документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійного листа заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи;
е) лист-підтвердження щодо того, що склад, виробництво та контроль лікарського засобу відповідають пропису, включеному до Переліку лікарських засобів, що виробляються згідно із затвердженими прописами, затвердженого наказом Міністерства охорони здоров’я України від 11 червня 2009 року № 423.
ХV. Окремі вимоги до лікарських засобів обмеженого застосування (препаратів-сиріт) та до їх матеріалів
реєстраційного досьє
1. Лікарські засоби обмеженого застосування (препарати-сироти) повинні відповідати таким умовам:
а) призначені для діагностики, профілактики або лікування рідкісного захворювання, що загрожує життю або серйозно ослаблює імунітет не більше ніж 5 осіб на 10000 жителів на дату подання заяви або призначені для діагностики, профілактики або лікування захворювання, що загрожує життю або серйозно ослаблює імунітет, або хронічних захворювань, та їх продаж без надання компенсації від держави або співтовариства не принесе значного прибутку, який компенсує витрати на лікарський засіб, понесені виробником:
б) не існує будь-якого іншого затвердженого задовільного методу діагностики, профілактики чи лікування даного захворювання або, за наявності такого методу, заявлений лікарський засіб принесе значну користь хворим.
2. Заява на реєстрацію лікарського засобу обмеженого застосування (препарату-сироти) подається відповідно до додатка 1 до цього Порядку. До заяви додаються:
підтвердження внесення заявленого лікарського засобу до реєстру Європейського співтовариства препаратів-сиріт або
підтвердження статусу препарату-сироти, надане компетентними органами країни заявника/виробника або країн, у яких лікарський засіб отримав цей статус, та відомості про реєстрацію його в інших країнах як препарату-сироти;
письмове зобов’язання про надання щорічного підтвердження статусу препарату-сироти, отримане від Комітету по препаратах-сиротах Європейського співтовариства або компетентних органів країни заявника/виробника або країн, у яких лікарський засіб отримав цей статус.
3. Матеріали реєстраційного досьє оформлюються відповідно до вимог розділів ІХ та Х цього Порядку (у форматі загального технічного документа).
При поданні матеріалів реєстраційного досьє на лікарські засоби обмеженого застосування (препарати-сироти) можуть бути застосовані положення глави 5 розділу Х цього Порядку. У такому випадку заявник у резюме доклінічних та клінічних даних повинен надати обґрунтування причин, за яких неможливо надати повну інформацію, та обґрунтування співвідношення користь/ризик для заявлених препаратів-сиріт.
4. Заява повинна охоплювати тільки ті терапевтичні показання, які відповідають умовам, викладеним у пункті 1 цього розділу. Це не може бути перешкодою для подання окремої заяви на нову реєстрацію такого лікарського засобу за іншими показаннями для застосування, що не відповідають вищезазначеним умовам (за іншим реєстраційним досьє).
ХVI. Вимоги до інструкції для медичного застосування лікарського засобу/лікарського засобу (медичного імунобіологічного препарату)
1. Інструкція для медичного застосування, що супроводжує лікарський засіб, повинна бути складена відповідно до короткої характеристики лікарського засобу, структуру якої наведено у додатку 14 (у разі наявності), або затверджена відповідно до нормативних вимог країни заявника/виробника або країни, регуляторний орган якої керується високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ, та/або результатів клінічних випробувань.
2. Інструкція для медичного застосування супроводжує лікарський засіб, якщо інформація, що має міститися в інструкції, не наведена безпосередньо на вторинній або первинній упаковці лікарського засобу.
3. Текст інструкції для медичного застосування лікарського засобу повинен бути викладений чітко, українською мовою. За бажанням виробника/заявника, поряд із текстом українською мовою додатково текст інструкції може бути викладений регіональною мовою або мовою меншин за умови, що в текстах різними мовами буде наведено ідентичну інформацію.
4. Стосовно певних лікарських засобів може бути відмінена вимога щодо необхідності вказувати в інструкції для медичного застосування деякі дані про застосування лікарського засобу за умови, що лікарський засіб не призначений для самостійного застосування пацієнтом.
5. Розмір шрифту повинен бути великим, наскільки це можливо для зручності, та читабельним. Мінімальні вимоги включають: шрифт розміром 8 пунктів Дідо з міжрядковим інтервалом не менше 1 мм.
6. У верхньому правому кутку інструкції для медичного застосування, яка вкладається у вторинну упаковку лікарського засобу, має бути зазначено номер та дата наказу МОЗ України про державну реєстрацію/перереєстрацію/внесення змін у реєстраційні матеріали лікарського засобу, яким даний лікарський засіб дозволено для медичного застосування в Україні, а також номер реєстраційного посвідчення.
7. Інструкція для медичного застосування повинна містити таку інформацію:
7.1. Назва лікарського засобу.
7.2. Повний якісний склад (стосовно діючих та допоміжних речовин) і кількісний склад (стосовно діючих речовин) з використанням загальноприйнятих назв для кожної форми випуску лікарського засобу (дозування та упаковка).
7.3. Лікарська форма.
Основні фізико-хімічні властивості.
7.4. Фармакотерапевтична група. Код АТХ.
7.5. Фармакологічні властивості та, якщо така інформація є корисною для лікування, фармакокінетичні характеристики або імунологічні і біологічні властивості.
Розділ «Фармакологічні властивості» на лікарські засоби, що відпускаються без рецепта, повинен бути викладений зрозумілою мовою для пацієнта та, за бажанням заявника, інформація може бути скорочена.
Розділ «Імунологічні і біологічні властивості» є розділом інструкції для медичного застосування певних категорій медичних імунобіологічних препаратів (вакцин, анатоксинів тощо).
7.6. Клінічні характеристики:
Терапевтичні показання.
7.7. Протипоказання.
7.8. Особливі заходи безпеки (у разі необхідності), яких потрібно дотримуватися особам, які працюють з лікарськими засобами та вводять їх пацієнтам, а також усі заходи безпеки, яких необхідно дотримуватися пацієнту.
7.9. Взаємодія з іншими лікарськими засобами та інші види взаємодій.
7.10. Особливості застосування:
7.10.1. Перерахувати та надати інформацію про всі допоміжні речовини, знання про наявність яких є важливими для безпечного та ефективного застосування лікарського засобу (згідно із додатком 16 до цього Порядку).
7.10.2. Застосування у період вагітності або годування груддю.
7.10.3. Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
7.11. Дози і спосіб введення для дорослих.
7.11.1. Діти.
7.12. Передозування (симптоми, невідкладні заходи лікування та антидоти).
7.13. Побічні реакції (частота та вираженість за умови наявності).
7.14. Термін придатності (у разі необхідності вказується термін зберігання після підготовки лікарського засобу для безпосереднього застосування, наприклад після розчинення або після першого відкриття первинної упаковки).
7.15. Умови зберігання.
7.16. Несумісність (основні види) за наявності інформації.
7.17. Тип і місткість первинної упаковки.
7.18. Особливі заходи безпеки при поводженні з невикористаними лікарськими засобами та відходами лікарських засобів у разі необхідності.
7.19. Категорія відпуску лікарського засобу, встановлена під час державної реєстрації/перереєстрації/внесення змін до матеріалів реєстраційного досьє з урахуванням вимог наказу Міністерства охорони здоров’я України від 17 травня 2001 року № 185 «Про затвердження критеріїв визначення категорій відпуску лікарських засобів», зареєстрованого у Міністерстві юстиції України 31 травня 2001 року за № 464/5655.
7.20. Найменування та місцезнаходження виробника та адреса його місця провадження діяльності (зазначається виробник, відповідальний за випуск серії лікарського засобу), а за бажанням заявника — найменування і місцезнаходження заявника та/або представника заявника.
7.21. Дата останнього перегляду інструкції для медичного застосування (зазначається дата наказу МОЗ про реєстрацію/перереєстрацію/внесення змін до інструкції для медичного застосування лікарського засобу під час дії реєстраційного посвідчення).
7.22. Для радіофармацевтичних лікарських засобів — повний докладний опис внутрішньої радіаційної дозиметрії, а також додаткові докладні інструкції щодо приготування для негайного застосування та контролю якості таких лікарських засобів, а у разі необхідності — максимальний термін зберігання, протягом якого будь-який проміжний лікарський засіб, такий, як елюат, або готовий для застосування фармацевтичний лікарський засіб буде відповідати власним специфікаціям.
Крім того, інструкція для медичного застосування радіофармацевтичного лікарського засобу повинна містити опис усіх заходів безпеки, яких необхідно дотримуватися медичним працівникам та пацієнтам під час приготування та застосування лікарського засобу, а також спеціальні заходи безпеки з утилізації упаковки та її невикористаного вмісту.
8. В інструкції для медичного застосування можуть бути розміщені символи або піктограми, які пояснюють інформацію, надану в ній, а також інша інформація, яка відповідає короткій характеристиці лікарського засобу і є корисною для санітарної просвіти, за винятком інформації рекламного характеру, яка сприяє просуванню лікарського засобу на ринку.
9. Інструкція для медичного застосування традиційного лікарського засобу рослинного походження повинна містити вказівки на те, що:
лікарський засіб є звичайним лікарським засобом рослинного походження для застосування відповідно до показань, підтверджених тривалим застосуванням;
користувач повинен проконсультуватися з лікарем, якщо симптоми захворювання не зникли під час застосування лікарського засобу або спостерігаються побічні реакції, не зазначені в інструкції для медичного застосування лікарського засобу.
10. Інструкція для медичного застосування гомеопатичних лікарських засобів, які відповідають вимогам, викладеним у розділі ХІІ цього Порядку, повинна містити:
формулювання «гомеопатичний лікарський засіб без затверджених терапевтичних показань для застосування»;
застереження для споживача про необхідність консультації з лікарем, якщо симптоми зберігаються при застосуванні лікарського засобу.
11. Власник реєстраційного посвідчення повинен гарантувати, що у разі необхідності зможе надати організаціям пацієнтів з обмеженим зором текст інструкції для медичного застосування у доступному для них форматі.
ХVІI. Вимоги до короткої характеристики лікарського засобу/лікарського засобу (медичного імунобіологічного препарату)
1. Коротка характеристика лікарського засобу (Summary of Product Characteristics — SPC) — інформація для спеціалістів щодо безпеки та ефективності застосування лікарського засобу, яка затверджується за бажанням заявника.
2. Текст короткої характеристики лікарського засобу повинен бути викладений чітко, українською мовою.
3. Коротка характеристика лікарського засобу, структуру якої наведено у додатку 14 до цього Порядку, повинна містити таку інформацію:
3.1. Назва лікарського засобу, до складу якої у разі необхідності може входити дозування та лікарська форма.
3.2. Повний якісний склад (стосовно діючих та допоміжних речовин) і кількісний склад стосовно діючих речовин з використанням загальноприйнятих назв для кожної форми випуску лікарського засобу (дозування та упаковка).
3.3. Лікарська форма.
3.4. Клінічна інформація:
3.4.1. Терапевтичні показання.
3.4.2. Нозологія і спосіб застосування для дорослих і, якщо необхідно, для дітей.
3.4.3. Протипоказання.
3.4.4. Особливі застереження та запобіжні заходи при застосуванні, а у випадку медичного імунобіологічного препарату — обов’язкові запобіжні заходи, що повинні бути вжиті лікарями, які працюють з цими засобами та вводять їх пацієнтам, а також усі запобіжні заходи, яких повинен дотримуватися пацієнт.
3.4.5. Взаємодія з іншими лікарськими засобами та інші види взаємодій.
3.4.6. Застосування під час вагітності та годування груддю.
3.4.7. Вплив на здатність керувати транспортними засобами або працювати з іншими автоматизованими системами.
3.4.8. Побічні реакції.
3.4.9. Передозування (симптоми, невідкладні заходи лікування, антидоти).
3.5. Фармакологічні властивості:
3.5.1. Фармакотерапевтична група. Код АТХ.
3.5.2. Фармакодинамічні властивості.
3.5.3. Фармакокінетичні властивості.
3.5.4. Доклінічні дані з безпеки.
3.6. Фармацевтична інформація:
3.6.1. Перелік допоміжних речовин.
3.6.2. Основні випадки несумісності.
3.6.3. Термін придатності, у разі необхідності вказується термін придатності після розчинення лікарського засобу або після першого відкриття первинної упаковки.
3.6.4. Особливі запобіжні заходи при зберіганні.
3.6.5. Тип та вміст первинної упаковки.
3.6.6. Спеціальні заходи безпеки при поводженні з невикористаним лікарським засобом або відходами лікарського засобу (у разі необхідності).
3.7. Власник реєстраційного посвідчення.
Виробник лікарського засобу (найменування та місцезнаходження виробника та адреса його місця провадження діяльності (зазначається виробник, відповідальний за випуск серії лікарського засобу).
3.8. Номер(и) реєстраційного(их) посвідчення(нь).
3.9. Дата першої реєстрації або перереєстрації лікарського засобу.
3.10. Дата перегляду тексту короткої характеристики.
3.11. Для радіофармацевтичних лікарських засобів — повний докладний опис внутрішньої радіаційної дозиметрії.
3.12. Для радіофармацевтичних лікарських засобів — додаткові вказівки для приготування лікарських засобів для негайного застосування та контролю якості такого лікарського засобу, а у разі необхідності — максимальний термін зберігання, протягом якого будь-який проміжний лікарський засіб, наприклад елюат, або готовий до застосування лікарський засіб буде відповідати власним специфікаціям.
ХVIII. Вимоги до маркування на упаковці лікарського засобу
1. Інформація, що наноситься на упаковку (етикетку).
На вторинній упаковці готового лікарського засобу, а за її відсутності — на первинній упаковці вказуються такі загальні відомості:
назва лікарського засобу;
найменування та місцезнаходження виробника;
номер реєстраційного посвідчення;
номер серії;
способи застосування;
доза діючої речовини в кожній одиниці та їх кількість в упаковці;
термін придатності;
умови зберігання;
запобіжні заходи.
2. Вимоги до маркування упаковки лікарського засобу.
2.1. Вимоги до відомостей, що вказуються на упаковці.
2.1.1. Вторинна упаковка лікарського засобу, а за її відсутності — первинна упаковка має містити такі відомості:
а) штрих-код лікарського засобу;
б) назву лікарського засобу, яка супроводжується зазначенням МНН або у разі її відсутності загальноприйнятої назви (коли лікарський засіб містить лише одну діючу речовину); якщо назва лікарського засобу може застосовуватися у декількох лікарських формах та/або мати різну силу дії, то необхідно вказати лікарську форму та/або силу дії із зазначенням того, чи призначений цей лікарський засіб для дітей віком до 1 року, старше 1 року або дорослих;
в) діючі речовини у якісному та кількісному вираженні із зазначенням їхнього вмісту в одиниці дози або, залежно від способу застосування, в одиниці об’єму чи маси, з використанням їх міжнародних непатентованих або загальноприйнятих назв;
г) лікарську форму із зазначенням маси, об’єму або кількості одиниць дозування, що містяться в упаковці;
ґ) перелік допоміжних речовин згідно з додатком 16 до цього Порядку, стосовно яких відомо, що вони спричиняють певну дію або ефект і їх назви зазначаються разом з формулюванням: «для детальної інформації див. інструкцію для медичного застосування». Однак на упаковці повинні бути вказані всі допоміжні речовини, якщо лікарський засіб використовується парентерально, або в офтальмологічній практиці, або для місцевого застосування (нашкірні та трансдермальні лікарські засоби, лікарські засоби для інгаляцій, для ротової порожнини, назальні, ректальні та вагінальні лікарські засоби, тобто такі, що мають місцеву або трансдермальну дію);
д) спосіб, а за необхідності — шлях введення лікарського засобу;
е) особливі застереження щодо того, чи слід зберігати лікарський засіб у недоступному для дітей місці і, за необхідності — поза полем зору дітей;
є) дату закінчення терміну придатності (місяць/рік) (зазначається останній місяць, коли термін придатності дійсний);
ж) за необхідності особливі вказівки відносно того, що робити з невикористаним лікарським засобом або відходами, які залишаються після використання такого лікарського засобу, а також, за бажанням заявника, посилання на будь-яку придатну систему збирання відходів на місці;
з) найменування та місцезнаходження виробника та адресу його місця провадження діяльності (зазначається виробник, відповідальний за випуск серій лікарського засобу) і за необхідності найменування та місцезнаходження заявника або представника заявника.
У разі здійснення виробництва лікарського засобу з пакування in bulk, поряд з найменуванням виробника зазначається: «виробництво з пакування in bulk»;
и) номер реєстраційного посвідчення;
і) номер серії лікарського засобу, присвоєний виробником;
ї) якщо лікарський засіб призначено для самостійного лікування, інформацію для його застосування;
й) за необхідності особливі застереження стосовно лікарського засобу;
к) умови зберігання, а за необхідності — особливі умови зберігання.
У разі відсутності відповідної площі на упаковці для нанесення повної інформації обов’язково наносяться дані, перелічені в підпунктах «а», «б», «в», «г», «є», «з», «и», «і», «й», «к» цього пункту, за умови наявності інструкції для медичного застосування.
2.1.2. На вторинній упаковці можуть бути також вміщені символи або піктограми, які дають змогу пояснити інформацію, вказану в підпункті 2.1.1 цього пункту, а також інша інформація, яка відповідає короткій характеристиці лікарського засобу та є корисною для пацієнта, за винятком будь-яких елементів рекламного характеру, які сприяють просуванню цього лікарського засобу на ринку.
2.1.3. Інформація, зазначена в підпунктах 2.1.1 та 2.1.2 цього пункту, вказується на всіх первинних упаковках, за винятком випадків, описаних у підпункті 2.1.4 цього пункту.
2.1.4. На первинній упаковці у формі блістера, стрипа тощо та на первинній упаковці невеликого розміру (ампулі, тюбику-крапельниці, шприці-тюбику тощо), що вкладається у вторинну упаковку, яка відповідає вимогам, викладеним у підпунктах пунктах 2.1.1, 2.1.2 цього пункту, зазначається як мінімум така інформація:
а) назва лікарського засобу;
б) маса, об’єм, концентрація або кількість одиниць дії лікарського засобу;
в) номер серії лікарського засобу;
г) дата закінчення терміну придатності;
ґ) найменування виробника та за необхідності заявника.
У разі відсутності відповідного місця на первинній упаковці для нанесення зазначеної інформації обов’язково зазначаються дані, перелічені у підпунктах «а», «б», «в» цього підпункту.
2.2. Вимоги до маркування упаковки лікарських засобів, що містять радіонукліди:
2.2.1. Вторинна упаковка та первинна упаковка лікарських засобів, що містять радіонукліди, повинні бути марковані відповідно до правил безпечного транспортування радіоактивних матеріалів та відповідати таким вимогам:
2.2.2. Етикетка на захисному контейнері повинна містити докладні відомості, вказані в підпункті 2.1.1 цього пункту. Додатково маркування на захисному контейнері повинно повністю пояснювати кодування на флаконі та за необхідності містити зазначення кількості одиниць радіоактивності лікарського засобу у дозі або цілому флаконі із зазначенням дати, а у разі необхідності часу, а також кількості капсул або для рідини — об’єм у мілілітрах вмісту флакона.
2.2.3. Маркування флакона повинно містити таку інформацію:
назву або код лікарського засобу, включаючи назву або хімічний символ радіонукліда;
номер серії та дату закінчення терміну придатності;
міжнародний символ радіоактивності;
найменування виробника;
кількість одиниць радіоактивності, як указано в підпункті 2.2.2 цього пункту.
2.3. Вимоги до маркування упаковки гомеопатичних лікарських засобів:
Для гомеопатичних лікарських засобів, які відповідають вимогам розділу ХІІ цього Порядку, на етикетці упаковки має бути наведена тільки така інформація:
штрих-код лікарського засобу;
наукова назва сировини або видів сировини із зазначенням ступеня розведення із застосуванням символів ДФУ або іншої фармакопеї (Європейська фармакопея, Німецька гомеопатична фармакопея (GHP), Гомеопатична фармакопея США (HPUS), Британська гомеопатична фармакопея (BHP), Гомеопатична фармакопея Швабе);
найменування та місцезнаходження виробника та заявника і, за необхідності, найменування представника заявника;
спосіб застосування та, за необхідності, шлях введення;
дата закінчення терміну придатності (місяць/рік) (зазначається останній місяць, коли термін придатності дійсний);
лікарська форма;
вміст упаковки для продажу;
за необхідності, особливі умови зберігання;
за необхідності, особливі застереження щодо лікарського засобу;
номер серії лікарського засобу, присвоєний виробником;
номер реєстраційного посвідчення;
формулювання «гомеопатичний лікарський засіб без затверджених терапевтичних показань для застосування»;
застереження для споживача про необхідність консультації з лікарем, якщо симптоми захворювання не зникли під час застосування лікарського засобу.
2.4. Вимоги до маркування упаковки традиційних лікарських засобів рослинного походження:
Для традиційних лікарських засобів рослинного походження етикетка повинна містити докладні відомості, вказані в підпунктах 2.1.1 та 2.1.2 цього пункту. Додатково повинні бути наведені вказівки про те, що:
лікарський засіб є традиційним лікарським засобом рослинного походження для використання відповідно до показань, підтверджених тривалим застосуванням;
користувач повинен проконсультуватися з лікарем, якщо симптоми захворювання не зникли під час застосування лікарського засобу або спостерігаються побічні реакції, не вказані в інструкції для медичного застосування.
2.5. Вимоги до маркування упаковки лікарських засобів, що містять один чи декілька наркотичних засобів та/або психотропних речовин:
На упаковці лікарських засобів, що містять один чи декілька наркотичних засобів та/або психотропних речовин, повинні міститись докладні відомості, вказані в підпунктах 2.1.1 та 2.1.2 цього пункту. Додатково первинна упаковка повинна бути позначена подвійною червоною смугою.
2.6. Вимоги до тексту упаковки лікарського засобу:
2.6.1. Дані на упаковці мають бути нанесені шрифтом не менше 7 пунктів Дідо.
2.6.2. Текст маркування викладається українською мовою. За бажанням виробника/заявника разом із текстом маркування українською мовою додатково текст маркування може бути викладений регіональною мовою або мовою меншин за умови, що в текстах різними мовами буде наведено ідентичну інформацію.
2.6.3. Допускається, за необхідності, нанесення на упаковку інформації за допомогою стикера, зокрема для оригінальних (інноваційних) лікарських засобів (строком на один рік з дати їх державної реєстрації), препаратів обмеженого застосування (препаратів-сиріт) або, за необхідності, в інших випадках.
Інформація, що міститься на стикері, має відповідати вимогам глав 1 та 2 цього розділу. Стикер затверджується у складі Методів контролю якості лікарського засобу при його реєстрації/перереєстрації/внесенні змін у реєстраційні матеріали.
2.6.4. На вторинній упаковці лікарських засобів (крім АФІ або діючих речовин та лікарських засобів в упаковці in bulk) також шрифтом Брайля зазначаються назва лікарського засобу, доза діючої речовини та лікарська форма відповідно до Порядку маркування лікарських засобів шрифтом Брайля, затвердженого наказом Міністерства охорони здоров’я України від 25 серпня 2010 року № 722, зареєстрованого у Міністерстві юстиції України 05 листопада 2010 року за № 1044/18339.
2.7. Вимоги щодо допоміжних речовин, які мають бути нанесені на упаковку:
2.7.1. Назви допоміжних речовин на упаковці повинні вказуватись у такий спосіб:
допоміжні речовини мають бути представлені за їхніми МНН відповідно до Європейської фармакопеї або за відсутності таких — загальноприйнятими назвами;
назву допоміжної речовини має супроводжувати номер «Е», якщо такий є. На упаковці може бути зазначений тільки номер «Е» за умови, що повна назва (МНН, а за її відсутності — загальноприйнята назва) та номер «Е» буде наведено в інструкції для медичного застосування у розділі «Склад»;
патентовані коригенти або ароматизатори мають бути надані у загальноприйнятих термінах (наприклад, «апельсиновий смак», «лимонний аромат»); мають бути зазначені будь-які відомі основні компоненти або такі, що мають певну визнану дію або реакцію;
хімічно модифіковані допоміжні речовини мають бути вказані у такий спосіб, щоб уникнути плутанини з немодифікованими допоміжними речовинами (наприклад, крохмаль попередньо желатинізований);
усі компоненти складних допоміжних речовин або сумішей мають бути заявлені, перелічені в описі загального складу із зазначенням основних складових речовин (наприклад, фарба для друку, що містить x, y, z). Загальна назва складної допоміжної речовини або суміші може бути використана на упаковці у випадку, якщо більш повна інформація надається в інструкції для медичного застосування. Будь-який компонент, здатний спричинити певну дію або реакцію, має бути вказаний на упаковці.
Ці вимоги не стосуються маркування на упаковках АФІ або діючих речовин.
2.7.2. Перелік допоміжних речовин, які мають бути обов’язково зазначені на упаковці, та інформація щодо впливу цих речовин залежно від шляху введення та вмісту допоміжної речовини, що має бути наведена в інструкції для медичного застосування лікарського засобу, наведено в додатку 16 до цього Порядку.
ХIX. Критерії проведення додаткових випробувань лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє
1. Додаткові випробування щодо фармацевтичної розробки при проведенні експертизи матеріалів реєстраційного досьє на лікарські засоби, що подаються на державну реєстрацію, а також експертизи матеріалів про внесення змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення проводяться у випадках, коли за результатами експертизи матеріалів реєстраційного досьє матеріалів було встановлено, що:
відсутні або недостатні дані, що мають бути представлені відповідно до вимог, передбачених частиною ІІ реєстраційного досьє розділів VII та VIII цього Порядку або Модулем 3 розділів ІХ та Х цього Порядку, для підтвердження того, що розроблена лікарська форма та запропонований склад задовольняють мету, вказану в заяві на проведення експертизи матеріалів реєстраційного досьє;
відсутні або недостатні дані, що мають бути представлені відповідно до вимог, передбачених частиною ІІ реєстраційного досьє розділів VII та VIII цього Порядку або Модулем 3 розділів ІХ та Х цього Порядку, щодо фізико-хімічних характеристик діючої речовини, які впливають на біодоступність;
відсутні або недостатні дані щодо досліджень для підтвердження еквівалентності генеричного лікарського засобу до референтного препарату на етапі фармацевтичної розробки, передбачені вимогами, викладеними у главі 4 розділу ХХ цього Порядку.
2. Додаткові випробування щодо підтвердження якості лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє на лікарські засоби проводяться у випадку, коли за результатами експертизи таких матеріалів було встановлено, що результати клінічного аудиту свідчать про недотримання умов зберігання досліджуваного лікарського засобу при проведенні клінічних випробувань, або виявлено невідповідність номерів серій зразків досліджуваного лікарського засобу при проведенні клінічних випробувань відповідно до вимог Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань, затвердженого наказом Міністерства охорони здоров’я України від 23 вересня 2009 року № 690, зареєстрованого у Міністерстві юстиції України 29 жовтня 2009 року за № 1010/17026 (у редакції наказу Міністерства охорони здоров’я України від 12 липня 2012 року № 523).
Додаткові випробування щодо підтвердження якості лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє на лікарські засоби не проводяться у випадках, зазначених у підпункті 5.2 пункту 5 розділу ХХ цього Порядку.
3. Додаткові випробування щодо доклінічних досліджень при проведенні експертизи матеріалів реєстраційного досьє на лікарські засоби проводяться у випадку, коли за результатами експертизи таких матеріалів було встановлено, що дані, представлені відповідно до вимог, передбачених частиною ІІІ реєстраційного досьє розділів VII та VIII цього Порядку або Модулем 4 розділів ІХ та Х цього Порядку, щодо фармакологічних, фармакокінетичних та токсикологічних досліджень, необґрунтовано відсутні або недостатні.
4. Додаткові випробування щодо ефективності та/або безпеки лікарського засобу при проведенні експертизи матеріалів реєстраційного досьє на лікарські засоби проводяться у випадку, коли за результатами експертизи звітів проведеного(их) клінічного(их) дослідження(ь), наданих заявником, встановлено, що:
клінічні дані, надані відповідно до вимог частини ІV реєстраційного досьє розділів VII та VIII цього Порядку або Модуля 5 розділів ІХ та Х цього Порядку, представлені не в повному обсязі або/та недостатньо обґрунтовують окремі показання для застосування або протипоказання, схеми призначення;
наявні негативні дані стосовно ефективності або безпеки лікарського засобу, представлені відповідно до вимог, передбачених частиною ІV реєстраційного досьє розділів VII та VIII цього Порядку або Модулем 5 розділів ІХ та Х цього Порядку, і ці дані потребують додаткових досліджень;
проведені клінічні випробування є неадекватними для підтвердження ефективності та безпеки лікарського засобу;
дані спричиняють сумнів щодо достовірності результатів проведених клінічних випробувань.
5. Додаткові випробування для комбінованих лікарських засобів, що містять у своєму складі відомі діючі речовини, які раніше не застосовувались у даному сполученні з терапевтичною метою, проводяться, якщо токсикологічні, фармакологічні або клінічні дослідження, викладені відповідно до вимог частини ІІІ реєстраційного досьє розділів VII та VIII цього Порядку або Модуля 4 розділів ІХ та Х цього Порядку, надані не у повному обсязі.
Це не стосується випадків, коли комбіновані лікарські засоби містять відомі діючі речовини, які раніше використовувались у медичній практиці як окремі препарати у тій самій лікарській формі, дозах та відповідному співвідношенні для досягнення тієї самої терапевтичної мети. При цьому заявник повинен надати результати досліджень, які підтверджують відсутність будь-якої взаємодії між інгредієнтами лікарського засобу, яка може вплинути на його ефективність та безпеку як на стадії виробництва, так і протягом терміну зберігання комбінованого лікарського засобу.
6. Додаткові випробування щодо біодоступності та еквівалентності генеричних лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє проводяться у випадку, коли:
дані представлено не в повному обсязі для підтвердження еквівалентності лікарського засобу відповідно до вимог, викладених у пункті 4 розділу ХХ цього Порядку;
дані, передбачені вимогами, викладеними у пункті 4 розділу ХХ цього Порядку, є неадекватними для підтвердження еквівалентності генеричного лікарського засобу;
існує ризик, що можливі відмінності у біодоступності можуть призвести до терапевтичної нееквівалентності.
7. Додаткові клінічні випробування МІБП (клініко-епідеміологічні випробування вакцин та анатоксинів) проводяться у випадку, коли за результатами експертизи звітів клінічних випробувань (клініко-епідеміологічних випробувань для вакцин та анатоксинів), наданих заявником, встановлено хоча б один із таких фактів:
у матеріалах реєстраційного досьє недостатньо даних для підтвердження показників ефективності МІБП;
дані клінічних випробувань (клініко-епідеміологічних випробувань для вакцин та анатоксинів) представлено не в повному обсязі або/та недостатньо обґрунтовують окремі показання для застосування або протипоказання, схеми призначення;
наявні дані стосовно ефективності або безпеки МІБП потребують додаткових перевірок;
дані викликають сумнів щодо достовірності результатів проведених клінічних випробувань.
ХХ. Порядок проведення додаткових випробувань лікарських засобів
1. Випробування щодо фармацевтичної розробки, підтвердження якості та відтворюваності методів контролю якості при проведенні експертизи матеріалів реєстраційного досьє проводяться в уповноважених лабораторіях відповідно до галузі акредитації за направленням Центру.
Додаткові випробування щодо фармацевтичної розробки лікарського засобу проводяться відповідно до вимог, зазначених у частині ІІ реєстраційного досьє розділів VII та VIII цього Порядку або Модулі 3 розділів ІХ та Х цього Порядку.
2. Додаткові випробування щодо доклінічних досліджень лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє проводяться відповідно до вимог Порядку проведення доклінічного вивчення лікарських засобів та експертизи матеріалів доклінічного вивчення лікарських засобів, затвердженого наказом Міністерства охорони здоров’я України від 14 грудня 2009 року № 944, зареєстрованого у Міністерстві юстиції України 19 січня 2010 року за № 53/17348.
3. Додаткові випробування щодо ефективності та/або безпеки лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє проводяться відповідно до вимог Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань (у редакції наказу Міністерства охорони здоров’я України від 12 липня 2012 року № 523), затвердженого наказом Міністерства охорони здоров’я України від 23 вересня 2009 року № 690, зареєстрованого в Міністерстві юстиції України 29 жовтня 2009 року за № 1010/17026.
4. Додаткові випробування щодо біодоступності та еквівалентності генеричних лікарських засобів при проведенні експертизи матеріалів реєстраційного досьє проводяться згідно з вимогами Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань (у редакції наказу Міністерства охорони здоров’я України від 12 липня 2012 року № 523), затвердженого наказом Міністерства охорони здоров’я України від 23 вересня 2009 року № 690, зареєстрованого в Міністерстві юстиції України 29 жовтня 2009 року за № 1010/17026 (у випадку дослідження еквівалентності за участю людини), та вимогами цього розділу.
Додаткові випробування щодо біодоступності та еквівалентності проводяться у закладах охорони здоров’я, а також лабораторіях фармакокінетики Центру або в інших уповноважених лабораторіях за направленням Центру.
4.1. Загальні положення:
4.1.1. Мета проведення додаткових досліджень встановлення еквівалентності (взаємозамінності) полягає в доведенні еквівалентності (взаємозамінності) біофармацевтичної якості між генеричним та референтним лікарськими засобами.
4.1.2. Для доведення еквівалентності (взаємозамінності) застосовуються такі методи:
порівняльні фармакокінетичні дослідження за участю людини, у яких діюча речовина та/або її метаболіти вимірюються як функція часу у доступній біологічній рідині, а саме: крові, плазмі, сироватці або сечі для отримання фармакокінетичних показників типу АUC і Смакс, що відображають системну дію;
порівняльні фармакодинамічні дослідження за участю людини;
порівняльні клінічні дослідження;
дослідження еквівалентності in vitro (процедура біовейвер).
4.1.3. Прийнятність кожного із зазначених методів для доведення еквівалентності (взаємозамінності) визначається на підставі комплексної експертизи факторів, які включають характеристики діючої речовини, лікарського засобу та часу дії активної(их) речовини(н).
4.1.4. При виборі методів доведення еквівалентності (взаємозамінності) застосовують такі підходи:
якщо можливо отримати вимірювані концентрації діючої речовини в наявній біологічній рідині, такій як плазма, проводяться порівняльні фармакокінетичні дослідження за участю людини;
якщо неможливо отримати вимірювані концентрації діючої речовини у відповідній біологічній рідині, проводяться порівняльні фармакодинамічні дослідження за участю людини;
якщо неможливо визначити фармакокінетичний профіль, знайти прийнятні фармакодинамічні кінцеві точки, проводяться порівняльні клінічні дослідження;
для лікарських засобів для орального застосування з негайним вивільненням може застосовуватися дослідження еквівалентності in vitro за процедурою біовейвер відповідно до БСК.
4.2. Відсутність необхідності проведення дослідження еквівалентності in vivo:
4.2.1. Генеричні лікарські засоби, які є фармацевтично еквівалентними до референтних, вважаються терапевтично еквівалентними (взаємозамінними) і не потребують подальшого підтвердження еквівалентності шляхом проведення порівняльних досліджень при дотриманні вимог до виробництва та стандартів якості, встановлених чинним законодавством України та міжнародними нормами, за умов, що:
а) лікарські засоби для парентерального застосування (наприклад, внутрішньовенно, підшкірно або внутрішньом’язово) у вигляді водних розчинів, які містять таку саму діючу речовину і в тій самій молярній концентрації, що і референтний препарат, і з такими самими або подібними допоміжними речовинами у порівнюваних з референтним препаратом концентраціях. Деякі допоміжні речовини (зокрема буферні розчини, консерванти, антиоксиданти) можуть відрізнятися за умови доведення у будь-який спосіб, що у даних концентраціях вони не впливають на безпеку та/або ефективність лікарського засобу;
б) лікарські засоби для орального застосування у формі розчинів (наприклад, сиропи, еліксири і настойки), які містять таку саму діючу речовину в тій самій молярній концентрації, що й референтний препарат, і містять, в основному, такі самі допоміжні речовини в порівнюваних концентраціях. Необхідно ретельно вивчити допоміжні речовини, про які відомо, що вони впливають на проходження шлунково-кишковим трактом (далі — ШКТ), проникність у ШКТ і, таким чином, на абсорбцію та/або стабільність діючої речовини у ШКТ;
в) лікарські засоби у формі порошків для приготування розчинів, якщо розчин відповідає вимогам підпунктів «а» або «б» цього пункту;
г) лікарські засоби, які є газами;
ґ) вушні або очні лікарські засоби, виготовлені у вигляді водних розчинів, які містять таку саму діючу речовину(и) в такій самій молярній концентрації(ях) і, по суті, такі самі допоміжні речовини у порівнюваних концентраціях, що й референтний препарат. Деякі допоміжні речовини (зокрема буферні розчини, консерванти, речовини, які коригують густину, або згущувачі) можуть відрізнятися за умови доведення у будь-який спосіб, що у даних концентраціях вони не впливають на безпеку та/або ефективність лікарського засобу;
д) лікарські засоби місцевої дії у вигляді водних розчинів, які містять таку саму діючу речовину(и) в такій самій молярній концентрації(ях) і, по суті, такі самі допоміжні речовини у порівнюваних концентраціях, що й референтний препарат;
е) лікарські засоби, які є водними розчинами у формі інгаляційно-розпилюваних небулайзером препаратів, або назальні спреї, що застосовуються за допомогою практично однакових пристроїв і які містять таку саму діючу речовину(и) в такій самій молярній концентрації(ях) і, по суті, такі самі допоміжні речовини у порівнюваних концентраціях, що й референтний препарат. Деякі допоміжні речовини можуть відрізнятися за умови доведення у будь-який спосіб, що у даних концентраціях вони не впливають на безпеку та/або ефективність лікарського засобу;
є) лікарські засоби системної дії у вигляді водних розчинів для ректального або вагінального застосування, які містять таку саму діючу(і) речовину(и) в такій самій молярній концентрації і, по суті, такі самі допоміжні речовини у порівнюваних концентраціях, що й референтний препарат. Деякі допоміжні речовини можуть відрізнятися за умови доведення у будь-який спосіб, що у даних концентраціях вони не впливають на безпеку та/або ефективність лікарського засобу.
4.2.2. Для випадків, наведених у підпунктах «б», «в», «ґ», «д», «е» та «є» підпункту 4.2.1 цього пункту, у разі відмінностей у складі допоміжних речовин заявник має довести, що при їх використанні не передбачається вплив на безпеку та/або ефективність лікарського засобу.
У випадку, коли заявник не може надати таку інформацію і не має доступу до відповідних даних, він повинен провести відповідні дослідження для доказу відсутності впливу різних допоміжних речовин або допоміжних пристроїв на безпеку та/або ефективність лікарського засобу.
4.3. Необхідність проведення дослідження еквівалентності in vivo:
Доведення еквівалентності in vivo потрібне у випадку, коли існує ризик того, що можливі відмінності у біодоступності можуть призвести до терапевтичної нееквівалентності генеричного лікарського засобу до референтного, наприклад, для:
а) лікарських засобів системної дії для орального застосування з негайним вивільненням, якщо до них застосовані один або декілька таких критеріїв:
застосування лікарського засобу для невідкладної допомоги;
вузький спектр терапевтичної дії (межа ефективність/безпека), крутий нахил кривої доза — відклик;
документально підтверджені проблеми щодо біодоступності або біонееквівалентності, які пов’язані з діючою речовиною або її формами (що не мають відношення до розчинення);
дані стосовно того, що на біоеквівалентність можуть впливати поліморфізм діючої речовини, допоміжні речовини та/або технологічні процеси;
б) лікарських засобів системної дії, що не призначені для орального або парентерального застосування (таких, як трансдермальні пластирі, супозиторії, нікотинові жувальні гумки, гелі тестостерону та трансдермальні контрацептиви);
в) лікарських засобів системної дії з модифікованим вивільненням;
г) лікарських засобів системної дії з фіксованою комбінацією, у яких як мінімум для однієї діючої речовини потрібно проведення дослідження in vivo;
ґ) лікарських засобів несистемної дії, зокрема для орального, назального, офтальмологічного, дерматологічного, ректального або вагінального застосування, без системної абсорбції, що не є розчинами. У такому разі еквівалентність доводять шляхом проведення, наприклад, порівняльних клінічних або фармакодинамічних, дерматофармакокінетичних досліджень та/або досліджень in vitro;
д) лікарських засобів, що містять відмінні від референтного: ефір, складний ефір, ізомер, рацемат, комплекси або продукти дериватизації діючої речовини, оскільки ці відмінності можуть спричинити різну біодоступність.
4.4. Умови проведення процедури біовейвер:
Застосування процедури біовейвер для доведення еквівалентності генеричних лікарських засобів обмежується швидкорозчинними діючими речовинами з відомою абсорбцією у людини, які не мають вузького терапевтичного індексу та критичного застосування.
Біовейвер може застосовуватися для лікарських засобів у твердих дозованих формах системної дії з негайним вивільненням для орального застосування. Процедура біовейвер не застосовується для сублінгвальних, букальних лікарських засобів і препаратів з модифікованим вивільненням. Для лікарських засобів, диспергованих у ротовій порожнині, підхід біовейвера на підставі БСК може застосовуватися лише тоді, коли абсорбція в ротовій порожнині може бути виключена.
4.4.1. Загальні вимоги:
Біовейвер на підставі БСК застосовується до лікарських засобів з негайним вивільненням, якщо:
було доведено, що діюча речовина проявляє високу розчинність і повну абсорбцію/високий ступінь проникнення (I клас за БСК);
лікарський засіб належить до дуже швидкорозчинних (>85% діючої речовини переходить у розчин за 15 хвилин) або швидкорозчинних (85% за 30 хвилин);
допоміжні речовини, які можуть вплинути на біодоступність, є якісно і кількісно подібними. В основному використання подібних допоміжних речовин у схожих кількостях є переважним
або:
було доведено, що діюча речовина проявляє високу розчинність і обмежену абсорбцію/низький ступінь проникнення (III клас за БСК);
лікарський засіб належить до дуже швидкорозчинних (>85% діючої речовини переходить у розчин за 15 хвилин);
допоміжні речовини, які можуть вплинути на біодоступність, є якісно і кількісно подібними, й інші допоміжні речовини є якісно подібними і кількісно дуже схожими.
В основному ризики невідповідного рішення щодо можливості проведення процедури біовейвер мають бути критичніше розглянуті для лікарських засобів, діюча речовина яких відноситься до ІІІ класу за БСК (наприклад, абсорбція, специфічна для певної ділянки, ризик взаємодії транспортних білків у ділянці абсорбції, склад допоміжних речовин і терапевтичні ризики).
4.4.2. Діюча речовина:
Процедура біовейвер застосовується тільки у випадках, коли діюча речовина у досліджуваному та референтному лікарських засобах є тією самою. У випадку використання діючої речовини у вигляді різних солей процедура біовейвер можлива, якщо діюча речовина відноситься до І класу за БСК.
Біовейвер не застосовується, якщо досліджуваний і референтний лікарські засоби містять різни ефіри, складні ефіри, ізомери, рацемати, комплекси або продукти дериватизації діючої речовини, оскільки означені відмінності можуть призвести до різної біодоступності, яка не може бути встановлена на підставі досліджень на основі БСК та результатів порівняльних досліджень in vitro з використанням тесту «Розчинення».
4.4.3. Розчинність:
Необхідно визначити та розглянути профіль рН-залежної розчинності діючої речовини. Діюча речовина вважається високорозчинною, якщо найвища одноразова доза повністю розчиняється в 250 мл або в меншому об’ємі кожного з трьох буферних розчинів із значенням рН в межах 1-6,8. Встановлення рівноважної розчинності необхідно проводити у трьох буферних розчинах при рекомендованих значеннях рН 1,2; 4,5 та 6,8, температурі (37±1)°С, та додатково враховуючи рКa, якщо її величина знаходиться у вищезазначеному діапазоні рН, де рКa — константа іонізації діючої речовини. Повторні дослідження при кожному значенні рН можуть бути необхідними для досягнення чіткої класифікації розчинності діючої речовини (наприклад, методом струшування колби або іншим обґрунтованим методом). Рекомендується вивчення розчинності діючої речовини проводити в трьох повторах при кожному із вибраних значень рН для кожного заявленого виробника діючої речовини. Значення рН кожного буферного розчину необхідно перевіряти до та після експерименту.
4.4.4. Всмоктування/абсорбція:
Демонстрація повної абсорбції у людини є переважною для проведення процедури біовейвер. З цією метою вважається, що повна абсорбція має бути встановлена, якщо вимірюваний рівень абсорбції складає більше 85%. Повна абсорбція в основному пов’язана з високою проникністю. Повна абсорбція препарату має бути обґрунтована на підставі досліджень у людини.
Для вивчення ступеня абсорбції можуть використовуватися дані досліджень абсолютної біодоступності або визначення масобалансу.
Якщо дані з встановлення масобалансу використовуються для підтвердження повної абсорбції, повинна бути гарантія, що метаболіти, які враховуються при визначенні абсорбованої фракції, формуються після абсорбції.
Дослідження проникності in vitro (in vivo або in situ кишкова перфузія з використанням моделей тварин; in vitro проникнення крізь ізольовані тканини тварин або моношар культури епітеліальних клітин) можуть також вважатися підтверджуючими для даних in vivo.
4.4.5. Готовий лікарський засіб:
Вивчення кінетики вивільнення діючої речовини генеричного та референтного лікарських засобів проводять у порівнянні з референтним препаратом за таких умов:
прилади — прилад із кошиком (100 об/хв) або прилад із лопаттю (50 об/хв);
об’єм середовища розчинення — 900 мл або менше, але не менше 500 мл;
температура середовища розчинення — (37±0,5)°С;
кількість досліджуваних зразків — 12 для досліджуваного лікарського засобу і 12 для референтного лікарського засобу;
точки контролю — не менше 3 точок (не враховуючи нульову), а саме: через 10 хв., 15 хв., 20 хв., 30 хв. та 45 хв., причому точка 15 хв. є визначальною;
відносне стандартне відхилення середнього результату або коефіцієнт варіації будь-якого лікарського засобу повинен бути менше 20% для першої точки і менше 10% від другої до останньої точки контролю;
середовища розчинення (буферні розчини): рН 1,0-1,2 (зазвичай 0,1N HCl або штучний шлунковий сік без ферментів), рН 4,5 і рН 6,8 (або штучна кишкова рідина без ферментів); (рН повинен гарантуватися впродовж всього дослідження; рекомендуються буферні розчини, описані у Державній фармакопеї України або Європейській фармакопеї); інші умови: відсутність поверхнево-активних речовин; у випадку желатинових капсул або таблеток з желатиновим покриттям може допускатися використання ферментів.
Необхідно надати результати порівняльних досліджень in vitro, що включають протокол випробовування та умови їх проведення, індивідуальні і середні результати та відповідну підсумкову статистику.
4.4.6. Оцінка результатів розчинення in vitro:
Лікарські засоби вважаються дуже швидкорозчинними, якщо не менше 85% від зазначеної у маркуванні кількості діючої речовини переходить у розчин за 15 хвилин або менше. У тому випадку, якщо це доведено для генеричного і референтного лікарських засобів, їх подібність може бути прийнята без будь-якого математичного обчислення.
Лікарські засоби вважаються швидкорозчинними, якщо не менше 85% від зазначеної у маркуванні кількості діючої речовини переходить у розчин за 30 хвилин.
Відсутність відповідних відмінностей (схожість) має бути продемонстрована у випадках, якщо потрібно більше 15 хвилин, але не більше 30 хвилин для досягнення майже повного розчинення (принаймні 85% від зазначеної у маркуванні кількості діючої речовини). Фактор подібності або інші відповідні тести повинні використовуватися для демонстрації профілю подібності досліджуваного та референтного лікарських засобів.
4.4.7. Допоміжні речовини:
Для лікарських засобів І класу за БСК рекомендується використовувати схожу кількість тих самих допоміжних речовин у складі генеричного лікарського засобу, як і у складі референтного лікарського засобу.
Якщо біовейвер застосовується до діючих речовин III класу за БСК, допоміжні речовини досліджуваного та референтного лікарських засобів мають бути якісно однаковими і кількісно дуже схожими, щоб виключити різні ефекти на мембранні білки-транспортери.
Допоміжні речовини, які можуть впливати на біодоступність (наприклад, сорбітол, манітол, натрію додецилсульфат або інші поверхнево-активні речовини), мають бути якісно і кількісно однаковими у досліджуваному та референтному лікарських засобах.
4.5. Загальні аспекти проведення тесту «Розчинення»:
Повинно бути достатньо точок відбору зразків для отримання значущих профілів розчинення, принаймні через кожні 15 хвилин. Рекомендується проводити частіший відбір зразків під час періоду найбільшої зміни в профілі розчинення. Для швидкорозчинних лікарських засобів, якщо повне розчинення відбувається протягом 30 хвилин, може бути необхідний відповідний профіль шляхом відбору зразків через кожні 5-10 хвилин.
Якщо більше ніж 85% лікарського засобу розчиняється протягом 15 хвилин, профілі розчинення можуть прийматися як подібні без подальшої математичної оцінки.
У разі якщо не менше 85% діючої речовини не переходить у розчин за 15 хвилин, а переходить у розчин за 30 хвилин, то потрібні принаймні три точки відбору зразків: перша точка відбору — до 15 хвилин, друга — 15 хвилин і третя, — коли вивільнення досягає близько 85%.
Для лікарських засобів з модифікованим вивільненням необхідно дотримуватися таких вимог:
профілі розчинення лікарських форм з відтермінованим вивільненням мають бути подібними в точках контролю, що відповідають механізму їх розчинення, тобто при рН=1,2 (протягом 2 год.) і при рН=6,8;
профілі розчинення форм з пролонгованим вивільненням мають бути подібними у трьох середовищах розчинення у точках контролю, які відповідають швидкості їх розчинення, наприклад, через 1 год., 2 год., 4 год., 6 год., 8 год. і 16 год.
Фактор подібності f2 обчислюють за формулою:
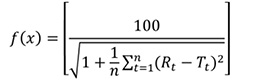
де :
f2 — фактор подібності;
n- кількість точок контролю,
R(t) — середнє значення кількості діючої речовини, що перейшла у розчин при кожній зазначеній точці контролю референтного препарату (у відсотках до значення, вказаного на упаковці),
T (t) — середнє значення кількості діючої речовини, що перейшла у розчин при кожній зазначений точці контролю досліджуваного препарату (у відсотках до значення, вказаного на упаковці).
Для референтного та генеричного лікарських засобів має визначатися кількість діючої речовини у відсотках від вказаного на етикетці.
Оцінка фактора подібності ґрунтується на таких умовах:
не менше трьох точок контролю (не враховуючи нульову);
точки контролю для досліджуваного і референтного лікарських засобів повинні збігатися;
кількість досліджуваних зразків — 12 повторюваностей для досліджуваного лікарського засобу і 12 — для референтного;
після розчинення 85% діючої речовини з референтного лікарського засобу до уваги беруться значення тільки в одній точці контролю;
відносне стандартне відхилення середнього значення для кожного з лікарських засобів має бути менше 20% у першій точці контролю і не більше 10%, починаючи з другої і до останньої точки контролю.
Профілі розчинення вважаються подібними, якщо фактор подібності f2≥50.
Необхідно надати задокументовані результати порівняльних досліджень in vitro, включаючи протокол випробовування, інформацію про досліджувану і референтну серії лікарських засобів, докладні дані щодо умов проведених випробувань, індивідуальні і середні результати та відповідну підсумкову статистику.
5. Додаткові випробування щодо підтвердження якості лікарського засобу та відтворюваності методів контролю проводяться відповідно до специфікації та методів контролю, наданих у матеріалах реєстраційного досьє.
5.1. Додаткові випробування щодо підтвердження якості лікарського засобу не проводяться у таких випадках:
після проведення клінічних випробувань (дослідження еквівалентності) за умови, що контроль якості вже проводився на досліджуваному лікарському засобі і специфікація, методи контролю якості та технологія виробництва лікарського засобу не зазнали змін;
при реєстрації додаткових доз лікарського засобу до вже зареєстрованого дозування за умови, що склад дозувань — пропорційноподібний, виробництво лікарського засобу відбувається на одному й тому самому обладнанні в умовах, передбачених ліцензією на виробництво цього лікарського засобу, проведено дослідження та надано звіт з валідації методів контролю та технологічного процесу щодо запропонованого додаткового дозування;
при одночасній реєстрації декількох дозувань лікарського засобу випробування з якості проводять лише для одного з дозувань за умови, що склад дозувань — пропорційноподібний, виробництво лікарського засобу відбувається на одному й тому самому обладнанні в умовах, передбачених ліцензією на виробництво цього лікарського засобу;
лікарський засіб має високу вартість (еквівалентну 500 і більше євро), при цьому постачання в Україну відбувається в обмежених об’ємах. У такому випадку контроль за показниками якості проводиться за процедурою «Lot Release» (шляхом експертизи матеріалів контролю якості серії лікарського засобу (протоколи контролю якості серії, аналітичні звіти), наданих виробником) (далі — «Lot Release»);
якщо для проведення випробування за окремими показниками якості (наприклад, «Стерильність», «Мікробіологічна чистота» тощо) необхідна велика кількість зразків, а вартість одного зразка — еквівалентна 100 і більше євро. У такому випадку контроль за цими показниками якості проводиться за процедурою «Lot Release»;
якщо для проведення випробування за окремими показниками якості у жодній з уповноважених лабораторій відсутнє необхідне обладнання для проведення контролю за такими показниками. У такому випадку контроль за цими показниками якості проводиться за процедурою «Lot Release»;
якщо лікарський засіб є лікарським засобом обмеженого застосування (препарат-сирота). У такому випадку контроль за показниками якості проводиться за процедурою «Lot Release»;
при реєстрації лікарського засобу під іншою назвою. У такому випадку до матеріалів реєстраційного досьє долучають результати лабораторного контролю якості, проведеного при реєстрації цього лікарського засобу з попередньою назвою;
при перереєстрації лікарського засобу, якщо протягом дії його реєстраційного посвідчення специфікація та методи контролю якості лікарського засобу не змінювались і заявником не вносились та не вносяться зміни до матеріалів реєстраційного досьє у частині, що стосується специфікації та методів контролю якості лікарського засобу;
при реєстрації (перереєстрації) АФІ або діючих речовин;
для лікарських засобів, виготовлених з лікарського засобу у формі in bulk шляхом фасування (пакування)/маркування, за умови, що для лікарського засобу у формі in bulk було проведено передреєстраційний контроль якості. У такому випадку до матеріалів реєстраційного досьє долучаються результати лабораторного контролю якості лікарського засобу у формі in bulk;
при внесенні змін до інструкції для медичного застосування лікарського засобу (зміни терапевтичних показань, дозуючого пристрою тощо);
при реєстрації лікарського засобу за процедурою «заява інформованої згоди» за умови наявності на ринку України власника реєстраційного посвідчення зареєстрованого раніше лікарського засобу. У такому випадку до матеріалів реєстраційного досьє долучаються результати лабораторного контролю якості зареєстрованого раніше лікарського засобу.
5.2. Додаткові випробування з контролю активності антибіотиків не проводяться у таких випадках:
у матеріалах реєстраційного досьє наведено власні дослідження in vitro з контролю активності антибіотиків;
у матеріалах реєстраційного досьє представлено доказ терапевтичної еквівалентності лікарського засобу шляхом проведення клінічних випробувань.
5.3. При реєстрації та перереєстрації медичного імунобіологічного препарату проводиться контроль однієї серії цього препарату на відповідність вимогам МКЯ.
6. Звіти щодо проведених додаткових випробувань заявник надає до Центру для проведення спеціалізованої експертизи.
XXI. Порядок видачі реєстраційного посвідчення (вкладки до реєстраційного посвідчення) на лікарський засіб
1. У разі складання Центром умотивованих висновків щодо ефективності, безпеки та якості, Центр протягом 10 робочих днів готує та надає заявнику для редакційного узгодження проекти таких документів:
при державній реєстрації (перереєстрації) лікарського засобу: реєстраційне посвідчення, інструкції для медичного застосування лікарського засобу та короткої характеристики лікарського засобу (за її наявності), МКЯ, що містить маркування первинної, вторинної (за наявності) упаковок лікарського засобу;
при внесенні змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення на лікарській засіб: вкладки до реєстраційного посвідчення, змін та оновленої редакції інструкції для медичного застосування лікарського засобу та короткої характеристики лікарського засобу, структуру якої наведено у додатку 14 до цього Порядку (за її наявності), змін та оновленої редакції МКЯ, що містить маркування первинної, вторинної (за наявності) упаковок лікарського засобу.
Інформація про видачу заявнику для редакційного узгодження проектів документів вноситься до електронної бази даних.
Якщо заявник не узгоджує редакцію проектів документів протягом 15 календарних днів, Центр передає ці документи до МОЗ.
Час, необхідний для узгодження заявником редакції документів, не входить до строку проведення експертизи матеріалів реєстраційного досьє.
2. Протягом 3 робочих днів після прийняття МОЗ рішення про державну реєстрацію (перереєстрацію) лікарського засобу або про внесення змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення Центр повідомляє заявника про прийняте рішення та передає узгоджені із заявником реєстраційні посвідчення або вкладки до реєстраційних посвідчень у МОЗ для їх засвідчення підписом керівника відповідального структурного підрозділу МОЗ.
3. Реєстраційне посвідчення (вкладка до реєстраційного посвідчення) протягом трьох робочих днів з дати отримання підписується керівником відповідального структурного підрозділу МОЗ та засвідчується печаткою. Оформлені реєстраційні посвідчення (вкладка до реєстраційного посвідчення) разом із затвердженими МОЗ реєстраційними документами (інструкцією для медичного застосування лікарського засобу або змінами до неї; короткою характеристикою лікарського засобу (за наявності — на бажання заявника); фармакопейною статтею або МКЯ чи змінами до них, що містять маркування первинної, вторинної (за наявності) упаковок на лікарський засіб) передаються для видачі заявнику за принципом «єдиного вікна».
4. Реєстраційне посвідчення із зазначенням строку, протягом якого дозволяється застосування лікарського засобу в Україні (вкладка до реєстраційного посвідчення), видається МОЗ у міру звернення заявників за наявності доручення від заявника на отримання зазначених документів.
XXII. Порядок розрахунків
1. Оплаті підлягають попередня експертиза та спеціалізована експертиза матеріалів реєстраційного досьє для державної реєстрації (перереєстрації), внесення змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення лікарського засобу, додаткові вивчення (випробування).
2. Оплата вищезазначених робіт проводиться відповідно до умов укладеного договору між заявником та Центром або між заявником та закладом охорони здоров’я/установою, що проводить такі вивчення (випробування).
XХIІІ. Захист конфіденційної інформації
1. Під час проведення експертизи матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення Центр зобов’язаний забезпечити захист конфіденційної реєстраційної інформації від розголошення і недобросовісного комерційного використання.
2. Не допускається ознайомлення третіх осіб з конфіденційною реєстраційною інформацією, зняття копій з таких матеріалів на паперових, електронних або інших носіях без письмової згоди власника такої інформації чи в інших випадках, встановлених чинним законодавством.
3. Обіг документів, що містять конфіденційну реєстраційну інформацію, здійснюється в Центрі.
4. Не допускаються до роботи з документами, що містять конфіденційну реєстраційну інформацію, особи, які можуть мати конфлікт інтересів із заявником.
| Начальник Управління лікарських засобів та медичної продукції |
Л.В. Коношевич |
Додаток 1
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення змін
до реєстраційних матеріалів протягом
дії реєстраційного посвідчення
ЗАЯВА
про державну реєстрацію лікарського засобу
Дата надходження заяви
«___» __________ 20__ року № ______________________
| Назва лікарського засобу | |
| Діюча(і) речовина(и) або комбінований лікарський засіб | |
| Сила(и) дії (дозування) | |
| Лікарська форма та упаковка | |
| Заявник | |
| Уповноважена особа, що виступає від імені заявника |
Я гарантую достовірність та відповідаю за інформацію, що міститься у наданих матеріалах реєстраційного досьє, відповідно до додатка ___.
Згоден, що у разі ненадання матеріалів реєстраційного досьє протягом трьох місяців відповідно до додатка ___ у розгляді цієї заяви буде відмовлено.
Усі дані одержано заявником в установленому законодавством порядку і не порушують права третьої сторони, захищені патентом, свідоцтвом про знак для товарів та послуг (пункти 4.16 — 4.18 цього додатка).
Також цим підтверджується, що всі передбачені збори сплачено/буде сплачено відповідно до вимог законодавства.
Доручення для ведення переговорів/підписання документів від імені заявника додається (пункт 4.4 цього додатка).
| Від імені заявника | |
|
____________________________________(підпис) _____________________________________(ім’я та прізвище) |
|
| М.П. | _____________________________________(посада) |
1. ЗАГАЛЬНІ ПУНКТИ ЗАЯВИ ПРО ДЕРЖАВНУ РЕЄСТРАЦІЮ
1.1. Ця заява подається відповідно до такого:
Примітка. Розділ повинен бути заповнений для будь-якої заяви, уключаючи заяви, на які є посилання у цьому розділі.
У разі надання заяви на відповідний тип вилучають перелік інших видів заяв, на які не поширюється дана заява.
Повна і незалежна заява/автономна заява
|
Медичний імунобіологічний препарат (абзаци сорок п’ятий та сорок шостий розділу ІІ Порядку) |
Інший лікарський засіб |
(надається реєстраційне досьє з адміністративними, фармацевтичними, доклінічними та клінічними даними)
Нова діюча речовина (далі — ДР)
Примітка . ДР не зареєстрована в Україні.
Відома ДР
Примітка. ДР була зареєстрована раніше.
Заява НА лікарський засіб, який має добре вивчене медичне застосування
(надається реєстраційне досьє з повними адміністративними, фармацевтичними даними та докладною науковою бібліографією, що свідчить про ефективність та безпеку діючої речовини (згідно з вимогами розділів ІХ та Х Порядку).
Заява інформованої згоди
(надається реєстраційне досьє з адміністративними, фармацевтичними, доклінічними та клінічними даними разом зі згодою власника реєстраційного посвідчення зареєстрованого раніше лікарського засобу використовувати його фармацевтичні, доклінічні та клінічні дані)
|
Медичний імунобіологічний препарат (абзаци сорок п’ятий та сорок шостий розділу ІІ Порядку) |
Інший лікарський засіб |
Примітка. Зареєстрований лікарський засіб і заява інформованої згоди можуть мати як одного, так і різного(их) власника(ів) реєстраційного посвідчення.
Зареєстрований лікарський засіб:
| назва лікарського засобу, сила дії, лікарська форма | |
| власник реєстраційного посвідчення | |
| номер(и) реєстраційного посвідчення |
Додайте інформовану згоду власника реєстраційного посвідчення на зареєстрований лікарський засіб (пункт 4.2 цього додатка).
Заява на генеричний лікарський засіб
| Однокомпонентний | Багатокомпонентний |
(надається реєстраційне досьє з адміністративними, фармацевтичними даними, уключаючи еквівалентність до референтного лікарського засобу, доклінічними (літературний огляд) та клінічними (власні дані або літературний огляд*) даними (згідно з вимогами розділів ІХ та Х Порядку)
____________
* Залежно від лікарської форми.
Референтний лікарський засіб:
| назва лікарського засобу, сила дії, лікарська форма | |
| власник реєстраційного посвідчення | |
| номер(и) реєстраційного посвідчення |
Лікарський засіб, що використовувався у дослідженнях еквівалентності (якщо такі проводилися):
| назва лікарського засобу, сила дії, лікарська форма | |
| власник реєстраційного посвідчення | |
| номер(и) реєстраційного посвідчення |
Примітка. Розділ необхідно заповнювати для кожного лікарського засобу, що використовувався у дослідженнях еквівалентності.
Заява НА Подібний біологічний лікарський засіб (біосиміляр)
|
Медичний імунобіологічний препарат (абзаци сорок п’ятий та сорок шостий розділу ІІ Порядку) |
Інший лікарський засіб |
(надається реєстраційне досьє з повними адміністративними, фармацевтичними, хімічними, біологічними даними та відповідні дані, що підтверджують належний рівень безпеки (токсикологічні чи інші доклінічні дані) та ефективності (клінічні дані (згідно з вимогами розділів ІХ та Х Порядку).
Референтний біологічний лікарський засіб:
| назва лікарського засобу, сила дії, лікарська форма | |
| власник реєстраційного посвідчення | |
| номер(и) реєстраційного посвідчення |
Відмінності порівняно із референтним біологічним лікарським засобом (якщо такі є):
відмінності у вихідному(их) матеріалі(ах);
відмінності у виробничому процесі;
інше терапевтичне застосування;
відмінності у лікарській формі;
інша(і) сила(и) дії (кількісні зміни ДР);
інший спосіб(оби) введення;
інші відмінності __________________________________________
__________________________________________
ГІБРИДНА ЗАЯВА
(у випадках, коли лікарський засіб не підпадає під визначення генеричного лікарського засобу, або коли біоеквівалентність не може бути продемонстрована під час досліджень біодоступності, або у випадку відмінностей у діючій речовині, дозуванні, лікарській формі, шляху введення тощо надаються матеріали реєстраційного досьє з повними адміністративними, фармацевтичними даними та Модулі 4 та 5, у яких містяться звіти про обмежені доклінічні дослідження та клінічні випробування, що проводилися заявником, і бібліографічні посилання.
Зареєстрований лікарський засіб:
| назва лікарського засобу, сила дії, лікарська форма | |
| власник реєстраційного посвідчення | |
| номер(и) реєстраційного посвідчення |
Відмінності порівняно із зареєстрованим лікарським засобом:
інша лікарська форма;
інша(і) сила(и) дії (кількісні зміни ДР);
інший спосіб(оби) введення;
інша фармакокінетика (уключаючи іншу біодоступність);
інше терапевтичне застосування;
інші відмінності __________________________________________
__________________________________________
Заява на фіксовану комбінацію
(надається реєстраційне досьє з адміністративними, фармацевтичними, доклінічними та клінічними даними тільки щодо цієї комбінації (згідно з вимогами розділів ІХ та Х Порядку).
Заява на ЛІКАРСЬКИЙ ЗАСІБ З ПРОДУКЦІЇ in bulk
|
Медичний імунобіологічний препарат (абзаци сорок п’ятий та сорок шостий розділу ІІ Порядку) |
Інший лікарський засіб |
(надається реєстраційне досьє з адміністративними, фармацевтичними (виробника продукції in bulk, доповненими документацією виробника щодо стадій, які ним виконуються), доклінічними (виробника продукції in bulk) та клінічними (виробника продукції in bulk) даними (згідно з вимогами розділів ІХ та Х Порядку)
Заява на зміни, що потребують нової реєстрації
(надаються відповідні розділи матеріалів реєстраційного досьє, які обґрунтовують указані зміни, і є достатніми для проведення експертизи)
Позначити необхідне:
Зміни активних речовин:
заміна активної речовини на іншу сіль, ефір, похідну тощо за умови, що характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
використання інших ізомерів або зміна ізомерного складу;
незначні зміни молекулярної структури речовин біологічного походження за винятком зміни в активній речовині вакцин для профілактики грипу;
зміна вектора, який використовується для виробництва антигена або вихідного матеріалу, включаючи новий головний банк клітин від іншого джерела, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
новий ліганд або механізм з’єднання для радіофармацевтичного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
застосування інших екстрагентів або співвідношення рослинна лікарська сировина/рослинний препарат для рослинного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих.
Зміни сили дії, лікарської форми та способу застосування:
зміна біодоступності;
зміна фармакокінетики;
зміна або введення додаткового дозування лікарського засобу (додаткова доза);
зміна або додавання нової лікарської форми;
зміна або додавання нового шляху введення (для парентеральних форм у зв’язку з відмінностями в ефективності та безпеці лікарського засобу при внутрішньоартеріальному, внутрішньовенному, внутрішньом’язовому та інших шляхах введення).
Лікарський засіб, зареєстрований в Україні, до якого вносяться відповідні зміни
| назва лікарського засобу, сила дії, лікарська форма | |
| власник реєстраційного посвідчення | |
| номер(и) реєстраційного посвідчення |
Заява на препарат обмеженого застосування (препарат-СИРОТА)
|
Медичний імунобіологічний препарат (абзаци сорок п’ятий та сорок шостий розділу ІІ Порядку) |
Інший лікарський засіб |
1. Чи надано лікарському засобу статус препарату обмеженого застосування (препарату-сироти)
Ні
У процесі розгляду
Так
| дата | |
| реєстраційний номер препарату обмеженого застосування (препарату-сироти) |
Відмовлено у присвоєнні статусу препарату обмеженого застосування (препарату-сироти)
| дата | |
| номер рішення |
Заяву на присвоєння статусу відкликано
| дата |
Додайте копію рішення щодо присвоєння статусу препарату обмеженого застосування (препарату-сироти) (якщо є) (пункт 4.15 цього додатка).
2. ОСОБЛИВІ ПУНКТИ ЗАЯВИ ПРО ДЕРЖАВНУ РЕЄСТРАЦІЮ
2.1. Назва та код АТХ
2.1. Назва та код АТХ
2.1.1. Назва лікарського засобу
_____________________________________________________________________________________________
2.1.2. Назва ДР або склад (указати)
______________________________________________________________________________________________
Примітка. Тільки одна назва повинна бути наведена в такому порядку: Міжнародна непатентована назва (далі — МНН)*, Державна фармакопея України (далі — ДФУ), Європейська фармакопея, загальноприйнята назва, наукова (хімічна) назва.
* Назву діючої речовини необхідно вказувати за її рекомендованою МНН з указанням її солей або гідратної форми, якщо необхідно
2.1.3. Фармакотерапевтична група (використовуйте діючий код АТХ)
Код АТХ ____________________ Група _____________________
Якщо код АТХ не присвоєно, зазначте чи була подана заява на присвоєння коду АТХ
Малюнок 1
2.2. Сила дії (дозування), лікарська форма та упаковка, шлях введення, об’єм контейнера та розмір упаковки
2.2.1. Сила дії (дозування) і лікарська форма (використовуйте перелік стандартних термінів ДФУ або Європейської фармакопеї)
| Лікарська форма | |
| Діюча(і) речовина(и) або комбінований лікарський засіб | |
| Сила(и) дії (дозування) |
2.2.2. Шлях(и) введення (використовуйте перелік стандартних термінів ДФУ або Європейської фармакопеї)____________________________________________________________________________________
2.2.3. Упаковка: контейнер, укупорювальна система та пристрої для введення, уключаючи опис матеріалу, з якого вони виготовлені (використовуйте перелік стандартних термінів ДФУ або Європейської фармакопеї)
_____________________________________________________________________________________________
Для кожного виду упаковки вкажіть:
2.2.3.1. Розмір(и) упаковки.
2.2.3.2. Пропонований термін придатності.
2.2.3.3. Пропонований термін придатності (після першого розкриття упаковки/контейнера).
2.2.3.4. Пропонований термін придатності (після відновлення/розчинення або розведення).
2.2.3.5. Пропоновані умови зберігання.
2.2.3.6. Пропоновані умови зберігання після першого розкриття упаковки.
Додайте пропозиції до маркування на упаковці (пункт 4.14 цього додатка)
2.3. Правовий статус
2.3.1. Пропонована категорія відпуску:
за рецептом
без рецепта
тільки в умовах стаціонару
2.3.2. Для лікарських засобів, пропонованих для рецептурного відпуску, заявник надає свої пропозиції щодо категорії відпуску лікарського засобу, однак право визначати категорію відпуску залишається за МОЗ України з урахуванням вимог наказу Міністерства охорони здоров’я України від 17 травня 2001 року № 185 «Про затвердження критеріїв визначення категорій відпуску лікарських засобів», зареєстрованого у Міністерстві юстиції України 31 травня 2001 року за № 464/5655.
2.3.3. Пропозиції щодо рекламування лікарських засобів, що відпускаються без рецепта:
рекламування тільки серед медичних працівників
рекламування для населення та медичних працівників
2.4. Заявник (власник реєстраційного посвідчення)/контактні особи
2.4.1. Заявник (власник реєстраційного посвідчення):
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| країна | |
| телефон/факс | |
2.4.2. Представник заявника (уповноважена особа, що виступає від імені заявника):
| прізвище, ім’я, по батькові уповноваженої особи представника заявника | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| країна | |
| телефон/факс | |
додайте доручення (пункт 4.4 цього додатка).
2.4.3. Представник заявника (уповноважена особа, що виступає від імені заявника) після реєстрації лікарського засобу, якщо вони відмінні від визначених у пункті 2.4.2:
| прізвище, ім’я, по батькові уповноваженої особи представника заявника | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| телефон/факс | |
У разі заповнення цього пункту додайте доручення (пункт 4.4 цього додатка).
2.4.4. Уповноважена кваліфікована особа заявника, відповідальна за фармаконагляд:
| прізвище, ім’я кваліфікованої особи заявника, відповідальної за фармаконагляд | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (заявника) | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця (заявника) | |
| країна | |
| цілодобовий телефон/факс | |
Додайте біографічну довідку уповноваженої кваліфікованої особи, відповідальної за фармаконагляд (пункт 4.5 цього додатка), та гарантійний лист заявника (пункт 4.21 цього додатка).
Укажіть місце проживання уповноваженої кваліфікованої особи заявника, відповідальної за фармаконагляд
2.4.5. Уповноважена кваліфікована особа заявника в Україні для здійснення фармаконагляду, якщо вона відмінна від зазначеної у пункті 2.4.4:
| прізвище, ім’я, по батькові уповноваженої кваліфікованої особи заявника, відповідальної за фармаконагляд | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (заявника) | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця (заявника) | |
| країна | |
| цілодобовий телефон/факс | |
Якщо відрізняється від пункту 2.4.4, додайте біографічну довідку уповноваженої кваліфікованої особи в Україні для здійснення фармаконагляду (пункт 4.5 цього додатка).
Укажіть місце проживання уповноваженої кваліфікованої особи в Україні для здійснення фармаконагляду
2.5. Виробники
Усі дані щодо виробничих дільниць, на яких здійснюються виробничі процедури, та дільниць, на яких проводиться контроль якості, що зазначаються у матеріалах реєстраційного досьє, мають збігатися щодо їх найменувань, місцезнаходжень та видів діяльності.
2.5.1. Виробник(и), що відповідає(ють) за випуск серії (як указано в інструкції про застосування лікарського засобу (інструкції для медичного застосування) та, за необхідності, у маркуванні)
| найменування юридичної особи або прізвище ім’я, по батькові фізичної особи — підприємця | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
Додайте копію ліцензії на виробництво (пункт 4.6 цього додатка).
Додайте копію сертифіката відповідності вимогам належної виробничої практики, передбаченого у пункті 4.8 цього додатка.
2.5.2. Лабораторія країни виробника з контролю якості препаратів крові та вакцин, відповідальна за контроль якості/випуск серії
| найменування лабораторії | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
2.5.3. Контактна особа, відповідальна за роботу з рекламаціями щодо дефектної продукції
| прізвище, ім’я контактної особи, відповідальної за роботу з рекламаціями щодо дефектної продукції | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| країна | |
| цілодобовий телефон/факс | |
2.5.4. Виробник(и) лікарського засобу і дільниця(і) виробництва:
Включаючи дільниці виробництва будь-якого розріджувача/розчинника в окремій упаковці/ємності, що є частиною лікарського засобу, дільниці, на яких здійснюється контроль якості/контроль у процесі виробництва, а також (за потреби) імпортерів
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
Короткий опис виконуваних функцій
_____________________________________________________________________________________________
Додайте технологічну схему виробництва (пункт 4.7 цього додатка).
Додайте копію сертифіката відповідності вимогам належної виробничої практики, передбаченого у пункті 4.8 цього додатка.
Додайте копію ліцензії на виробництво (пункт 4.6 цього додатка).
Зазначте прізвище та ім’я уповноваженої особи виробника (якщо не вказано у ліцензії на виробництво)
______________________________________________________________________________________________
Чи проводилася перевірка дільниці на дотримання вимог належної виробничої практики (далі — GMP) уповноваженим органом України або органами країн відповідно до міжнародних договорів про взаємне визнання результатів інспектування на відповідність виробництва вимогам GMP?
Ні Так
Якщо так, укажіть:
| дату останньої перевірки GMP | |
| найменування уповноваженого органу, що проводив перевірку | |
| вид перевірки | |
| категорію лікарських засобів, що перевіряються, і діючих речовин |
висновок:
відповідає GMP: Ні Так
Додайте для кожної дільниці виробництва висновок про відповідність вимогам GMP відповідно до пункту 4.8 цього додатка.
2.5.5. Виробник(и) ДР та виробничі дільниці
Повинні бути зазначені всі дільниці виробництва, задіяні у виробничому процесі кожного джерела діючої речовини, включаючи дільниці, на яких проводиться контроль якості/контроль у процесі виробництва. Для високотехнологічних (біотехнологічних) лікарських засобів мають бути включені усі дільниці зберігання головного та робочого банків клітин та дільниці приготування робочого банку клітин.
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
Короткий опис етапів виробництва, виконуваних на виробничій дільниці
______________________________________________________________________________________________
Додайте технологічну схему виробництва (пункт 4.7 цього додатка).
Для кожної ДР додайте заяву уповноваженої особи виробника згідно з пунктом 4.22 цього додатка.
Чи проводилася перевірка дільниці на дотримання вимог GMP уповноваженим органом України або органами країн відповідно до міжнародних договорів про взаємне визнання результатів інспектування на відповідність виробництва вимогам GMP?
Ні Так
Додайте для кожної дільниці виробництва документ, передбачений пунктом 4.6 цього додатка,
та, за наявності,
Додайте копію сертифіката відповідності вимогам належної виробничої практики відповідно до пункту 4.8 цього додатка.
• Чи видано сертифікат відповідності Європейській фармакопеї для ДР?
Ні Так
Якщо так:
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| номер сертифіката | |
| дата останнього перегляду |
Додайте копію сертифіката відповідності (пункт 4.9 цього додатка).
• Чи є матеріали реєстраційного досьє (мастер-файл) на ДР, посилання на які буде використовуватися у досьє, оригінальними?
Ні Так
Якщо так:
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| ідентифікаційний номер або реєстраційний номер облікової картки платника податків, або серія та номер паспорта (для фізичних осіб, які через свої релігійні переконання відмовляються від прийняття реєстраційного номера облікової картки платника податків та повідомили про це відповідний орган державної податкової служби і мають відмітку у паспорті) | |
| дата подання на розгляд | |
| дата останнього перегляду |
Додайте лист(и) виробника(ів) ДР, що дає(ють) право використання мастер-файла на ДР (пункт 4.9 цього додатка).
Додайте копію письмового зобов’язання від виробника(ів) ДР інформувати заявника про зміни у виробничому процесі або специфікаціях (пункт 4.10 цього додатка).
• Чи видано сертифікат на загальний файл на вакцинний антиген (далі — ВАЗФ), що використовується у цій заяві, або на нього подано заявку?
Ні Так
Якщо так:
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (власника сертифіката на ВАЗФ) | |
| номер заявки/сертифіката | |
| дата подання (якщо на розгляді) | |
| дата затвердження або останнього перегляду (якщо наявний) |
Додайте копію сертифіката на ВАЗФ (пункт 4.19 цього додатка)
2.5.6. Заклади охорони здоров’я/установи, що залучалися до клінічного(их) випробування(нь) для дослідження біодоступності та/або біоеквівалентності, або такі, що залучалися для валідації процесів виробництва продуктів крові
Для кожного закладу охорони здоров’я/установи вкажіть, де проводилися аналітичні тестування та де зібрано клінічні дані, а також:
| назва дослідження | |
| код протоколу | |
| номер EudraCT (за наявності) | |
| найменування закладу охорони здоров’я/установи | |
| місцезнаходження закладу охорони здоров’я/установи | |
| країна | |
| телефон/факс | |
Обов’язки відповідно до договору____________________________________________________________
2.6. Якісний та кількісний склад лікарського засобу
2.6.1. Якісний та кількісний склад лікарського засобу (ДР та допоміжні речовини):
Необхідно вказати, на яку кількість розраховано склад (наприклад, 1 капсула).
Перерахуйте діючі речовини окремо від допоміжних речовин:
| Назва ДР* | Кількість | Одиниця | Посилання/монографія |
| 1. | |||
| 2. | |||
| 3. | |||
| Тощо |
| Назва допоміжної(их) речовини(н) | Кількість | Одиниця | Посилання/монографія |
| 1. | |||
| 2. | |||
| 3. | |||
| Тощо |
Примітка. *Тільки одну назву для кожної ДР необхідно вказати в такій послідовності: МНН**, ДФУ, Європейська фармакопея, загальноприйнята назва, наукова (хімічна) назва.
**Назву діючої речовини необхідно вказувати за її рекомендованою МНН з указанням її солей або гідратної форми, якщо необхідно.
Інформацію про надлишкову кількість не слід указувати в колонках щодо складу, а необхідно викласти нижче:
діюча(і) речовина(и) _________________________________________________________________________
допоміжна(і) речовина(и)___________________________________________________________________
2.6.2. Перелік матеріалів тваринного та/або людського походження, що входять до складу або використовуються у процесі виробництва лікарського засобу
ВІДСУТНІ
| Назва | Функція | Тваринного походження, сприятливі до ГЕ4 | Інші тваринного походження | Людського походження | Сертифікат відповідності ЄФ5 щодо ГЕ4 (укажіть номер) | ||
| ДР1 | ДОП.Р2 | Р3 | |||||
| 1. | |||||||
| 2. | |||||||
| 3. | |||||||
| Тощо | |||||||
Якщо є сертифікат відповідності ЄФ5 щодо ГЕ4 або документ, що видано уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків ГЕ4, він повинен бути доданий у пункті 4.11 цього додатка.
1 ДР — діюча речовина.
2 ДОП.Р — допоміжна речовина (уключаючи вихідні матеріали, які використовуються у виробництві діючої/допоміжної речовини).
3 Р — реагент/середовище культивування (уключаючи використовувані для приготування головного та робочого банків клітин).
4 ГЕ — губчата енцефалопатія.
5 ЄФ — Європейська фармакопея.
2.6.3. Чи видано сертифікат на матеріали реєстраційного досьє (мастер-файл) на плазму (далі — ПМФ), що використовується у цій заяві на реєстрацію, або на нього подано заявку?
Ні Так
Якщо так:
| речовина (субстанція) з посиланням на ПМФ |
Функція
| ДР1 | ДОП.Р2 | Р3 | ||
| найменування власника/заявника ПМФ | ||||
| номер сертифіката/заявки | ||||
| дата подання заявки (якщо на розгляді) | ||||
| дата затвердження або останнього перегляду (якщо сертифікат наявний) | ||||
1 ДР — діюча речовина.
2ДОП.Р — допоміжна речовина (уключаючи вихідні матеріали, які використовуються у виробництві діючої/допоміжної речовини).
3 Р — реагент/середовище культивування (уключаючи використовувані для приготування головного та робочого банків клітин).
Додайте копію сертифіката (пункт 4.20 цього додатка).
2.6.4. Чи містить або складається лікарський засіб з генетично модифікованих організмів (ГМО)?
НіТак
Якщо так, то чи відповідає лікарський засіб встановленим вимогам?
Зробіть необхідне посилання
НіТак
Додайте копію письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою (пункт 4.12 цього додатка).
3. ІНШІ ВІДОМОСТІ
3.1. Чи захищено лікарський засіб патентами на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну?
Ні Так
Якщо так, то наведіть таку інформацію:
| Номер патенту | Дата видачі | Діє до | Власник патенту |
Додайте копії патентів згідно з пунктом 4.16 цього додатка.
Для державної реєстрації лікарських засобів, що базуються або мають відношення до об’єктів інтелектуальної власності, на які відповідно до законів України видано патент, заявник подає засвідчену відповідно копію патенту або ліцензії, якою дозволяється виробництво та продаж зареєстрованого лікарського засобу, а також документ, що підтверджує чинність патенту в Україні. Заявники подають лист, в якому вказується, що права третьої сторони, захищені патентом або передані за ліцензією, не порушуються у зв’язку з реєстрацією лікарського засобу.
3.2. Чи захищена торговельна марка в Україні?
Ні Так
Якщо так, то наведіть таку інформацію:
| Номер документа | Дата видачі | Діє до | Власник документа |
Додайте копії документів, що передбачені в пункті 4.17 цього додатка.
3.3. Чи зареєстровано лікарський засіб у країні виробника та інших країнах?
Ні Так
Якщо так:
Додайте копію реєстраційного посвідчення (пункт 4.3 цього додатка).
Зазначте перелік країн, де лікарський засіб зареєстрований/перереєстрований.
3.4. Чи була проведена попередня наукова консультація щодо цього лікарського засобу в Україні?
Ні Так
Якщо так:
| дата проведення | |
| посилання на наукові рекомендації |
Чи була проведена попередня наукова консультація щодо цього лікарського засобу в іншій країні?
Ні Так
Якщо так:
| країна | |
| дата проведення | |
| посилання на наукові рекомендації |
Додайте копію консультаційного листа (пункт 4.13 цього додатка)
4. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхідне)
4.1. Документ, що підтверджує сплату реєстраційного збору.
4.2. Інформована згода власника реєстраційного посвідчення на зареєстрований лікарський засіб (у довільній формі).
4.3. Копія реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розміщено лікарський засіб, із зазначенням назви документа та назви компетентного органу, що його видав, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу (для продукції in bulk — підтвердження реєстрації лікарського засобу, виготовленого із заявленої продукції in bulk, у споживчій упаковці в країні виробника або іншій країні, регуляторний орган якої керується високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розмішено зазначений лікарський засіб). Копія має бути засвідчена печаткою заявника/представника заявника в Україні. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано.
4.4. Доручення для ведення переговорів/підписання документів від імені заявника (власника реєстраційного посвідчення).
4.5. Біографічна довідка уповноваженої кваліфікованої особи, відповідальної за здійснення фармаконагляду (у довільній формі).
4.6. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника. Копія повинна бути засвідчена печаткою заявника (для фізичної особи за наявності)/представника заявника в Україні.
4.7. Проект технологічного регламенту або відомості про технологію виробництва (технологічна схема виробництв із зазначенням усіх дільниць виробництва, задіяних у процесі виробництва лікарського засобу та діючої речовини (уключаючи дільниці для відбору зразків і контролю серій лікарського засобу). Копія повинна бути засвідчена печаткою заявника (для фізичної особи за наявності)/представника заявника в Україні.
4.8. Засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи. За необхідності — висновки щодо інших інспекцій, які проводилися. Копії документів мають бути засвідчені печаткою заявника (для фізичної особи за наявності)/представника заявника в Україні.
4.9. Письмова згода на використання матеріалів реєстраційного досьє (мастер-файл) на діючу речовину (у довільній формі) від її виробника або копія сертифіката відповідності Європейській фармакопеї.
4.10. Копія письмового зобов’язання виробника діючої речовини (у довільній формі) інформувати заявника про зміни у виробничому процесі або специфікаціях.
4.11. Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків губчатої енцефалопатії.
4.12. Копія письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою.
4.13. Копія консультаційного листа до проведеної попередньої наукової консультації щодо лікарського засобу.
4.14. Пропозиції до маркування упаковки лікарського засобу.
4.15. Копія рішення про присвоєння статусу препарату обмеженого застосування (препарату-сироти).
4.16. Копії патентів на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну.
4.17. Копії документів щодо захисту торговельної марки в Україні.
4.18. Лист, форма якого наведена у додатку 15 до цього Порядку.
4.19. Копія сертифіката на ВАЗФ.
4.20. Копія сертифіката на ПМФ.
4.21. Гарантійний лист заявника (у довільній формі) щодо забезпечення функціонування належної системи нагляду за безпекою лікарських засобів при їх медичному застосуванні, у тому числі в Україні.
4.22. Для кожної діючої речовини дільниць виробництва, задіяних у виробничому процесі, включаючи дільниці, на яких проводиться контроль якості/контроль у процесі виробництва та випуск серій, заява уповноваженої особи щодо відповідності виробництва вимогам належної виробничої практики та Керівництвам з належної виробничої практики щодо вихідних матеріалів.
Додаток 2
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
Заява
про державну реєстрацію (перереєстрацію) лікарського засобу
Дата надходження заяви
«___» ____________ 20___ року № ________________
1. Назва лікарського засобу
_______________________________________________________________________________
2. Лікарська форма, сила(и) дії (дозування)
_______________________________________________________________________________
3. Упаковка:
первинна
_______________________________________________________________________________
вторинна
_______________________________________________________________________________
4. Заявник (власник реєстраційного посвідчення) (для вітчизняних заявників — українською, для зарубіжних — українською та англійською мовами).
Найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця
_______________________________________________________________________________
Місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця
_______________________________________________________________________________
Телефон/факс
_______________________________________________________________________________
_______________________________________________________________________________
Керівник
_______________________________________________________________________________
4.1. Представник заявника (уповноважена особа, що виступає від імені заявника) (українською мовою):
Прізвище, ім’я, по батькові уповноваженої особи представника заявника
_______________________________________________________________________________
Найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця
_______________________________________________________________________________
Місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця
_______________________________________________________________________________
Телефон/факс
_______________________________________________________________________________
_______________________________________________________________________________
4.2. Уповноважена кваліфікована особа заявника для здійснення фармаконагляду в Україні (українською мовою):
Прізвище, ім’я, по батькові кваліфікованої особи заявника, відповідальної за фармаконагляд
_______________________________________________________________________________
Найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (заявника)
_______________________________________________________________________________
Місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця (заявника)
_______________________________________________________________________________
Цілодобовий телефон/факс
_______________________________________________________________________________
_______________________________________________________________________________
5. Виробник(и) (для вітчизняних виробників — українською, для зарубіжних — українською та англійською мовами)
_______________________________________________________________________________
5.1. Виробник(и) лікарського засобу
Найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця
_______________________________________________________________________________
Адреса місця провадження діяльності
_______________________________________________________________________________
Телефон/факс
_______________________________________________________________________________
_______________________________________________________________________________
Керівник
_______________________________________________________________________________
Виробництво лікарського засобу (потрібне позначити):
| повністю даним виробником | |
| частково даним виробником | |
| повністю іншим виробником |
5.2. Виробник(и) діючої(их) речовини(н)
Діюча речовина
_______________________________________________________________________________
Найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника)
_______________________________________________________________________________
Адреса місця провадження діяльності
_______________________________________________________________________________
Телефон/факс
_______________________________________________________________________________
_______________________________________________________________________________
Керівник
_______________________________________________________________________________
6. Якісний і кількісний склад лікарського засобу (діючі та допоміжні речовини)
Укажіть діючу(і) речовину(и) окремо від допоміжних
| Назва речовини* | Кількість на одиницю лікарської форми** | Посилання/монографія |
____________
* Повинна вказуватися тільки одна назва в такому порядку: міжнародна непатентована назва, Державна фармакопея України, Європейська фармакопея, загальноприйнята назва, наукова (хімічна) назва. Діюча речовина повинна бути заявлена за рекомендованою міжнародною непатентованою назвою із зазначенням форми солі або гідрату, якщо необхідно.
** В одиницях ваги чи біологічних одиницях на 1 одиницю лікарської форми: драже, таблетки, супозиторії, ампули, флакони; у відсотках чи мг/мл, мг/г: мазі, креми, розчини, неподільні порошки, збори.
7. Фармакотерапевтична група
_______________________________________________________________________________
8. Код АТХ або пропозиції щодо нього
_______________________________________________________________________________
9. Захищеність патентами на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну (потрібне зазначити): Так Ні
Якщо так, то навести інформацію:
| Номер патенту | Дата видачі | Діє до | Власник патенту |
10. Захищеність торговельної марки в Україні (потрібне зазначити): Так Ні
Якщо так, то навести інформацію:
| Номер документа | Дата видачі | Діє до | Власник документа |
11. Пропонований термін придатності
_______________________________________________________________________________
12. Пропоновані умови зберігання
_______________________________________________________________________________
13. Пропонована категорія відпуску:
за рецептом
_______________________________________________________________________________
без рецепта
_______________________________________________________________________________
тільки в умовах стаціонару
_______________________________________________________________________________
Для лікарських засобів, пропонованих для рецептурного відпуску, заявник надає свої пропозиції щодо категорії відпуску лікарського засобу, однак право визначати категорію відпуску залишається за МОЗ України з урахуванням вимог наказу Міністерства охорони здоров’я України від 17 травня 2001 року № 185 «Про затвердження критеріїв визначення категорій відпуску лікарських засобів», зареєстрованого у Міністерстві юстиції України 31 травня 2001 року за № 464/5655.
Заявник є відповідальним за якість, безпеку та ефективність лікарського засобу в порядку, визначеному чинним законодавством, гарантує достовірність інформації, що міститься в матеріалах реєстраційного досьє, а також підтверджує, що в матеріалах реєстраційного досьє наявні дані щодо якості, безпеки та ефективності лікарського засобу.
Усі дані одержано заявником в установленому порядку і права третьої сторони, захищені патентом або передані за ліцензією, не порушуються у зв’язку з реєстрацією лікарського засобу.
ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхідне):
1. Доручення для ведення переговорів/підписання документів від імені заявника (власника реєстраційного посвідчення).
2. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника (для фізичної особи — за наявності), або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника. Копія повинна бути засвідчена печаткою заявника (для фізичної особи — за наявності)/представника заявника в Україні.
3. Копії патентів на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну.
4. Копії документів щодо захисту торговельної марки в Україні.
5. Копія реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розмішено лікарський засіб, із зазначенням назви документа та назви компетентного органу, що його видав, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу. Копія має бути засвідчена печаткою заявника (для фізичної особи — за наявності)/представника заявника в Україні. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано.
6. Копії реєстраційного(их) посвідчення(нь) та вкладки до них (у разі перереєстрації).
7. Засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи. За необхідності — висновки щодо інших інспекцій, які проводилися. Копії документів мають бути засвідчені печаткою заявника (для фізичної особи — за наявності)/представника заявника в Україні.
|
Дата заповнення: «___» ____________ 20____ року |
Підпис керівника заявника або уповноваженої особи, що виступає від імені заявника ________________________ М. П. |
Додаток 3
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення змін
до реєстраційних матеріалів протягом дії
реєстраційного посвідчення
ЗАЯВА
про державну реєстрацію (перереєстрацію) АФІ або діючої речовини
Дата надходження заяви
«___» __________ 20__ року № ______________________
| Назва АФІ або діючої речовини | |
| Заявник | |
| Представник заявника (уповноважена особа, що виступає від імені заявника) |
Назва повинна бути наведена в такому порядку: міжнародна непатентована назва*, Державна фармакопея України (далі — ДФУ), Європейська фармакопея, загальноприйнята назва, наукова (хімічна) назва.
* Назву АФІ або діючої речовини необхідно вказувати за його рекомендованою міжнародною непатентованою назвою з указанням її солей або гідратної форми, якщо необхідно.
Я гарантую достовірність та відповідаю за інформацію, що міститься у наданих матеріалах реєстраційного досьє.
Згоден, що у разі ненадання матеріалів реєстраційного досьє протягом трьох місяців у розгляді цієї заяви буде відмовлено.
Також цим підтверджується, що всі передбачені збори сплачено/буде сплачено відповідно до вимог законодавства.
Додайте доручення для ведення переговорів/підписання документів від імені заявника (пункт 3.2 цього додатка).
| Від імені заявника | |
|
____________________________________ (підпис) _____________________________________ (ім’я та прізвище) |
|
| М. П. |
_____________________________________ (посада) |
1. ОСОБЛИВІ ПУНКТИ ЗАЯВИ
| Медичний імунобіологічний препарат(абзаци сорок п’ятий та сорок шостийрозділу ІІ Порядку) | Інший лікарський засіб |
1.1. Технологічна форма (форма випуску)
(рідина, порошок, гранули, пелети тощо)
____________________________________________________________________________________________
1.2. Упаковка, уключаючи опис матеріалу, з якого вони виготовлені (використовуйте перелік стандартних термінів ДФУ або Європейської фармакопеї)
____________________________________________________________________________________________
Для кожного виду упаковки укажіть:
1.2.1. Розмір(и) упаковки.
1.2.2. Пропонований термін придатності.
1.2.3. Пропонований період проведення переконтролю.
1.2.4. Пропоновані умови зберігання.
1.3. Заявник (власник реєстраційного посвідчення)/контактна особа
1.3.1. Заявник (власник реєстраційного посвідчення) (для вітчизняних заявників — українською, для зарубіжних — українською та англійською мовами):
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| країна | |
| телефон/факс | |
1.3.2. Представник заявника (уповноважена особа, що виступає від імені заявника):
| прізвище, ім’я, по батькові уповноваженої особи представника заявника | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| країна | |
| телефон/факс | |
У разі заповнення цього пункту додайте доручення (пункт 3.2 цього додатка)
1.4. Виробники
1.4.1. Виробник, що відповідає за випуск серії АФІ або діючої речовини (для вітчизняних заявників — українською, для зарубіжних — українською та англійською мовами):
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
1.4.2. Виробник(и) та виробничі дільниці (для вітчизняних заявників — українською, для зарубіжних — українською та англійською мовами).
Повинні бути зазначені всі дільниці виробництва, задіяні у виробничому процесі кожного джерела АФІ або діючої речовини, включаючи дільниці, на яких проводиться контроль якості/контроль у процесі виробництва. Для високотехнологічних (біотехнологічних) лікарських засобів мають бути включені усі дільниці зберігання головного та робочого банків клітин і дільниці приготування робочого банку клітин.
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
Короткий опис етапів виробництва, виконуваних на виробничій дільниці
____________________________________________________________________________________________
Додайте технологічну схему виробництва (пункт 3.4 цього додатка).
• Чи видано сертифікат відповідності Європейській фармакопеї для діючої речовини (далі — ДР)?
Ні Так
Якщо так:
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| номер сертифіката | |
| дата останнього перегляду |
Додайте копію сертифіката відповідності (пункт 3.5 цього додатка).
• Чи є матеріали реєстраційного досьє (мастер-файл) на ДР оригінальними?
Ні Так
Якщо так:
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| ідентифікаційний номер або реєстраційний номер облікової картки платника податків, або серія та номер паспорта (для фізичних осіб, які через свої релігійні переконання відмовляються від прийняття реєстраційного номера облікової картки платника податків та повідомили про це відповідний орган державної податкової служби і мають відмітку у паспорті) | |
| дата подання на розгляд | |
| дата останнього перегляду |
Для АФІ або діючих речовин тваринного походження, якщо є сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії (далі — ГЕ) або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків ГЕ, він повинен бути доданий у пункті 3.6 цього додатка.
1.5. Чи містить або складається АФІ або діюча речовина з генетично модифікованих організмів (ГМО)?
НіТак
Якщо так, то чи відповідає АФІ або діюча речовина встановленим вимогам?
Зробіть необхідне посилання
НіТак
Додайте копію письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою (пункт 3.7 цього додатка).
2. ІНШІ ВІДОМОСТІ
2.1. Чи захищений АФІ або діюча речовина патентами на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну?
Ні Так
Якщо так, то наведіть таку інформацію:
| Номер патенту | Дата видачі | Діє до | Власник патенту |
Додайте копії патентів згідно з пунктом 3.8 цього додатка.
Для державної реєстрації АФІ або діючих речовин, що базуються або мають відношення до об’єктів інтелектуальної власності, на які відповідно до законів України видано патент, заявник подає засвідчену відповідно копію патенту або ліцензії, якою дозволяється виробництво та продаж АФІ або лікарського засобу, а також документ, що підтверджує чинність патенту в Україні. Заявники подають лист, в якому вказується, що права третьої сторони, захищені патентом або передані за ліцензією, не порушуються у зв’язку з реєстрацією АФІ або діючої речовини.
2.2. Чи захищена торговельна марка в Україні?
Ні Так
Якщо так, то наведіть таку інформацію:
| Номер документа | Дата видачі | Діє до | Власник документа |
Додайте копії документів, що передбачені в пункті 3.9 цього додатка.
3. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхідне)
3.1. Підтвердження оплати (за наявності).
3.2. Доручення для ведення переговорів/підписання документів від імені заявника (власника реєстраційного посвідчення).
3.3. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника (для фізичної особи за наявності) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника. Копія повинна бути засвідчена печаткою заявника (для фізичної особи за наявності)/представника заявника в Україні.
3.4. Проект технологічного регламенту або відомості про технологію виробництва (технологічна схема виробництва із зазначенням усіх дільниць виробництва, задіяних у процесі виробництва АФІ або діючої речовини (уключаючи дільниці для відбору зразків і контролю якості). Копія повинна бути засвідчена печаткою заявника (для фізичної особи — за наявності)/представника заявника в Україні.
3.5. Копія сертифіката відповідності Європейській фармакопеї (за наявності).
3.6. Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків губчатої енцефалопатії.
3.7. Копія письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою.
3.8. Копії патентів на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну.
3.9. Копії документів щодо захисту торговельної марки в Україні.
3.10. Копії реєстраційного(их) посвідчення(нь) та вкладки до них (у разі перереєстрації).
Додаток 4
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення змін
до реєстраційних матеріалів протягом дії
реєстраційного посвідчення
Заява
про державну перереєстрацію лікарського засобу
Дата надходження заяви
«___» __________ 20___ року № ______________________
Цим я подаю заяву на перереєстрацію.
Я заявляю, що якість лікарського засобу, методи виготовлення та контролю регулярно оновлювалися відповідно до процедури внесення змін з огляду на технічний і науковий прогрес.
Я підтверджую, що до звіту про лікарський засіб не було внесено змін, крім тих, які схвалено МОЗ.
Я гарантую достовірність та відповідаю за інформацію, що міститься у наданих матеріалах реєстраційного досьє. Згоден, що у разі ненадання матеріалів реєстраційного досьє протягом
трьох місяців у розгляді цієї заяви буде відмовлено.
Усі передбачені збори сплачено/буде сплачено відповідно до вимог законодавства.
| Від імені заявника | |
|
____________________________________ (підпис) _____________________________________ (ім’я та прізвище) |
|
| М. П. |
_____________________________________ (посада) |
| Назва лікарського засобу | |
| Діюча(і) речовина(и) або комбінований лікарський засіб | |
|
Фармакотерапевтична група (використовуйте діючий код АТХ) |
|
| Сила(и) дії (дозування) | |
| Шлях(и) введення | |
| Лікарська форма та упаковка | |
| Номер(и) реєстраційного посвідчення | |
| Заявник | |
| Уповноважена особа, що виступає від імені заявника | |
| Дата першої реєстрації в Україні | |
| Дата закінчення строку дії реєстраційного посвідчення | |
| Пропонована дата продовження реєстрації |
1. ОСОБЛИВІ ПУНКТИ ЗАЯВИ
1.1. Пропозиції щодо рекламування лікарських засобів, що відпускаються без рецепта:
рекламування тільки серед медичних працівників
рекламування для населення та медичних працівників
1.2. Заявник (власник реєстраційного посвідчення)/контактна особа
1.2.1. Заявник (власник реєстраційного посвідчення) (для вітчизняних виробників — українською, для зарубіжних — українською та англійською мовами):
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця | |
| країна | |
| телефон/факс | |
1.2.2. Представник заявника (уповноважена особа, що виступає від імені заявника):
| прізвище, ім’я, по батькові уповноваженої особи представника заявника | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| місцезнаходження юридичної особи або місце проживання фізичної особи-підприємця | |
| країна | |
| телефон/факс | |
1.3. Затверджені зміни або зміни, що перебувають на розгляді для внесення або розширення заяви, які мали місце з дати видачі реєстраційного посвідчення або останнього продовження дії
Укажіть у хронологічному порядку список затверджених змін або змін, що перебувають на розгляді. Укажіть дату подачі, дату затвердження (якщо затверджено) і короткий опис зміни.
1. Дата подання
номер процедури, якщо необхідно
дата затвердження
короткий опис зміни
2. Дата подання
номер процедури, якщо необхідно
дата затвердження
короткий опис зміни
3. Дата подання
номер процедури, якщо необхідно
дата затвердження
короткий опис зміни
Усі інші зміни мають бути зазначені в такому самому форматі
1.4. Виробники (для вітчизняних виробників — українською, для зарубіжних — українською та англійською мовами)
1.4.1. Виробник(и), що відповідає(ють) за випуск серії
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
1.4.2. Дільниці, на яких проводиться контроль серії, якщо вони відмінні від визначених у пункті 1.4.1:
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
1.4.3. Виробник(и) лікарського засобу і дільниця(і) виробництва:
Включаючи дільниці виробництва будь-якого розріджувача/розчинника в окремій упаковці/ємності, що є частиною лікарського засобу, дільниці, на яких здійснюється контроль якості/контроль у процесі виробництва, а також (за потреби) імпортерів
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
Короткий опис виконуваних функцій
_____________________________________________________________________________
Додайте копію ліцензії на виробництво (пункт 2.2 цього додатка).
Додайте копію сертифіката відповідності вимогам належної виробничої практики, передбаченого у пункті 2.10 цього додатка.
1.4.4. Виробник(и) ДР та виробничі дільниці
Повинні бути зазначені всі дільниці виробництва, задіяні у виробничому процесі кожного джерела діючої речовини, включаючи дільниці, на яких проводиться контроль якості/контроль у процесі виробництва. Для високотехнологічних (біотехнологічних) лікарських засобів мають бути включені усі дільниці зберігання головного та робочого банків клітин та дільниці приготування робочого банку клітин.
| діюча речовина | |
| найменування юридичної особи або прізвище, ім’я, по батькові фізичної особи — підприємця (виробника) | |
| адреса місця провадження діяльності | |
| країна | |
| телефон/факс | |
Короткий опис етапів виробництва, виконуваних на виробничій дільниці
____________________________________________________________________________________________
Для кожної ДР додайте заяву уповноваженої особи виробника (пункт 2.13 додатка).
• Чи видано сертифікат відповідності Європейській фармакопеї для ДР?
Ні Так
Якщо так:
Додайте копію сертифіката відповідності (пункт 2.6 цього додатка).
• Чи є матеріали реєстраційного досьє (мастер-файл) на ДР, посилання на які буде використовуватися у досьє, оригінальними?
НіТак
Якщо так:
Додайте лист(и) виробника ДР, що дає(ють) право використання матеріалів реєстраційного досьє (мастер-файл) на ДР (пункт 2.6 цього додатка).
Додайте копію письмового зобов’язання від виробника ДР інформувати заявника про зміни у виробничому процесі або специфікаціях (пункт 2.7 цього додатка).
• Чи видано сертифікат на загальний файл на вакцинний антиген (далі — ВАЗФ), що використовується у даній заяві, або на нього подано заявку?
НіТак
Якщо так:
Додайте копію сертифіката на ВАЗФ (пункт 2.8 цього додатка).
1.5. Якісний та кількісний склад
1.5.1. Якісний та кількісний склад лікарського засобу (ДР та допоміжні речовини):
Необхідно вказати, на яку кількість розрахований склад (наприклад, 1 капсула)
Перерахуйте діючі речовини окремо від допоміжних речовин:
| Назва ДР* | Кількість | Одиниця | Посилання/монографія |
| 1. | |||
| 2. | |||
| 3. | |||
| Тощо |
| Назва допоміжної(их) речовини(н) | Кількість | Одиниця | Посилання/монографія |
| 1. | |||
| 2. | |||
| 3. | |||
| Тощо |
* Назву для кожної ДР необхідно вказати в такій послідовності: міжнародна непатентована назва**, Державна фармакопея України, Європейська фармакопея, загальноприйнята назва, наукова (хімічна) назва.
**Назву ДР необхідно вказувати за її рекомендованою міжнародною непатентованою назвою з указанням її солей або гідратної форми, якщо необхідно.
Інформацію про надлишкову кількість не слід указувати в колонках щодо складу, а необхідно викласти нижче:
діюча(і) речовина(и) _________________________________________________________________________
допоміжна(і) речовина(и) ___________________________________________________________________
1.5.2. Перелік матеріалів тваринного та/або людського походження, що входять до складу або використовуються у процесі виробництва лікарського засобу
ВІДСУТНІ
| Назва | Функція | Тваринного походження, сприятливі до ГЕ4 | Інші тваринного походження | Людського походження | Сертифікат відповідності ЄФ5 щодо ГЕ4 (укажіть номер) | ||
| ДР1 | ДОП.Р2 | Р3 | |||||
| 1. | |||||||
| 2. | |||||||
| 3. | |||||||
| Тощо | |||||||
Якщо є сертифікат відповідності ЄФ5 щодо ГЕ4 або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків ГЕ4, він повинен бути доданий у пункті 2.11 цього додатка.
1ДР — діюча речовина.
2ДОП.Р — допоміжна речовина (уключаючи вихідні матеріали, які використовуються у виробництві діючої/допоміжної речовини).
3Р — реагент/середовище культивування (уключаючи використовувані для приготування головного та робочого банків клітин).
4ГЕ — губчата енцефалопатія.
5ЄФ — Європейська фармакопея.
1.5.3. Чи видано сертифікат на матеріали реєстраційного досьє (мастер-файл) на плазму (далі — ПМФ), що використовується у цій заяві, або на нього подано заявку?
НіТак
Якщо так:
Додайте копію сертифіката (пункт 2.9 цього додатка).
1.5.4. Чи містить або складається лікарський засіб з генетично модифікованих організмів (ГМО)?
НіТак
Якщо так, то чи відповідає лікарський засіб встановленим вимогам?
Зробіть необхідне посилання
НіТак
Додайте копію письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою (пункт 2.12 цього додатка).
2. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхідне):
2.1. Доручення для ведення переговорів/підписання документів від імені заявника (власника реєстраційного посвідчення).
2.2. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника. Копія повинна бути засвідчена печаткою заявника/представника заявника в Україні.
2.3. Копії патентів на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну.
2.4. Копії документів щодо захисту торговельної марки в Україні.
2.5. Копія реєстраційного(их) посвідчення(ь) в Україні разом із вкладками до реєстраційного(их) посвідчення(ь) та копія реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розмішено лікарський засіб, із зазначенням назви документа та назви компетентного органу, що його видав, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу. Копія має бути засвідчена печаткою заявника/представника заявника в Україні. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано.
2.6. Письмова згода на використання матеріалів реєстраційного досьє на діючу речовину (у довільній формі) від її виробника або копія сертифіката відповідності Європейській фармакопеї.
2.7. Копія письмового зобов’язання виробника діючої речовини (у довільній формі) інформувати заявника про зміни у виробничому процесі або специфікаціях.
2.8. Копія сертифіката на ВАЗФ.
2.9. Копія сертифіката на ПМФ.
2.10. Засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійного листа заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи. За необхідності — висновки щодо інших інспекцій, які проводилися. Копії документів мають бути засвідчені печаткою заявника/представника заявника в Україні.
2.11. Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків губчатої енцефалопатії.
2.12. Копія письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивільнення ГМО у навколишнє середовище при використанні з дослідницькою метою.
2.13. Для кожної діючої речовини дільниць виробництва, задіяних у виробничому процесі, включаючи дільниці, на яких проводиться контроль якості/контроль у процесі виробництва та випуск серій, заява уповноваженої особи щодо відповідності виробництва вимогам належної виробничої практики та керівництвам з належної виробничої практики щодо вихідних матеріалів.
Додаток 5
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
СТРУКТУРА
реєстраційного досьє
(формат загального технічного документа)
Повне реєстраційне досьє складається з п’яти Модулів:
Модуль 1. Адміністративна інформація
1.1. Зміст.
1.2. Форма заяви.
1.3. Коротка характеристика лікарського засобу, маркування та інструкція для медичного застосування:
1.3.1. Копія короткої характеристики лікарського засобу/інструкції про застосування лікарського засобу (інструкції для медичного застосування), затвердженої відповідно до нормативних вимог країни заявника/виробника або інших країн, регуляторні органи яких керуються високими стандартами до якості, що відповідають стандартам, рекомендованим ВООЗ.
1.3.2. Маркування.
1.3.3. Інструкція для медичного застосування (на паперовому та електронному носіях).
1.3.4. Коротка характеристика лікарського засобу.
1.4. Інформація про незалежних експертів:
1.4.1. Інформація про експерта з якості.
1.4.2. Інформація про експерта з доклінічних даних.
1.4.3. Інформація про експерта з клінічних даних.
1.5. Спеціальні вимоги до різних типів заяв.
Додаток до Модуля 1. Оцінка небезпеки для довкілля.
Модуль 2. Резюме загального технічного документа
2.1. Зміст Модулів 2–5.
2.2. Вступ.
2.3. Загальне резюме з якості.
2.4. Огляд доклінічних даних.
2.5. Огляд клінічних даних.
2.6. Резюме доклінічних даних:
2.6.1. Резюме фармакологічних даних у текстовому форматі.
2.6.2. Резюме фармакологічних даних у вигляді таблиць.
2.6.3. Резюме фармакокінетичних даних у текстовому форматі.
2.6.4. Резюме фармакокінетичних даних у вигляді таблиць.
2.6.5. Резюме токсикологічних даних у текстовому форматі.
2.6.6. Резюме токсикологічних даних у вигляді таблиць.
2.7. Резюме клінічних даних:
2.7.1. Резюме біофармацевтичних досліджень та пов’язаних з ними аналітичних методів.
2.7.2. Резюме досліджень з клінічної фармакології.
2.7.3. Резюме з клінічної ефективності.
2.7.4. Резюме з клінічної безпеки.
2.7.5. Посилання на джерела літератури.
2.7.6. Короткі огляди індивідуальних досліджень.
Модуль 3. Якість. Хімічна, фармацевтична та біологічна інформація про лікарські засоби, що містять хімічні та/або біологічні діючі речовини
3.1. Зміст.
3.2. Основні дані.
3.2.S. Діюча(і) речовина(и).
3.2.S.1. Загальна інформація:
3.2.S.1.1. Назва.
3.2.S.1.2. Структура.
3.2.S.1.3. Загальні властивості.
3.2.S.2. Процес виробництва діючої(их) речовини(н):
3.2.S.2.1. Виробник(и).
3.2.S.2.2. Опис виробничого процесу та його контролю.
3.2.S.2.3. Контроль матеріалів.
3.2.S.2.4. Контроль критичних стадій і проміжної продукції.
3.2.S.2.5. Валідація процесу та/або його оцінка.
3.2.S.2.6. Розробка виробничого процесу.
3.2.S.3. Опис характеристик діючої(их) речовини(н):
3.2.S.3.1. Доказ структури та інші характеристики.
3.2.S.3.2. Домішки.
3.2.S.4. Контроль діючої(их) речовини(н):
3.2.S.4.1. Специфікація.
3.2.S.4.2. Аналітичні методики.
3.2.S.4.3. Валідація аналітичних методик.
3.2.S.4.4. Аналізи серій.
3.2.S.4.5. Обґрунтування специфікації.
3.2.S.5. Стандартні зразки або препарати.
3.2.S.6. Система упаковка/укупорка.
3.2.S.7. Стабільність:
3.2.S.7.1. Резюме щодо стабільності та висновки.
3.2.S.7.2. Протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності.
3.2.S.7.3. Дані про стабільність.
3.2.P. Готовий лікарський засіб:
3.2.P.1. Опис і склад лікарського засобу.
3.2.P.2. Фармацевтична розробка:
3.2.P.2.1. Компоненти лікарського засобу.
3.2.P.2.1.1. Діюча(і) речовина(и).
3.2.P.2.1.2. Допоміжні речовини.
3.2.P.2.2. Лікарський засіб.
3.2.P.2.2.1. Розробка складу.
3.2.P.2.2.2. Надлишки.
3.2.P.2.2.3. Фізико-хімічні та біологічні властивості.
3.2.P.2.3. Розробка виробничого процесу.
3.2.P.2.4. Система упаковка/укупорка.
3.2.P.2.5. Мікробіологічні характеристики.
3.2.P.2.6. Сумісність.
3.2.P.3. Процес виробництва лікарського засобу:
3.2.P.3.1. Виробник(и).
3.2.P.3.2. Склад на серію.
3.2.P.3.3. Опис виробничого процесу та контролю процесу.
3.2.P.3.4. Контроль критичних стадій і проміжної продукції.
3.2.P.3.5. Валідація процесу та/або його оцінка.
3.2.P.4. Контроль допоміжних речовин:
3.2.P.4.1. Специфікації.
3.2.P.4.2. Аналітичні методики.
3.2.P.4.3. Валідація аналітичних методик.
3.2.P.4.4. Обґрунтування специфікацій.
3.2.P.4.5. Допоміжні речовини людського або тваринного походження.
3.2.P.4.6. Нові допоміжні речовини.
3.2.P.5. Контроль лікарського засобу:
3.2.P.5.1. Специфікація(ї).
3.2.P.5.2. Аналітичні методики.
3.2.P.5.3. Валідація аналітичних методик.
3.2.P.5.4. Аналізи серій.
3.2.P.5.5. Характеристика домішок.
3.2.P.5.6. Обґрунтування специфікації(й).
3.2.P.6. Стандартні зразки та препарати.
3.2.P.7. Система упаковка/укупорка.
3.2.P.8. Стабільність:
3.2.P.8.1. Резюме щодо стабільності та висновки.
3.2.P.8.2. Протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності.
3.2.P.8.3. Дані про стабільність.
Додаток — інформація щодо:
Приміщення та обладнання.
Оцінка безпеки щодо сторонніх агентів.
Нові допоміжні речовини.
Додаткова інформація.
3.3. Посилання на джерела літератури.
Модуль 4. Звіти про доклінічні дослідження
4.1. Зміст.
4.2. Звіти про дослідження.
4.2.1. Фармакологія:
4.2.1.1. Первинна фармакодинаміка.
4.2.1.2. Вторинна фармакодинаміка.
4.2.1.3. Фармакологія безпеки.
4.2.1.4. Фармакодинамічні взаємодії.
4.2.2. Фармакокінетика:
4.2.2.1. Аналітичні методи та звіти щодо їх валідації.
4.2.2.2. Усмоктування.
4.2.2.3. Розподіл.
4.2.2.4. Метаболізм.
4.2.2.5. Виведення.
4.2.2.6. Фармакокінетичні взаємодії (доклінічні).
4.2.2.7. Інші фармакокінетичні дослідження.
4.2.3. Токсикологія:
4.2.3.1. Токсичність при одноразовому введенні.
4.2.3.2. Токсичність при повторних введеннях.
4.2.3.3. Генотоксичність.
4.2.3.4. Канцерогенність.
4.2.3.5. Репродуктивна токсичність та токсичний вплив на розвиток потомства.
4.2.3.6. Місцева переносимість.
4.2.3.7. Додаткові дослідження токсичності.
4.3. Посилання на джерела літератури.
Модуль 5. Звіти про клінічні випробування
5.1. Зміст.
5.2. Перелік усіх клінічних випробувань у вигляді таблиць.
5.3. Звіти про клінічні випробування:
5.3.1. Звіти про біофармацевтичні дослідження.
5.3.2. Звіти, що стосуються дослідження фармакокінетики при використанні біоматеріалів людського походження.
5.3.3. Звіти про фармакокінетичні дослідження у людини.
5.3.4. Звіти про фармакодинамічні дослідження у людини.
5.3.5. Звіти про дослідження ефективності та безпеки.
5.3.5.1. Звіти про контрольовані клінічні дослідження щодо підтвердження заявлених показань для застосування.
5.3.5.2. Звіти про неконтрольовані клінічні дослідження, звіти про аналізи даних за кількома дослідженнями і звіти про інші клінічні дослідження.
5.3.6. Звіти про післяреєстраційний досвід застосування.
5.3.7. Зразки індивідуальних реєстраційних форм та індивідуальні списки пацієнтів.
5.4. Посилання на джерела літератури.
Додаток 6
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
ПЕРЕЛІК
документів для проведення експертизи матеріалів реєстраційного досьє для державної реєстрації АФІ або діючої речовини
1. Адміністративні дані.
2. Зміст реєстраційного досьє.
3. Форма заяви.
4. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої АФІ або діючої речовини у країні виробника. Копія повинна бути засвідчена печаткою заявника/представника заявника в Україні.
5. Сертифікати якості на три промислові серії АФІ або діючої речовини або один сертифікат на одну промислову серію у супроводі зобов’язання надати сертифікати на дві інші серії, як тільки вони стануть доступними (усі сертифікати повинні подаватися за кожним заявленим місцем виробництва).
6. Пропозиції до маркування.
7. Загальна інформація.
8. Виробництво:
8.1. Виробник(и).
8.2. Опис виробничого процесу та його контролю.
8.3. Контроль матеріалів.
8.4. Контроль критичних стадій і проміжної продукції.
8.5. Валідація процесу та/або його оцінка.
9. Опис характеристик:
9.1. Доказ структури та інші характеристики.
9.2. Домішки.
10. Контроль:
10.1. Специфікація.
10.2. Аналітичні методики.
10.3. Валідація аналітичних методик.
10.4. Обґрунтування специфікації.
11. Система упаковки/укупорки.
12. Стабільність:
12.1. Протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності (за необхідності).
12.2. Дані про стабільність (за необхідності).
Для апробації методів аналізу якості АФІ або діючої речовини можуть бути запитані зразки, еталонні зразки АФІ або діючої речовини із сертифікатами якості на серію, включаючи дату виробництва, терміни та умови зберігання.
Додаток 7
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
ПЕРЕЛІК
документів для проведення експертизи матеріалів для державної перереєстрації лікарського засобу
1. Супровідний лист.
2. Повний зміст.
3. Заява про державну перереєстрацію лікарського засобу (додаток 4).
4. Інформація про уповноважених кваліфікованих осіб заявника в Україні для здійснення фармаконагляду.
5. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника.
6. Засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи.
7. Копія реєстраційного(их) посвідчення(ь) в Україні разом із вкладками до реєстраційного(их) посвідчення(ь) та копія реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розміщено лікарський засіб, із зазначенням назви документа та назви компетентного органу, що його видав, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу. Копія має бути засвідчена печаткою заявника/представника заявника в Україні. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано.
8. Перелік одержаних за останні 5 років в Україні рекламацій на лікарський засіб (у хронологічному порядку) з докладним аналізом причин, що стали підставами для видачі рекламацій, та заходів, вжитих заявником для запобігання їх у майбутньому.
9. Перелік гарантій заявника та зобов’язань, наданих після реєстрації/перереєстрації (у хронологічному порядку) із зазначенням характеру, стану виконання, дати подання та дати, коли проблема була вирішена (рекомендації щодо післяреєстраційних досліджень, усунення будь-яких недоліків, що надавалися контролюючими та іншими органами тощо).
10. Оновлена коротка характеристика лікарського засобу/інструкція для медичного застосування лікарського засобу, затверджені в країні виробника/заявника; діючі в Україні інструкція для медичного застосування лікарського засобу та коротка характеристика лікарського засобу (за наявності); проект оновленої інструкції для медичного застосування лікарського засобу (на паперовому та електронному носіях).
11. Діючі в Україні методи контролю якості готового лікарського засобу, зміни до них (за наявності) та оновлені методи контролю якості готового лікарського засобу.
12. Діюча версія частини ІІ або Модуля 3 реєстраційного досьє (залежно від обраного заявником формату матеріалів реєстраційного досьє).
13. Звіт за результатами післяреєстраційного нагляду (регулярно оновлюваний звіт з безпеки) та/або узагальнюючий звіт.
14. Зведені дані виробника/заявника про стан безпеки медичного застосування лікарського засобу в Україні за період дії останнього реєстраційного посвідчення згідно з формою, передбаченою вимогами Порядку здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування, затвердженого наказом МОЗ від 27 грудня 2006 року № 898, зареєстрованого у Міністерстві юстиції України 29 січня 2007 року за № 73/13340.
__________
Додаток 8
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
ПЕРЕЛІК
документів для проведення експертизи матеріалів для державної перереєстрації традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами
1. Заява про державну реєстрацію (перереєстрацію) лікарського засобу згідно з додатком 2 до Порядку.
2. Інформація про уповноважених кваліфікованих осіб заявника для здійснення фармаконагляду в Україні.
3. Перелік одержаних за останні 5 років в Україні рекламацій на лікарський засіб (у хронологічному порядку) з докладним аналізом причин, що стали підставами для видачі рекламацій, та заходів, вжитих заявником для запобігання їм у майбутньому.
4. Копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника.
5. Засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи.
6. Копія реєстраційного(их) посвідчення(ь) в Україні разом із вкладками до реєстраційного(их) посвідчення(ь) та копія реєстраційного посвідчення або іншого дозвільного документа (Marketing Authorization, Сertificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстраційного посвідчення (заявника) та/або виробника, або іншої країни, регуляторний орган якої керується високими стандартами якості, що відповідають стандартам, рекомендованим ВООЗ, на ринку якої розміщено лікарський засіб, із зазначенням назви документа та назви компетентного органу, що його видав, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу. Копія має бути засвідчена печаткою заявника/представника заявника в Україні. Зазначити перелік країн, де даний лікарський засіб зареєстровано/перереєстровано.
7. Проект інструкції для медичного застосування лікарського засобу.
8. Діючі в Україні методи контролю якості готового лікарського засобу, зміни до них (за наявності) та оновлені методи контролю якості готового лікарського засобу.
9. Відомості про технологію виробництва (при перереєстрації традиційних лікарських засобів).
10. Лист-підтвердження стосовно того, що склад, виробництво та контроль лікарського засобу відповідають пропису, включеному до Переліку лікарських засобів, що виробляються згідно із затвердженими прописами, затвердженого наказом Міністерства охорони здоров’я України від 11 червня 2009 року № 423.
Додаток 9
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
СТРУКТУРА
реєстраційного досьє
(у форматі чотирьох частин)
Повне реєстраційне досьє складається з чотирьох частин:Частина I. Резюме досьє
I. A. Адміністративні дані.
Зміст реєстраційного досьє.
Форма заяви.
I. B. Коротка характеристика лікарського засобу, інструкція для медичного застосування лікарського засобу:
I. B. 1. Копія короткої характеристики лікарського засобу/інструкції для медичного застосування, затвердженої відповідно до нормативних вимог країни заявника/виробника або інших країн, регуляторні органи яких застосовують високі стандарти до якості, що відповідають стандартам, рекомендованим ВООЗ.
I. B. 2. Пропозиції до маркування на упаковці (первинній та вторинній (за наявності)), інструкції для медичного застосування лікарського засобу (на паперовому та електронному носіях).
I. B. 3. Коротка характеристика лікарського засобу.
I. C. Звіти незалежних експертів:
I. C. 1. Звіт експерта щодо хімічної, фармацевтичної та біологічної документації.
I. C. 2. Звіт експерта щодо фармако-токсикологічної документації.
I. C. 3. Звіт експерта щодо клінічної документації.
Частина II. Хімічна, фармацевтична та біологічна документація
Зміст.
II. A. Склад:
II. A. 1. Склад лікарського засобу.
II. A. 2. Упаковка (короткий опис).
II. A. 3. Склад для клінічних випробувань.
II. A. 4. Фармацевтична розробка.
II. B. Метод виготовлення (відомості про технологію виробництва):
II. B. 1. Виробнича формула.
II. B. 2. Виробничий процес.
II. B. 3. Валідація процесу.
II. C. Методи контролю вихідних матеріалів*:
II. C. 1. Діюча речовина*:
II. C. 1.1. Специфікації та стандартні методики*.
II. C. 1.2. Наукові дані*:
II. C. 1.2.1. Номенклатура*.
II. C. 1.2.2. Опис*.
II. C. 1.2.3. Виробництво*.
II. C. 1.2.4. Контроль якості в процесі виробництва.
II. C. 1.2.5. Хімічна розробка.
II. C. 1.2.6. Домішки*.
II. C. 1.2.7. Аналіз серії*.
__________
* Мінімальний обсяг відомостей, які необхідно надавати у розділі II. C. 1.
II. C. 2. Допоміжні речовини (ексципієнти):
II. C. 2.1. Специфікації та встановлені методи контролю якості.
II. C. 2.2. Наукові дані.
II. C. 3. Пакувальний матеріал (внутрішня/зовнішня упаковка):
II. C. 3.1. Специфікації та встановлені методи контролю якості.
II. C. 3.2. Наукові дані.
II. D. Методи контролю якості проміжних продуктів (за необхідності).
II. E. Методи контролю якості готового лікарського засобу:
II. E. 1. Специфікації та встановлені методи контролю якості:
II. E. 1.1. Специфікації продукту та методи контролю якості в процесі виробництва, специфічні стандарти.
II. E. 1.2. Методи контролю якості:
II. E. 1.2.1. Методи для ідентифікації та кількісного визначення діючої речовини(н).
II. E. 1.2.2. Ідентифікація та визначення ексципієнта(ів).
II. E. 2. Наукові дані:
II. E. 2.1. Валідація аналітичних методів та коментарі, стандарти (робочі стандарти).
II. E. 2.2. Аналіз серії.
II. F. Стабільність:
II. F. 1. Методики визначення стабільності діючої речовини(н).
II. F. 2. Методики визначення стабільності готового лікарського засобу.
II. G. Біодоступність/біоеквівалентність.
Дайте посилання на відповідні розділи частини IV, якщо це необхідно.
II. H. Дані щодо вірогідної небезпеки для навколишнього середовища лікарських засобів, які містять генетично модифіковані мікроорганізми (ГМО).
II. Q. Інша інформація.
Частина III. Фармакологічна та токсикологічна документація
Зміст.
III. A. Токсичність при однократному введенні та введенні повторних доз:
III. A. 1. Дослідження токсичності при однократному введенні.
III. A. 2. Дослідження токсичності при повторних введеннях.
III. B. Репродуктивна функція (фертильність та загальна характеристика репродуктивної функції).
III. C. Дані щодо ембріотоксичності та тератогенності.
III. D. Мутагенний потенціал.
III. E. Канцерогенний потенціал.
III. F. Фармакодинаміка:
III. F.1. Фармакодинамічні ефекти відносно пропонованих показань.
III. F.2. Загальна фармакодинаміка.
III. F.3. Лікарські взаємодії.
III. G. Фармакокінетика:
III. G.1. Фармакокінетика при введенні однократної дози.
III. G.2. Фармакокінетика при повторних введеннях.
III. G.3. Розподіл в інтактних та вагітних тваринах.
III. G.4. Біотрансформація.
III. H. Місцева переносність.
III. Q. Інша інформація (дані щодо алергенності тощо).
III. R. Експертиза вірогідного ризику для навколишнього середовища/екотоксичність (не обумовлена ГМО).
Частина IV. Клінічна документація
Зміст.
IV. A. Клінічна фармакологія:
IV. A.1. Фармакодинаміка.
IV. A.2. Фармакокінетика.
IV. B. Клінічний досвід:
IV. B.1. Клінічні випробування.
IV. B.2. Післяреєстраційний досвід.
IV. B.3. Опубліковані та неопубліковані дані щодо клінічного досвіду.
IV. C. Надання результатів.
IV. D. Клінічна фармакологія.
IV. E. Біодоступність/біоеквівалентність.
IV. F. Клінічна ефективність та безпека.
IV. G. Матеріали реєстраційного досьє, які супроводжують заяву у виняткових випадках.
IV. H. Післяреєстраційний досвід.
IV. Q. Інша інформація.
Якщо окремі частини документації не включені до матеріалів реєстраційного досьє, то слід у відповідному місці зазначити причину під відповідним заголовком.
Для лікарських засобів тваринного походження у розділі II.C.1 має бути надана така додаткова інформація:
дані щодо виду, віку, раціону тварин, від яких отримано сировину;
дані про характер (категорію) тканини, з якої одержується сировина для виробництва лікарського засобу, з точки зору її небезпеки щодо вмісту пріонів;
технологічна схема обробки сировини із зазначенням екстрагентів, температурного режиму тощо;
методи контролю вихідної сировини, уключаючи методи виявлення пріонів у кінцевому продукті (за потреби).
Додаток 10
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
СТРУКТУРА
інструкції для медичного застосування лікарського засобу/лікарського засобу (медичного імунобіологічного препарату)
Назва лікарського засобу (укр.)
Англійською (за бажанням виробника)
Склад:
діюча(і) речовина(и): (МНН або в разі її відсутності скорочена хімічна назва)
склад на 1 одиницю лікарської форми;
допоміжні речовини:
Лікарська форма.
Основні фізико-хімічні властивості:
Фармакотерапевтична група. Код АТХ.
Фармакологічні властивості/Імунологічні і біологічні властивості
Фармакодинаміка.
Фармакокінетика.
Клінічні характеристики.
Показання.
Протипоказання.
Особливі заходи безпеки (за наявності).
Взаємодія з іншими лікарськими засобами та інші види взаємодій.
Особливості застосування.
Застосування у період вагітності або годування груддю.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Спосіб застосування та дози.
Діти.
Передозування.
Побічні реакції.
Термін придатності.
Умови зберігання.
Несумісність (за наявності).
Упаковка.
Категорія відпуску.
Виробник/заявник.
Місцезнаходження виробника та його адреса місця провадження
діяльності/місцезнаходження заявника та/або представника заявника.
Дата останнього перегляду.
Додаток 11
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення змін
до реєстраційних матеріалів протягом дії
реєстраційного посвідчення
МАТЕРІАЛИ РЕЄСТРАЦІЙНОГО ДОСЬЄ,
що подаються для експертизи при внесенні змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
I. АДМІНІСТРАТИВНІ ЗМІНИ
| 1.1. Зміна найменування та/або місцезнаходження заявника (власника реєстраційного посвідчення) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1,2 | IА |
Умови
1. Власником реєстраційного посвідчення повинна залишатися одна й та сама юридична особа або фізична особа — підприємець.
Документація
1. Документ відповідного компетентного уповноваженого органу, в якому зазначено нове найменування та/або нове місцезнаходження заявника (власника реєстраційного посвідчення).
2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці лікарського засобу (за необхідності).
| 1.2. Зміна торговельної назви лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1,2 | IБ |
Умови
1. Запропонована назва не порушує права третіх сторін.
Документація
1. Обґрунтування зміни назви лікарського засобу.
2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці лікарського засобу.
| 1.3. Зміна назви АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2 | IА |
Умови
1. АФІ або діюча речовина повинна залишатися тією самою.
Документація
1. Підтвердження рекомендованих ВООЗ міжнародних непатентованих назв або копія їх переліку. Для рослинних лікарських засобів — заява про те, що назва відповідає чинним вимогам або чинному Керівництву з якості рослинних лікарських засобів та керівним принципам для декларування рослинних субстанцій та рослинних препаратів, що входять до складу (традиційних) рослинних лікарських засобів.
2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці лікарського засобу.
| 1.4. Зміна найменування та/або місцезнаходження виробника (включаючи за необхідності місце проведення контролю якості) або постачальника АФІ або діючої речовини/вихідного матеріалу/реагенту/проміжного продукту, що застосовуються у виробництві АФІ або діючої речовини (за відсутності сертифіката відповідності Європейській фармакопеї у затвердженому реєстраційному досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2, 3 | IА |
Умови
1. Виробнича дільниця та усі виробничі операції залишаються незмінними.
Документація
1. Документ відповідного уповноваженого органу, у якому зазначено нове найменування та/або місцезнаходження.
2. Зміни до відповідних розділів матеріалів реєстраційного досьє.
3. У разі зміни у найменуванні власника мастер-файла на діючу речовину — поновлений дозвіл власника на користування даними, що містяться у мастер-файлі.
| 1.5. Зміна найменування та/або місцезнаходження виробника готового лікарського засобу, включаючи місце проведення контролю якості | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) виробнича дільниця випуску серій | 1 | 1, 2 | IА |
| б) усі інші дільниці | 1 | 1, 2 | IА |
Умови
1. Виробнича дільниця та всі виробничі операції залишаються незмінними.
Документація
1. Копія оновленої ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника, у якому зазначено нове найменування та/або місцезнаходження, та засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи.
2. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи оновлені коротку характеристику лікарського засобу, інструкцію для медичного застосування та пропозиції до маркування на упаковці лікарського засобу (за необхідності).
| 1.6. Зміна коду АТХ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2 | IА |
Умови
1. Присвоєння нового або зміна ВООЗ коду АТХ.
Документація
1. Підтвердження присвоєння ВООЗ коду АТХ або копія переліку кодів АТХ.
2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці лікарського засобу (за необхідності).
| 1.7. Вилучення виробничої дільниці (включаючи дільниці для АФІ або діючої речовини, проміжного продукту або готового лікарського засобу, дільниці для проведення пакування, виробника, відповідального за випуск серій, місце проведення контролю серії) або постачальника вихідного матеріалу, реагенту або допоміжної речовини (якщо зазначено у реєстраційному досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | 1, 2 | IА |
Умови
1. Необхідно залишити принаймні одну затверджену дільницю/виробника, що виконують таку саму функцію, що й вилучені.
2. Вилучення не обумовлено непередбаченими обставинами у виробничому процесі.
Документація
1. Заява про внесення змін має чітко визначати затвердженого та запропонованого виробників, як зазначено в пункті 2.5 заяви про державну реєстрацію лікарського засобу, форма якої наведена у додатку 1 до Порядку.
2. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці лікарського засобу (за необхідності).
ІІ. ЗМІНИ ЩОДО ЯКОСТІ
2.1. АФІ або діюча речовина
2.1.1. Виробництво
| 2.1.1.1. Зміна виробника вихідного/проміжного продукту/реагенту, що використовується у виробничому процесі АФІ або діючої речовини, або зміна виробника (включаючи, де необхідно, місце проведення контролю якості) АФІ або діючої речовини (за відсутності сертифіката відповідності Європейській фармакопеї у матеріалах реєстраційного досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) запропонована додаткова виробнича дільниця є дільницею одного виробника | 1, 2, 3 | 1, 2, 3, 4, 5, 6, 7 | IА |
| б) введення нового виробника АФІ або діючої речовини з наданням матеріалів реєстраційного досьє (мастер-файла) на діючу речовину | II | ||
| в) запропонований виробник використовує спосіб синтезу, що суттєво відрізняється від попереднього, або умови виробництва, які потенційно можуть змінити важливі характеристики якості АФІ або діючої речовини, такі як якісний та/або кількісний профіль домішок, що потребує кваліфікації, або фізико-хімічні властивості АФІ або діючої речовини, що впливають на біодоступність | ІІ | ||
| г) новий виробник вихідного продукту, для якого вимагається попередня оцінка вірусної безпеки та/або ризику передачі збудників губчатої енцефалопатії | II | ||
| ґ) зміна у АФІ або діючої речовини біологічного походження або вихідному матеріалі/реагенті/ проміжному продукті, що використовується для виробництва лікарського засобу біологічного походження | II | ||
| д) зміни у методах контролю якості АФІ або діючої речовини або додавання дільниці для проведення контролю серії/випробування | 2, 4 | 1, 5 | IА |
Умови
1. Для вихідних матеріалів та реагентів специфікації (включаючи контроль під час виробництва, методи аналізу всіх матеріалів) ідентичні затвердженим. Для проміжних продуктів та АФІ або діючих речовин специфікації (включаючи контроль під час виробництва, методи аналізу всіх матеріалів) спосіб виробництва (включаючи розмір серії) та детальний опис методу синтезу — ідентичні затвердженим.
2. АФІ або діюча речовина не є стерильною або речовиною біологічного/імунологічного походження.
3. Якщо у процесі виробництва використовуються матеріали людського або тваринного походження, виробник не використовує будь-якого нового постачальника, для якого потрібна оцінка вірусної безпеки або відповідності виробництва Керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами.
4. Перенесення способу виробництва з попередньої дільниці до нової було успішно виконано.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє (за необхідності).
2. Заява від власника реєстраційного посвідчення або від власника мастер-файла на діючу речовину (за необхідності), що метод синтезу (або у разі лікарських засобів рослинного походження за потреби метод приготування, місце походження, виробництво рослинної субстанції та спосіб виробництва), методи контролю якості та специфікації на АФІ або діючу речовину та вихідний матеріал/реагент/проміжний продукт у виробництві АФІ або діючої речовини не відрізняються від затверджених за потреби.
3. Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії для нового вихідного продукту або, якщо необхідно, документальне підтвердження того, що вихідний продукт для отримання АФІ або діючої речовини, який має ризик передачі збудників губчатої енцефалопатії, попередньо оцінений уповноваженим органом країни виробника та продемонстрована його відповідність Керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами (чинне видання). Інформація повинна містити: найменування виробника, види та тканини тварин, з яких одержано вихідний продукт, назву країни походження тваринної сировини, її використання та попередній документ.
4. Дані аналізів (у вигляді таблиць порівняння) принаймні для двох серій (мінімум дослідно-промислових) АФІ або діючої речовини від затвердженого та запропонованого виробників/дільниць.
5. Заява про внесення змін має містити чітку інформацію щодо «затвердженого» та «запропонованого» виробників, як зазначено в пункті 2.5 заяви про державну реєстрацію лікарського засобу, форма якої наведена у додатку 1 до Порядку.
6. Заява уповноваженої особи (далі — УО) кожного з виробників, перелічених у заяві, якщо АФІ або діюча речовина використовується як вихідний матеріал, та заява УО кожного з перелічених у заяві виробників, відповідальних за випуск серії. У заявах УО необхідно зазначити, що виробник(и) АФІ або діючої речовини діє відповідно до настанов та керівництв з належної виробничої практики щодо вихідних матеріалів. Узагальнена заява може бути прийнятною за умов, приведених у примітці до підпункту 2.2.2.1 цього додатка.
7. Зобов’язання виробника АФІ або діючої речовини інформувати власника реєстраційного посвідчення про будь-які зміни у процесі виробництва, специфікаціях та методах аналізу АФІ або діючої речовини (за необхідності).
| 2.1.1.2. Зміни в процесі виробництва АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначна зміна у процесі виробництва АФІ або діючої речовини | 1, 2, 3, 4, 5, 6, 7 | 1, 2, 3 | IА |
| б) значна зміна у процесі виробництва АФІ або діючої речовини, що може мати істотний вплив на якість, безпеку або ефективність лікарського засобу | II | ||
| в) зміна стосується АФІ або діючої речовини біологічного/імунологічного походження або використання хімічних субстанцій у виробництві лікарського засобу біологічного/імунологічного походження і не відноситься до протоколу виробництва | II | ||
| г) зміна у лікарському засобі рослинного походження, що стосується однієї з таких характеристик: джерело походження сировини, спосіб виробництва або виготовлення | II | ||
| ґ) незначна зміна у закритій частині матеріалів реєстраційного досьє (мастер-файла) на діючу речовину | 1, 2, 3, 4 | IБ |
Умови
1. Не повинно бути жодних змін у якісному та кількісному складі домішок або змін фізико-хімічних властивостей АФІ або діючої речовини.
2. Шлях синтезу не змінюється, а саме проміжні продукти залишаються незмінними, і не використовуються нові реагенти, каталізатори або розчинники. Для рослинних лікарських засобів — джерело походження сировини, виробництво субстанції та спосіб виробництва залишаються незмінними.
3. Специфікації на АФІ або діючу речовину або проміжні продукти залишаються незмінними.
4. Зміна цілком описана у відкритій для заявника частині мастер-файла на діючу речовину (за необхідності).
5. АФІ або діюча речовина не є речовиною біологічного/імунологічного походження.
6. Зміна не стосується джерела походження та способу виробництва рослинного лікарського засобу.
7. Зміна не стосується закритої частини мастер-файла на діючу речовину.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє та затвердженого мастер-файла на діючу речовину (за можливості), включаючи результати порівняння узгодженого та запропонованого виробничого процесу.
2. Дані аналізів (у вигляді таблиці порівняння) принаймні для двох серій (мінімум дослідно-промислових) лікарського засобу (АФІ або діючої речовини), виготовлених відповідно до узгодженого та запропонованого виробничого процесу.
3. Копія затверджених специфікацій на АФІ або діючу речовину.
4. Заява від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину (за необхідності), що будь-які зміни у якісному та кількісному профілі домішок або у фізико-хімічних властивостях — відсутні, метод синтезу не змінюється, а також не змінюються специфікації на АФІ або діючу речовину або проміжні продукти.
Примітка до підпункту «б» підпункту 2.1.1.2 цього додатка. Для хімічних АФІ або діючих речовин це стосується значних змін у методі синтезу або умовах виробництва, що потенційно можуть змінювати важливі характеристики АФІ або діючої речовини, такі як якісний та/або кількісний профіль домішок, що потребує кваліфікації, або фізико-хімічні властивості АФІ або діючої речовини, що впливають на біодоступність.
| 2.1.1.3. Зміна розміру серії (включаючи діапазони) АФІ або діючої речовини або проміжного продукту | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) збільшення до 10 разів порівняно із затвердженим розміром серії | 1, 2, 3, 4, 6, 7, 8 | 1, 2, 5 | IА |
| б) зменшення обсягу виробництва | 1, 2, 3, 4, 5 | 1, 2, 5 | IА |
| в) зміна розміру серії, що потребує доведення подібності АФІ або діючої речовини біологічного/імунологічного походження порівняно із затвердженою | II | ||
| г) збільшення у понад 10 разів порівняно із затвердженим розміром серії | 1, 2, 3, 4 | IБ | |
| ґ) розмір серії АФІ або діючої речовини біологічного/імунологічного походження збільшився/зменшився без зміни параметрів процесу (наприклад, дублювання лінії) | 1, 2, 3, 4 | IБ |
Умови
1. Зміни у процесі виробництва обумовлені тільки збільшенням або зменшенням обсягу виробництва, наприклад, використанням обладнання іншої продуктивності.
2. Наявність результатів аналізу принаймні двох серій запропонованого розміру серії відповідно до специфікацій.
3. АФІ або діюча речовина не є речовиною біологічного/імунологічного походження.
4. Зміна не повинна впливати на відтворюваність процесу виробництва.
5. Зміна не повинна бути обумовлена непередбаченими обставинами в процесі виробництва або проблемами, пов’язаними зі стабільністю.
6. Специфікації на АФІ або діючі речовини/проміжні продукти залишаються незмінними.
7. АФІ або діюча речовина не є стерильною.
8. Затверджений розмір серії не був схвалений при внесенні змін типу IА.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Номери серій запропонованого розміру серії, які проходять випробування.
3. Дані аналізів (у вигляді таблиці порівняння) принаймні однієї промислової серії АФІ або діючої речовини або проміжного продукту для затвердженого та запропонованого розмірів серії (за необхідності). Дані про наступні дві повні промислові серії мають бути надані на вимогу, а також заява про те, що у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією).
4. Копія затверджених специфікацій на АФІ або діючу речовину (та проміжні продукти за потреби).
5. Заява від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину про те, що зміни у процесі виробництва обумовлені тільки збільшенням або зменшенням обсягу виробництва, наприклад, використанням обладнання іншої продуктивності, що зміна не впливає на відтворюваність процесу і не обумовлена непередбаченими обставинами в процесі виробництва або проблемами, пов’язаними зі стабільністю, і що специфікації на АФІ або діючу речовину/проміжні продукти залишаються незмінними.
| 2.1.1.4. Зміни випробувань або допустимих меж, що встановлені у специфікаціях, у процесі виробництва АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) додавання нового випробування та допустимих меж | 1, 2, 5, 6 | 1, 2, 3, 4, 6 | IА |
| в) вилучення несуттєвого випробування | 1, 2 | 1, 2, 5 | IА |
| г) розширення затверджених допустимих меж для показників, які можуть істотно вплинути на якість АФІ або діючої речовини | II | ||
| ґ) вилучення випробування, що може мати істотний влив на загальну якість АФІ або діючої речовини | II | ||
| д) додавання або заміна випробування за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 6 | IБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду показників, зазначених у специфікації (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II).
2. Зміна не обумовлена непередбаченими обставинами в процесі виробництва, наприклад, появою нової некваліфікованої домішки; зміною меж загального вмісту домішок.
3. Будь-які зміни не повинні виходити за допустимі межі затвердженої специфікації.
4. Методи випробувань залишилися незмінними або такі зміни є незначними.
5. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
6. Новий метод випробувань не належить до біологічних/імунологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу АФІ або діючої речовини біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця щодо затверджених та запропонованих методів випробувань.
3. Опис нового нефармакопейного методу випробування та дані з його валідації (за необхідності).
4. Дані аналізів для двох промислових серій (трьох промислових серій для АФІ або діючої речовини біологічного походження, якщо не обґрунтовано інше) АФІ або діючої речовини за всіма показниками, що зазначені у специфікації.
5. Обґрунтування/оцінка ризику від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину (за необхідності), що підтверджує незначність зміненого параметра.
6. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину (за необхідності) введення нового методу випробування або допустимих меж, визначених у специфікаціях.
| 2.1.1.5. Зміни у АФІ або діючій речовині для сезонних, передпандемічних або пандемічних вакцин для профілактики грипу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) заміна штаму(ів) у сезонних, передпандемічних або пандемічних вакцинах для профілактики грипу | II |
2.1.2. Контроль АФІ або діючої речовини
| 2.1.2.1. Зміна у параметрах специфікацій та/або допустимих меж, визначених у специфікаціях на АФІ або діючу речовину, або вихідний/проміжний продукт/реагент, що використовуються у процесі виробництва АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж, визначених у специфікації на лікарські засоби, що підлягають випуску серії | 1, 2, 3, 4 | 1, 2 | IA |
| б) звуження допустимих меж, визначених у специфікації | 1, 2, 3, 4 | 1, 2 | IA |
| в) доповнення специфікації новим показником якості та відповідним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 7 | IA |
| г) вилучення незначного показника якості (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 6 | IА |
| ґ) вилучення параметра специфікації, який може мати суттєвий влив на якість АФІ або діючої речовини та/або готового лікарського засобу | II | ||
| д) розширення допустимих меж, визначених у специфікації на АФІ або діючу речовину | II | ||
| е) розширення допустимих меж, визначених у специфікаціях на вихідні матеріали/проміжні продукти, які мають істотний вплив на якість АФІ або діючої речовини та/або готового лікарського засобу | II | ||
| є) доповнення або заміна параметра специфікації за результатами досліджень з безпеки або якості (за винятком АФІ або діючої речовини біологічного або імунологічного походження) | 1, 2, 3, 4, 5, 7 | IБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду показників, зазначених у специфікації (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II).
2. Зміна не обумовлена непередбаченими обставинами в процесі виробництва, наприклад, появою нової некваліфікованої домішки; зміною меж загального вмісту домішок.
3. Будь-які зміни не повинні виходити за допустимі межі затвердженої специфікації.
4. Методи випробувань залишилися незмінними або такі зміни є незначними.
5. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
6. Новий метод випробувань не належить до біологічних/імунологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу АФІ або діючої речовини біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
7. Зміна не стосується домішки, яка має генотоксичну дію.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця щодо затверджених та запропонованих специфікацій.
3. Опис нового аналітичного методу та дані з його валідації (за необхідності).
4. Результати аналізу для двох промислових серій (трьох промислових серій для АФІ або діючих речовин біологічного походження, якщо не обґрунтовано інше) АФІ або діючої речовини за всіма показниками, що зазначені у специфікації.
5. Порівняльні дані щодо профілю розчинення готового лікарського засобу для принаймні однієї дослідно-промислової серії, що містить АФІ або діючу речовину за затвердженою та запропонованою специфікаціями (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.
6. Обґрунтування/оцінка ризику від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину (за необхідності), що підтверджує незначність зміненого параметра.
7. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину (за необхідності) введення нового методу випробування або допустимих меж, визначених у специфікаціях.
| 2.1.2.2. Зміна у методах випробування АФІ або діючої речовини або вихідного/проміжного продукту/реагенту, що використовується у процесі виробництва АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробування | 1, 2, 3, 4, | 1, 2 | IА |
| б) видалення методу випробування для АФІ або діючої речовини /реагенту/проміжного продукту, якщо альтернативний метод вже затверджений | 7 | 1 | IА |
| в) інші зміни в методах випробування (включаючи заміну або доповнення) для реагенту, що не спричиняє істотного впливу на якість АФІ або діючої речовини | 1, 2, 3, 5, 6 | 1, 2 | IA |
| г) зміна у біологічному/імубіонологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент для АФІ або діючої речовини біологічного походження, наприклад, пептидні карти, глік-карти тощо | II | ||
| ґ) інші зміни у методах випробування (включаючи заміну або доповнення) АФІ або діючої речовини або вихідного/проміжного продукту | 1, 2 | IБ |
Умови
1. Дослідження з валідації, які були проведені відповідно до чинних фармакопейних вимог або настанов та керівних принципів з валідації, підтверджують, що результати аналізу, отримані за затвердженою та запропонованою методиками, — ідентичні.
2. Не відбулося жодних змін меж загального вмісту домішок, не виявлено нових некваліфікованих домішок.
3. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу, але тип колонки або методика — незмінні).
4. Новий метод випробувань не належить до біологічних/імунологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу АФІ або діючої речовини біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
6. АФІ або діюча речовина не є речовиною біологічного/імунологічного походження.
7. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні зміни типу ІА.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи опис методів контролю, звіт про дані з валідації, переглянуті допустимі межі для домішок (за необхідності).
2. Порівняльні дані з валідації або, якщо обґрунтовано, порівняльні результати аналізу, які підтверджують, що затверджена та запропонована методики випробування — ідентичні. Ця вимога не стосується випадку додавання нової методики випробування.
2.1.3. Система упаковка/укупорка
| 2.1.3.1. Зміна у безпосередній упаковці АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) якісні та/або кількісні зміни складу | 1, 2, 3 | 1, 2, 3, 4, 6 | IА |
| б) якісні та/або кількісні зміни складу для стерильних та незаморожених АФІ або діючих речовин біологічного/імунологічного походження | II | ||
| в) рідких АФІ або діючих речовин (нестерильних) | 1, 2, 3, 5, 6 | IБ |
Умови
1. Запропонований пакувальний матеріал має бути ідентичним затвердженому за відповідними показниками якості.
2. Дослідження стабільності розпочато відповідно до настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) принаймні для двох дослідно-промислових або промислових серій АФІ або діючої речовини і на дату випуску у розпорядженні заявника були задовільні дані зі стабільності принаймні за три місяці. Однак, якщо запропонована упаковка стійкіша, ніж затверджена, дані щодо стабільності за три місяці ще можуть бути недоступні. При цьому мають бути надані гарантії, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності невідповідності специфікаціям наприкінці терміну придатності/періоду повторного випробування дані будуть подані до Центру (із запропонованими заходами).
3. Не стосується стерильних рідких АФІ або діючих речовин та АФІ або діючих речовин біологічного/імунологічного походження.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Відповідні дані щодо нової упаковки (наприклад, порівняльні дані про проникність, наприклад, для О2, СО2, вологи), включаючи підтвердження того, що пакувальний матеріал відповідає чинним фармакопейним вимогам щодо пакувальних матеріалів або законодавству ЄС щодо безпеки взаємодії пластикових пакувальних матеріалів та упаковок з харчовими продуктами.
3. Підтвердження відсутності будь-якої взаємодії між АФІ або діючою речовиною та пакувальним матеріалом (наприклад, відсутнє перенесення компонентів запропонованого пакувального матеріалу до АФІ або діючої речовини та немає жодних втрат компонентів упаковки), включаючи підтвердження того, що пакувальний матеріал відповідає чинним фармакопейним вимогам щодо пакувальних матеріалів або законодавству ЄС щодо безпеки взаємодії пластикових пакувальних матеріалів та упаковок з харчовими продуктами.
4. Підтвердження від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину (за необхідності), що дослідження стабільності розпочато відповідно до настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням кількості та номерів серій АФІ або діючої речовини) та підтвердження (за необхідності), що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності та наявні дані не вказують на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності невідповідності специфікаціям наприкінці терміну придатності/періоду повторного випробування дані будуть подані до Центру (із запропонованими заходами).
5. Результати досліджень стабільності, проведених згідно з настановою щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) за відповідними параметрами принаймні для двох дослідно-промислових або промислових серій АФІ або діючої речовини із задовільними показниками щодо стабільності принаймні за три місяці. При цьому мають бути надані гарантії, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності невідповідності специфікаціям наприкінці терміну придатності/періоду повторного випробування дані будуть подані до Центру (із запропонованими заходами).
6. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій (за необхідності).
| 2.1.3.2. Зміни у параметрах специфікацій та/або допустимих меж, зазначених у специфікаціях, для первинної упаковки АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж, зазначених у специфікаціях | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфікації новим показником та відповідним методом випробування | 1, 2, 5 | 1, 2, 3, 4, 6 | IА |
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 5 | IА |
| г) доповнення або заміна показника за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 6 | IБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду показників, зазначених у специфікації (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II), якщо раніше вона була розглянута і затверджена як частина послідовних змін.
2. Зміна не обумовлена непередбаченими обставинами, що виникли в процесі виробництва пакувального матеріалу або зберігання АФІ або діючої речовини.
3. Будь-які зміни не повинні виходити за допустимі межі затвердженої специфікації.
4. Методи випробувань залишилися незмінними або такі зміни є незначними.
5. Новий метод випробування не належить до нестандартного методу або стандартного методу, що використовується у новий спосіб.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій.
3. Опис нового методу випробування та дані з його валідації (за необхідності).
4. Результати аналізу для двох серій первинної упаковки за всіма показниками, зазначеними у специфікації.
5. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину, що показник якості не є суттєвим (за необхідності).
6. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на діючу речовину нових показників якості та допустимих меж, зазначених у специфікації.
| 2.1.3.3. Зміна в методах випробування первинної упаковки АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробування | 1, 2, 3 | 1, 2 | IА |
| б) інші зміни в методах випробування (включаючи заміну або доповнення) | 1, 3, 4 | 1, 2 | IА |
| в) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | 5 | 1 | IА |
Умови
1. Відповідні дослідження з валідації, які були проведені відповідно до чинних фармакопейних вимог або керівних принципів та настанов з валідації (чинне видання), та результати підтверджують, що затверджена та запропонована методики — ідентичні.
2. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу, але тип колонки або методика — незмінні).
3. Новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
4. АФІ або діюча речовина не є речовиною біологічного/імунологічного походження.
5. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні змін типу ІА.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, які включають опис методу випробування та звіт про дані з валідації.
2. Порівняльні дані з валідації або, якщо необхідно, порівняльні результати аналітичних випробувань, які підтверджують ідентичність результатів, отриманих за затвердженим та новим методами. Це не стосується випадків додавання нового методу випробування.
2.1.4. Стабільність
| 2.1.4.1. Зміна періоду повторних випробувань/терміну придатності або умов зберігання АФІ або діючої речовини (за наявності у матеріалах реєстраційного досьє сертифіката відповідності Європейській фармакопеї, включає період повторного випробування) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) період повторного випробування/термін придатності | |||
| зменшення | 1 | 1, 2, 3 | ІА |
| збільшення періоду повторного випробування на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності до настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (крім АФІ або діючих речовин біологічного/імунологічного походження) | ІІ | ||
| збільшення терміну придатності АФІ або діючої речовини біологічного/імунологічного походження на основі результатів досліджень, виконаних не у відповідності до затвердженого протоколу стабільності | ІІ | ||
| збільшення або введення періоду повторного випробування/терміну придатності на основі результатів досліджень у реальному часі | 1, 2, 3 | ІБ | |
| б) умови зберігання | |||
| обмеження умов зберігання | 1 | 1, 2, 3 | ІА |
| зміна умов зберігання АФІ або діючої речовини біологічного/імунологічного походження, якщо дослідження стабільності ще не проводились відповідно до затвердженого протоколу стабільності | II | ||
| зміна умов зберігання АФІ або діючої речовини | 1, 2, 3 | ІБ |
Умови
1. Зміна не обумовлена непередбаченими обставинами у процесі виробництва або проблемами щодо стабільності.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє повинні містити результати досліджень стабільності, представлені у реальному часі і проведені відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання), принаймні для двох (трьох — для лікарських засобів біологічного походження) дослідно-промислових або промислових серій АФІ або діючої речовини у затвердженій упаковці з відповідним періодом повторного випробування у визначених умовах зберігання.
2. Підтвердження, що дослідження стабільності виконані відповідно до затвердженого протоколу стабільності. Дослідження повинні підтвердити відповідність специфікацій.
3. Копії затверджених специфікацій на АФІ або діючу речовину.
2.1.5. Проектний простір
| 2.1.5.1. Введення нового проектного простору або розширення затвердженого для АФІ або діючої речовини щодо: | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) одного елементу (блок, частина) виробничого процесу АФІ або діючої речовини, включаючи контроль у процесі виробництва та/або методи випробування | 1, 2, 3 | II | |
| б) методів випробувань для вихідних матеріалів/реагентів/проміжних продуктів та/або АФІ або діючої речовини | 1, 2, 3 | II |
Документація
1. Проектний простір був розроблений відповідно до європейських та міжнародних наукових керівництв. Результати досліджень розробки лікарського засобу, виробничого процесу або методів випробувань (наприклад, необхідно вивчити взаємодію різних параметрів, що формують проектний простір, включаючи дослідження оцінки ризику та багатомірні дослідження, якщо необхідно), які демонструють, що функціональна взаємодія властивостей матеріалу та параметрів процесу, які можуть впливати на критичні характеристики якості АФІ або діючої речовини, була досягнута.
2. Опис проектного простору у формі таблиці, включаючи змінні (властивості матеріалу та параметри процесу, якщо необхідно) та їх запропоновані діапазони.
3. Зміни до відповідних розділів матеріалів реєстраційного досьє.
| 2.1.5.2. Внесення змін після затвердження протоколу управління для АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | II |
Документація
1. Детальний опис запропонованої зміни.
2. Протокол управління змінами для АФІ або діючої речовини.
| 2.1.5.3. Вилучення затвердженого протоколу управління змінами для АФІ або діючої речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1 | IА |
Умови
1.Вилучення, не спричинене непередбаченими обставинами або результатами випробувань, що виходять за межі специфікацій під час внесення зміни, описаної у протоколі.
Документація
1. Обґрунтування запропонованого вилучення.
2.2. Готовий лікарський засіб
2.2.1. Опис та склад
| 2.2.1.1. Зміна або додавання штампів, потовщень або інших маркувань, уключаючи заміну або додавання фарб для маркування лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміни штампів, потовщень або інших маркувань | 1, 2, 3 | 1, 2 | IА |
| б) зміна риски, призначеної для поділу таблетки на рівні дози | 1, 2, 3 | IБ |
Умови
1. Специфікації при випуску і наприкінці терміну придатності готового лікарського засобу не змінилися (крім зовнішнього вигляду).
2. Будь-яка фарба повинна відповідати вимогам до фармацевтичної продукції.
3. Риски не призначено для розділу на рівні дози.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи детальне зображення або опис затвердженого та запропонованого вигляду лікарського засобу, а також оновлену коротку характеристику лікарського засобу (за необхідності).
2. Зразки готового лікарського засобу (за необхідності).
3. Результати фармакопейних випробувань, що підтверджують відповідність характеристик/ правильного дозування лікарського засобу.
| 2.2.1.2. Зміна форми або розмірів лікарської форми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) таблетки з негайним вивільненням, капсули, супозиторії та песарії | 1, 2, 3, 4 | 1, 4 | IА |
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим або пролонгованим вивільненням та ділимі таблетки, призначені для поділу на рівні дози | 1, 2, 3, 4, 5 | IБ |
Умови
1. Порівняння профілів розчинення лікарського засобу з новими та затвердженими формою або розмірами (за необхідності). Для рослинних лікарських засобів, коли випробування на розчинення не може бути проведене, — дані щодо розпадання.
2. Специфікації при випуску та наприкінці терміну придатності лікарського засобу не змінюються (крім розміру).
3. Якісний та кількісний склад, середня маса лікарського засобу залишаються незмінними.
4. Зміни не стосуються таблеток з рискою, призначеною для розподілу на рівні дози.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи детальне зображення затверджених та запропонованих форми або розміру, а також оновлену коротку характеристику лікарського засобу (за необхідності).
2. Порівняльні дані досліджень для принаймні однієї дослідно-промислової серії лікарського засобу, які підтверджують відсутність змін у профілі розчинення (згідно з настановами та керівними принципами щодо дослідження біодоступності) для нових та затверджених розміру або форми. Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.
3. Обґрунтування відсутності необхідності проведення нового дослідження з еквівалентності відповідно до вимог настанов та керівних принципів щодо дослідження біодоступності.
4. Зразки готового лікарського засобу з новими розміром або формою (за потреби).
5. Результати фармакопейних випробувань, що підтверджують відповідність характеристик/правильного дозування лікарського засобу.
| 2.2.1.3. Зміна у складі (допоміжних речовинах) готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) смакові добавки або барвники | |||
| додавання, вилучення або заміна | 1, 2, 3, 4, 5, 6, 7, 9 | 1, 2, 4, 5, 6 | IА |
| збільшення або зменшення | 1, 2, 3, 4 | 1, 2, 4 | IА |
| б) інші допоміжні речовини | II | ||
| будь-яка незначна зміна кількісного складу допоміжних речовин у готовому лікарському засобі | 1, 2, 4, 8, 9, 10 | 1, 2, 7 | IА |
| якісні або кількісні зміни щодо однієї або декількох допоміжних речовин, які можуть значно вплинути на безпеку, якість або ефективність готового лікарського засобу | II | ||
| зміна у лікарському засобі біологічного/ імунологічного походження | II | ||
| будь-яка нова допоміжна речовина, що включає використання матеріалів людського або тваринного походження, для яких вимагається оцінка даних з вірусної безпеки або ризику передачі збудників губчатої енцефалопатії | II | ||
| зміна, яка підтверджується дослідженнями з еквівалентності | II | ||
| заміна однієї допоміжної речовини на іншу з тими самими функціональними характеристиками та на тому самому рівні | 1, 3, 4, 5, 6, 7, 8, 9 | IБ |
Умови
1. Не відбулося будь-яких змін функціональних характеристик лікарської форми, наприклад часу розпадання, профілю розчинення.
2. Будь-яке незначне коригування складу для збереження загальної маси повинно проводитись щодо допоміжної речовини, що становить основну частину складу готового лікарського засобу.
3. Специфікації готового лікарського засобу змінилися тільки у частині зовнішнього вигляду/запаху/смаку і за потреби вилучено показник якості щодо ідентифікації.
4. Розпочато дослідження стабільності відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням номерів серій) для принаймні двох дослідно-промислових або промислових серій із задовільними результатами стабільності протягом трьох місяців (на час подання змін типу IA або IБ), які демонструють подібність профілю стабільності до вже затвердженого. Заявник гарантує, що дослідження будуть завершені і одразу після завершення досліджень у разі невідповідності специфікаціям або за наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (з відповідною пропозицією). Крім того, за потреби мають бути проведені дослідження фотостабільності готового лікарського засобу.
5. Будь-які нові компоненти повинні відповідати встановленим вимогам.
6. Будь-який новий компонент не повинен передбачати використання матеріалів людського або тваринного походження, для яких необхідна оцінка вірусної безпеки або ризику передачі збудників губчатої енцефалопатії.
7. Зміна не впливає на відмінності між дозуваннями та не спричиняє негативного впливу на смакову прийнятність педіатричних форм (за потреби).
8. Профіль розчинення принаймні двох дослідно-промислових серій запропонованого складу та затвердженого складу лікарського засобу демонструє відсутність будь-яких відмінностей (згідно з настановами та керівними принципами щодо дослідження біодоступності). Для рослинних лікарських засобів, коли випробування на розчинення не може бути проведене, — дані щодо розпадання.
9. Зміна не обумовлена проблемами зі стабільністю та/або не повинна спричиняти можливі проблеми, пов’язані з безпекою, тобто розходження між дозуваннями.
10. Лікарський засіб не належить до препаратів біологічного/імунологічного походження.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє (включаючи метод ідентифікації будь-якого нового барвника за потреби), а також оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці (за необхідності).
2. Підтвердження, що дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням номерів серій) та підтвердження за необхідності, що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності, а також наявні дані не вказували на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами).
3. Результати дослідження стабільності, розпочаті відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання), за відповідними параметрами принаймні для двох дослідно-промислових або промислових серій АФІ або діючої речовини із задовільними показниками стабільності принаймні за три місяці. При цьому мають бути надані гарантії, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами).
4. Зразок готового лікарського засобу з новим складом (за потреби).
5. Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії на будь-який новий вихідний матеріал тваринного походження або, якщо необхідно, документальне підтвердження того, що вихідне джерело одержання допоміжної речовини, яке має ризик передачі збудників губчатої енцефалопатії, попередньо оцінено вповноваженими органами країни-виробника та відповідає керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами. Інформація повинна містити: назву виробника, види та тканини тварин, з яких було одержано вихідний матеріал, назву країни-постачальника тваринної сировини, її використання.
6. Дані, які підтверджують, що нова допоміжна речовина не впливає на методи контролю готового лікарського засобу, визначені у специфікації (за потреби).
7. Обґрунтування зміни/вибору допоміжних речовин тощо, що має бути представлено відповідно до фармацевтичної розробки (включаючи стабільність та антимікробні консерванти, за необхідності).
8. Для твердих лікарських форм порівняльні дані щодо профілю розчинення принаймні для двох дослідно-промислових серій лікарського засобу з новим та затвердженим складом. Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.
9. Обґрунтування відсутності необхідності проведення нового дослідження еквівалентності відповідно до вимог настанов або керівництв з дослідження біодоступності та біоеквівалентності (чинне видання).
| 2.2.1.4. Зміна маси покриття лікарських форм для перорального застосування або зміна маси оболонки капсул | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) тверді лікарські форми для перорального застосування | 1, 2, 3, 4 | 1, 2 | IА |
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим вивільненням або пролонгованої дії, для яких покриття є вирішальним чинником механізму вивільнення | II |
Умови
1. Відсутність змін у профілі розчинення принаймні двох дослідно-промислових серій лікарського засобу із запропонованим та затвердженим складом. Для рослинних лікарських засобів, коли випробування на розчинення не може бути проведене, — дані щодо розпадання.
2. Покриття не є вирішальним чинником механізму вивільнення.
3. Специфікації готового лікарського засобу змінено тільки щодо маси і розміру (за необхідності).
4. Дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) для принаймні двох дослідно-промислових або промислових серій із задовільними результатами стабільності протягом трьох місяців або більше. Заявник гарантує, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або за наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (з відповідною пропозицією).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Підтвердження, що дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням номерів серій), та підтвердження за необхідності, що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами). Крім того, у відповідних випадках мають бути проведені дослідження фотостабільності готового лікарського засобу.
| 2.2.1.5. Зміна концентрації в окремій дозі багатодозового лікарського засобу для парентерального застосування, коли кількість діючої речовини на одиницю дози (тобто сила дії) не змінюється | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| II | |||
| 2.2.1.6. Вилучення контейнера з розчинником з упаковки | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
Документація
1. Обґрунтування вилучення, включаючи заяву щодо альтернативних шляхів отримання розчинника, необхідного для безпечного та ефективного застосування лікарського засобу.
2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці.
2.2.2. Виробництво
| 2.2.2.1. Заміна або введення додаткової дільниці виробництва для частини або всього виробничого процесу готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) дільниця для вторинного пакування | 1, 2 | 1, 3, 8 | IА |
| б) дільниця для первинного пакування | 1, 2, 3, 4, 5 | 1, 2, 3, 4, 8, 9 | IА |
| в) дільниця, на якій проводяться будь-які виробничі стадії, за винятком випуску серій, проведення контролю якості та вторинного пакування, для лікарських засобів біологічного/імунологічного походження | II | ||
| г) дільниця, яка вимагає проведення первинної перевірки виробництва або перевірки виробництва конкретного готового лікарського засобу | II | ||
| ґ) дільниця, на якій проводяться будь-які виробничі стадії, за винятком випуску серій, контролю якості, первинного та вторинного пакування, для нестерильних лікарських засобів | 1, 2, 3, 4, 5, 6, 7, 8, 9 | IБ | |
| д) дільниця, на якій проводяться будь-які виробничі стадії, за винятком випуску серії, контролю якості та вторинного пакування для стерильних лікарських засобів, що вироблені з використанням асептичного методу, за винятком лікарських засобів біологічного/імунологічного походження | 1, 2, 3, 4, 5, 6, 7, 8 | IБ |
Умови
1. Задовільні результати перевірки виробництва за останні три роки, проведеної компетентними уповноваженими органами держав, які входять до PIC/s, Інспекцією ВООЗ або уповноваженим органом України.
2. Дільниця має дозвіл (ліцензію) на виробництво відповідних лікарських форм або лікарського засобу.
3. Цей лікарський засіб є нестерильним.
4. У разі необхідності, наприклад, для суспензій та емульсій — проведена схема валідації процесу або успішно виконана валідація виробництва на новій дільниці відповідно до затвердженого протоколу принаймні на трьох промислових серіях.
5. Цей лікарський засіб не є лікарським засобом біологічного/імунологічного походження.
Документація
1. Підтвердження, що запропонована дільниця виробництва має відповідний дозвіл (ліцензію) на виробництво лікарської форми або лікарського засобу, а саме:
для виробничої дільниці в Україні — копія чинної ліцензії на виробництво;
копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника;
засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійний лист заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи.
2. Інформація щодо кількості (≥ 3), номерів, розміру та дати виробництва серій, які використовуються у дослідженнях з валідації, дані з валідації або протокол (схема) валідації (за потреби).
3. Заява про внесення змін має чітко визначати затвердженого та запропонованого виробників готового лікарського засобу, як зазначено в пункті 2.5 заяви про державну реєстрацію лікарського засобу, форма якої наведена у додатку 1 до Порядку.
4. Копії затверджених специфікацій при випуску та наприкінці терміну придатності (за потреби).
5. Порівняльні дані аналізів однієї промислової та двох дослідно-промислових серій (або двох промислових серій), вироблених на запропонованій дільниці, та трьох серій, вироблених на затвердженій дільниці. Дані про наступні дві промислові серії мають бути надані до Центру на вимогу або у разі невідповідності специфікаціям (з відповідною пропозицією).
6. Для м’яких та рідких лікарських форм, у яких АФІ або діюча речовина присутня у нерозчиненій формі, мають бути надані необхідні дані з валідації, мікроскопічне зображення розподілу та морфологія часток.
7. Якщо нова виробнича дільниця використовує АФІ або діючу речовину як вихідний матеріал — заява УО, яка відповідає за випуск серії на дільниці, що АФІ або діюча речовина виробляється згідно з вимогами належної виробничої практики щодо вихідних матеріалів (чинне видання).
8. Зміни до відповідних розділів матеріалів реєстраційного досьє.
9. Якщо виробництво готового лікарського засобу та його первинне пакування здійснюється на різних виробничих дільницях, слід визначити умови транспортування та зберігання готового лікарського засобу у формі in bulk та провести дослідження з їх валідації.
Примітка.
Виробники повинні використовувати у якості вихідних матеріалів тільки ті АФІ або діючі речовини, що виробляються відповідно до вимог належної виробничої практики, тому кожний виробник, що використовує АФІ або діючі речовини у якості вихідного матеріалу, повинен надати таку заяву. Крім того, оскільки УО, відповідальна за випуск серії, несе відповідальність за серію, у разі, коли контроль серії відбувається на іншій дільниці, необхідна додаткова заява щодо відповідності вимогам належної виробничої практики від УО, відповідальної за контроль.
У випадках, коли виробником виступає одна особа, слід надавати тільки одну заяву.
Якщо виробників більше одного, заявник може надати тільки одну заяву, підписану однією УО, за умови, що:
із заяви зрозуміло, що вона складена від імені всіх задіяних УО;
є у наявності письмовий контракт між заявником та виробником, у якому чітко визначені обов’язки кожної зі сторін, а УО, що надає заяву, зазначена у контракті як особа, що несе відповідальність за дотримання вимог GMP при виробництві АФІ або діючої речовини.
Заявником підтверджується, що він користується послугами принаймні однієї УО. Не можуть бути прийняті заяви від персоналу, який найнятий виробниками в інших країнах, включаючи країни з міжнародними договорами про взаємне визнання результатів інспектування на відповідність виробництва вимогам належної виробничої практики.
Заява не вимагається для крові або компонентів крові, що повинні відповідати вимогам Директиви 2002/98/ЄС Європейського парламенту та Ради від 27 січня 2003 року про встановлення стандартів якості та безпеки для збору, тестування, обробки, зберігання та розподілу людської крові та її компонентів, а також поправок до Директиви 2001/83/ЄС.
| 2.2.2.2. Зміни, що стосуються випуску серії та контролю якості готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) заміна або додавання дільниці, на якій здійснюється контроль/випробування серії | 2, 3, 4 | 1, 2, 5 | IА |
| б) заміна або додавання виробника, відповідального за випуск серії | |||
| не включаючи контроль/випробування серії | 1, 2 | 1, 2, 3, 4, 5 | IА |
| включаючи контроль/випробування серії | 1, 2, 3, 4 | 1, 2, 3, 4, 5 | IА |
| включаючи контроль/випробування серії для лікарського засобу біологічного/імунологічногопоходження та один з методів аналізу, що застосовується на дільниці, що є біологічним/імунологічним/імунохімічним методом | II |
Умови
1. Виробник (дільниця) повинен мати відповідний дозвіл (ліцензію).
2. Лікарський засіб не є лікарським засобом біологічного/імунологічного походження.
3. Перенесення із затвердженої до нової дільниці або лабораторії має бути успішно виконано.
Документація
1. Підтвердження, що запропонована дільниця виробництва має відповідний дозвіл (ліцензію) на виробництво лікарської форми або лікарського засобу, а саме:
для виробничої дільниці в Україні — копія чинної ліцензії на виробництво;
копія ліцензії на виробництво (якщо згідно із законодавством країни виробника ліцензія на виробництво існує лише в електронному вигляді (наприклад, США), має бути надана роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом/печаткою заявника) або іншого дозвільного документа на виробництво заявленої лікарської форми у країні виробника;
засвідчена копія документа, що підтверджує відповідність виробництва лікарського засобу вимогам GMP, виданого Держлікслужбою відповідно до вимог наказу Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130 «Про затвердження Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики», зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665, або гарантійного листа заявника щодо надання такого документа протягом строку проведення спеціалізованої експертизи.
2. Заява про внесення змін має чітко визначати затвердженого та запропонованого виробників готового лікарського засобу, як зазначено в пункті 2.5 заяви про державну реєстрацію лікарського засобу, форма якої наведена у додатку 1 до Порядку.
3. Заява Уповноваженої особи, яка відповідає за контроль якості серії, що АФІ або діюча речовина виробляється згідно з вимогами належної виробничої практики щодо вихідних матеріалів (чинне видання). Узагальнена заява може бути надана за умов, викладених у підпункті 2.2.2.1 цього додатка.
4. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи оновлену коротку характеристику лікарського засобу, інструкцію для медичного застосування та пропозиції до маркування на упаковці (за необхідності).
| 2.2.2.3. Зміни у процесі виробництва готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначна зміна у процесі виробництва твердої лікарської форми для перорального застосування з негайним вивільненням або розчинів для перорального застосування | 1, 2, 3, 4, 5, 6, 7 | 1, 3, 4, 6, 7, 8 | ІА |
| б) зміна у процесі виробництва, яка може мати істотний влив на якість, безпеку та ефективність лікарського засобу | II | ||
| в) лікарський засіб є лікарським засобом біологічного/імунологічного походження та зміна вимагає проведення порівняльних досліджень | ІІ | ||
| г) ведення нестандартного методу кінцевої стерилізації | ІІ | ||
| ґ) введення або збільшення припустимого надлишку діючої речовини | ІІ | ||
| д) незначна зміна у процесі виробництва водної суспензії для перорального застосування | 1, 2, 4, 6, 7, 8 | IБ |
Умови
1. Немає жодних змін у якісних або кількісних показниках профілю домішок або у фізико-хімічних властивостях.
2. Цей лікарський засіб не є лікарським засобом біологічного/імунологічного або рослинного походження.
3. Виробничий процес, який включає окремі стадії, залишається таким самим, наприклад, обробка проміжних продуктів, і немає жодних змін у будь-якому розчиннику, що використовується у процесі виробництва.
4. Затверджений виробничий процес має контролюватися відповідними методами і ці методи не потребують жодних змін (розширення або вилучення допустимих меж).
5. Специфікації на готовий лікарський засіб або проміжні продукти залишаються незмінними.
6. Новий виробничий процес повинен забезпечити виготовлення аналогічного попередньому лікарського засобу за усіма показниками якості, безпеки та ефективності.
7. Розпочато дослідження стабільності відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) для принаймні однієї дослідно-промислової або промислової серії із задовільними результатами стабільності протягом трьох місяців або більше. Заявник гарантує, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або за наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (з відповідною пропозицією).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи порівняння затвердженого та запропонованого виробничого процесу.
2. Для м’яких та рідких лікарських форм, у яких діюча речовина міститься у нерозчинній формі, відповідні дані з валідації змін, включаючи мікроскопію часток для перевірки видимих змін у морфології; порівняльні дані гранулометричного складу за відповідним методом.
3. Для твердих лікарських форм — дані, що підтверджують відсутність змін у профілі розчинення однієї промислової серії, виробленої за зміненою технологією порівняно з останніми трьома серіями, виробленими за узгодженою технологією. Дані про наступні дві повні промислові серії мають бути надані на вимогу або у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією). Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.
4. Обґрунтування відсутності необхідності проведення досліджень еквівалентності згідно з настановами або керівництвами з дослідження біодоступності (чинне видання).
5. У разі зміни у процесі стерилізації необхідно надати дані з валідації.
6. Копія затверджених специфікацій при випуску та наприкінці терміну придатності.
7. Дані аналізів (у вигляді порівняльної таблиці) принаймні однієї промислової серії, виробленої за узгодженою та запропонованою технологіями. Дані про наступні дві повні промислові серії мають бути надані на вимогу або у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією).
8. Підтвердження, що дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням номерів серій) для принаймні однієї дослідно-промислової або промислової серії із задовільними результатами стабільності протягом трьох місяців або більше та профілем стабільності, подібним до профілю стабільності лікарського засобу, виробленого за узгодженою технологією. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами).
| 2.2.2.4. Зміна розміру серії (включаючи діапазон розміру серії) готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) збільшення до 10 разів порівняно із затвердженим розміром серії | 1, 2, 3, 4, 5, 7 | 1, 4 | IА |
| б) зменшення до 10 разів порівняно із затвердженим розміром серії | 1, 2, 3, 4, 5, 6 | 1, 4 | IА |
| в) зміна вимагає оцінки порівнянності (проведення порівняльних досліджень) лікарського засобу біологічного/імунологічного походження | ІІ | ||
| г) зміна стосується всіх інших лікарських форм сукупного (комплексного) виробничого процесу | ІІ | ||
| ґ) збільшення розміру серії більш ніж у 10 разів порівняно із затвердженим розміром для твердих лікарських форм з негайним вивільненням | 1, 2, 3, 4, 5, 6 | ІБ | |
| д) масштаб для лікарського засобу біологічного/імунологічного походження збільшився/зменшився без зміни виробничого процесу (наприклад, дублювання лінії) | 1, 2, 3, 4, 5, 6 | ІБ |
Умови
1. Зміна не впливає на відтворюваність та/або постійність лікарського засобу.
2. Зміна стосується тільки твердих лікарських форм з негайним вивільненням для перорального застосування або нестерильних рідких лікарських форм.
3. Будь-які зміни методу виробництва та/або контролю у процесі виробництва спричинені зміною розміру серії, наприклад, використанням обладнання іншої продуктивності.
4. Існує схема валідації або валідація виробництва була успішно проведена згідно із затвердженим протоколом з використанням принаймні трьох серій лікарського засобу нового розміру відповідно до настанов або керівництв з валідації виробництва (чинне видання).
5. Цей лікарський засіб не є лікарським засобом біологічного/імунологічного походження.
6. Зміна не обумовлена непередбаченими обставинами у процесі виробництва або пересторогами щодо стабільності.
7. Попередній розмір серії не був затверджений під час внесення зміни типу ІА.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Дані аналізів (у вигляді порівняльної таблиці) принаймні однієї промислової серії затвердженого та запропонованого розмірів. Дані про наступні дві повні промислові серії мають бути надані на вимогу або у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією).
3. Копії затверджених специфікацій при випуску та наприкінці терміну придатності.
4. Інформація щодо кількості (≥ 3), номерів, розмірів серій лікарських засобів та дат їх виробництва, що використовуються у дослідженнях з валідації, або схема валідації.
5. Дані з валідації.
6. Підтвердження, що дослідження стабільності розпочаті відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням кількості та номерів серій) для принаймні однієї дослідно-промислової або промислової серії із задовільними результатами стабільності протягом трьох місяців або більше. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами). Для лікарських засобів біологічного/імунологічного походження — підтвердження відсутності необхідності проведення порівняльних досліджень.
| 2.2.2.5. Зміни випробувань або допустимих меж, встановлених у специфікаціях, під час виробництва готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення нового методу випробування та допустимих меж | 1, 2, 5, 6 | 1, 2, 3, 4, 5, 7 | IА |
| в) вилучення несуттєвого випробування | 1, 2 | 1, 2, 6 | IА |
| г) вилучення випробування у процесі виробництва, яке може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | ||
| ґ) розширення затверджених допустимих меж для показників, які можуть мати істотний вплив на загальну якість готового лікарського засобу | ІІ | ||
| д) доповнення або заміна випробування за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 5, 7 | ІБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду допустимих меж специфікацій (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II).
2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва, наприклад, утворенням нової некваліфікованої домішки, зміною допустимих меж загального вмісту домішок.
3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.
4. Метод випробування залишається незмінним або такі зміни є незначними.
5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
6. Новий метод випробування не належить до біологічного/імунологічного/імунохімічного методу або методу, у якому використовується біологічний реактив для субстанції біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця щодо затвердженого та запропонованого випробування та допустимих меж.
3. Опис будь-якого нового методу випробування та дані з його валідації (за необхідності).
4. Дані аналізів для двох промислових серій (трьох промислових серій для лікарських засобів біологічного походження, якщо не обґрунтовано інше) готового лікарського засобу за всіма показниками специфікацій.
5. Порівняльні дані щодо профілю розчинення для готового лікарського засобу принаймні однієї дослідної серії, виготовленої за затверджених та запропонованих умов (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.
6. Обґрунтування/оцінка ризику, що підтверджує незначність зміненого параметра.
7. Обґрунтування введення нового випробування у процесі виробництва та допустимих меж.
2.2.3. Контроль допоміжних речовин
| 2.2.3.1. Зміна параметрів специфікацій та/або допустимих меж для допоміжної речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | ІА |
| б) доповнення специфікації новим показником з відповідним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 6, 8 | ІА |
| в) вилучення із специфікації незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 7 | ІА |
| г) зміна знаходиться поза затвердженими допустимими межами специфікацій | ІІ | ||
| ґ) вилучення із специфікації показника, який може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | ||
| д) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за винятком лікарських засобів біологічного та імунологічного походження) | 1, 2, 3, 4, 5, 6, 8 | ІБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду допустимих меж специфікацій (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II).
2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва, наприклад, утворення нової некваліфікованої домішки; зміна допустимих меж загального вмісту домішок.
3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.
4. Метод випробування залишається незмінним або такі зміни є незначними.
5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
6. Новий метод випробування не належить до біологічного/імунологічного/імунохімічного методу або методу, у якому використовується біологічний реактив для субстанції біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
7. Зміни не стосуються домішки, яка має генотоксичну дію.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця щодо затвердженої та запропонованої специфікацій.
3. Опис нового методу випробування та дані з його валідації (за необхідності).
4. Дані аналізів для двох промислових серій (трьох промислових серій для лікарських засобів біологічного походження, якщо не обґрунтовано інше) готового лікарського засобу за всіма показниками специфікацій.
5. Порівняльні дані профілю розчинення для готового лікарського засобу принаймні однієї дослідної серії з допоміжними речовинами, які відповідають затвердженій та запропонованій специфікаціям (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.
6. Обґрунтування відсутності нових даних з еквівалентності згідно з вимогами настанов та керівними принципами щодо дослідження біодоступності (чинне видання) (за необхідності).
7. Обґрунтування/оцінка ризику, що підтверджує незначність зміненого параметра.
8. Обґрунтування введення нового показника специфікації та допустимих меж.
| 2.2.3.2. Зміна у методах випробування допоміжної речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробувань | 1, 2, 3, 4 | 1, 2 | IА |
| б) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | 5 | 1 | ІА |
| в) заміна біологічного/імунологічного/імунохімічного методу випробування або методу, у якому використовується біологічний реагент | ІІ | ||
| г) інші зміни у методах випробування (включаючи заміну або додавання) | 1, 2 | IБ |
Умови
1. Дослідження з валідації були проведені відповідно до чинних фармакопейних вимог або настанов та керівних принципів з валідації (чинне видання) та результати проведених досліджень підтверджують, що нова методика — ідентична затвердженій.
2. Не було жодних змін меж загального вмісту домішок, не виявлено нових некваліфікованих домішок.
3. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу, але тип колонки або методика — незмінні).
4. Новий метод випробувань не належить до біологічних/імунологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу субстанції біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
5. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні зміни типу ІА.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи опис методу аналізу, звіт про дані з валідації, оновлені специфікації щодо домішок (за необхідності).
2. Порівняльні дані з валідації або, якщо обґрунтовано, порівняльні результати аналізу, які підтверджують, що затверджений та запропонований методи випробування є ідентичними. Ця вимога не стосується випадку додавання нового методу випробування.
| 2.2.3.3. Заміна джерела одержання допоміжної речовини або реактиву, що становить ризик передачі збудників губчатої енцефалопатії | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) матеріалу, що становить ризик передачі збудників губчатої енцефалопатії, на матеріал рослинного або синтетичного походження | |||
| для допоміжних речовин або реактивів, які не використовуються у виробництві активної субстанції біологічного/імунологічного походження або лікарського засобу біологічного/імунологічного походження | 1 | 1 | IА |
| для допоміжних речовин або реактивів, які використовуються у виробництві активної субстанції біологічного/імунологічного походження або лікарського засобу біологічного/імунологічного походження | 1, 2 | IБ | |
| б) заміна або додавання речовини, що становить ризик передачі збудників губчатої енцефалопатії, або заміна речовини, що становить ризик передачі збудників губчатої енцефалопатії, на іншу речовину, що становить ризик передачі збудників губчатої енцефалопатії, для якої немає сертифіката відповідності Європейській фармакопеї щодо губчатої енцефалопатії | II |
Умови
1. Специфікації при випуску та наприкінці терміну придатності на допоміжну речовину та готовий лікарський засіб залишаються незмінними.
Документація
1. Заява від виробника або заявника, яка підтверджує, що допоміжна речовина є речовиною виключно рослинного або синтетичного походження.
2. Результати дослідження еквівалентності матеріалів, їх впливу на виробництво кінцевої речовини та впливу на характеристики (наприклад, параметри розчинення) готового лікарського засобу.
| 2.2.3.4. Зміни в методі синтезу або регенерації нефармакопейної допоміжної речовини (за наявності в досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у методі синтезу або регенерації | 1, 2 | 1, 2, 3, 4 | IА |
| б) зміни у специфікації або зміна фізико-хімічних властивостей допоміжної речовини, що може мати вплив на якість готового лікарського засобу | ІІ | ||
| в) допоміжна речовина є речовиною біологічного/імунологічного походження | ІІ |
Умови
1. Метод синтезу та специфікації залишаються незмінними, відсутні зміни у якісних і кількісних показниках профілю домішок (за винятком залишкових розчинників, за умови, що вони відповідають керівним принципам ІСН) або фізико-хімічних властивостях.
2. За винятком ад’ювантів для вакцин.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Дані аналізів (у формі порівняльної таблиці) принаймні двох серій (мінімум дослідно-промислових) з використанням допоміжної речовини, виготовленої за узгодженим та новим методами виробництва.
3. Порівняльні дані щодо профілю розчинення для готового лікарського засобу принаймні двох серій (мінімум дослідно-промислових) з використанням допоміжної речовини, виготовленої за узгодженим та новим методами виробництва (за необхідності). Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.
4. Копії затверджених та нових (за необхідності) специфікацій на допоміжну речовину.
2.2.4. Контроль готового лікарського засобу
| 2.2.4.1. Зміна параметрів специфікацій та/або допустимих меж готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) звуження допустимих меж для лікарського засобу, що підлягає офіційному випуску серії | 1, 2, 3, 4 | 1, 2 | IА |
| в) доповнення специфікації новим показником з відповідним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 5, 7 | IА |
| г) вилучення незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 6 | IА |
| ґ) зміна знаходиться поза затвердженими допустимими межами | ІІ | ||
| д) вилучення показника, який може мати істотний вплив на якість готового лікарського засобу | ІІ | ||
| е) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за винятком лікарських засобів біологічного/імунологічного походження) | 1, 2, 3, 4, 5, 7 | ІБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду допустимих меж специфікацій (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II).
2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва, наприклад, утворенням нової некваліфікованої домішки; зміною допустимих меж загального вмісту домішок.
3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.
4. Метод випробування залишається незмінним або такі зміни є незначними.
5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
6. Новий метод випробування не належить до біологічного/імунологічного/імунохімічного методу або методу, у якому використовується біологічний реактив для АФІ або діючої речовини біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
7. Зміни не стосуються домішки, яка має генотоксичну дію.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця щодо затверджених та запропонованих специфікацій.
3. Опис нового методу випробування та дані з його валідації (за необхідності).
4. Дані аналізів для двох промислових серій (трьох промислових серій для лікарських засобів біологічного походження, якщо не обґрунтовано інше) готового лікарського засобу за всіма показниками специфікацій.
5. Порівняльні дані профілю розчинення для готового лікарського засобу принаймні однієї дослідної серії, які відповідають затвердженій та запропонованій специфікаціям (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.
6. Обґрунтування/оцінка ризику, що підтверджує незначність зміненого параметра.
7. Обґрунтування введення нового показника та допустимих меж.
| 2.2.4.2. Зміна у методах випробування готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначна зміна у затверджених методах випробування | 1, 2, 3, 4 | 1, 2 | IА |
| б) вилучення методу випробування, якщо вже затверджено альтернативний метод | 4 | 1 | IА |
| в) зміна у біологічному/імунологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент | ІІ | ||
| г) інші зміни у методах випробувань (включаючи заміну або доповнення) | 1, 2 | IБ |
Умови
1. Дослідження з валідації були проведені відповідно до чинних фармакопейних вимог або настанов, або керівних принципів з валідації (чинне видання) та результати проведених досліджень підтверджують, що нова методика — ідентична затвердженій.
2. Не було жодних змін меж загального вмісту домішок, не виявлено нових некваліфікованих домішок.
3. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу, але тип колонки або методика — незмінні).
4. Новий метод випробувань не належить до біологічних/імунологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу АФІ або діючої речовини біологічного походження (за винятком стандартних фармакопейних мікробіологічних методів).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи опис методу аналізу, звіт про дані з валідації, оновлені специфікації щодо домішок (якщо необхідно).
2. Порівняльні дані валідації або, якщо обґрунтовано, порівняльні результати аналізу, які підтверджують, що затверджений та запропонований методи випробування є ідентичними. Ця вимога не стосується випадку додавання нового методу випробування.
| 2.2.4.3. Зміни, які стосуються виробничого процесу у реальному часі або випуску за параметрами для готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| ІІ |
2.2.5. Система упаковка/укупорка
| 2.2.5.1. Зміна у первинній упаковці готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| а) якісний та кількісний склад | |||
| тверді лікарські форми | 1, 2, 3 | 1, 2, 3, 4, 6 | IА |
| м’які та нестерильні рідкі лікарські форми | 1, 2, 3, 5, 6 | IБ | |
| стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | ||
| зміна стосується зниження ступеня захисту оновленої упаковки, якщо наявні відповідні зміни в умовах зберігання та/або скорочення терміну придатності | ІІ | ||
| б) тип контейнера | |||
| тверді, м’які та нестерильні рідкі лікарські форми | 1, 2, 3, 5, 6, 7 | ІБ | |
| стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ |
Умови
1. Зміна стосується лише одного типу упаковки/контейнера (наприклад, заміна блістера на блістер).
2. Запропонований пакувальний матеріал повинен бути ідентичним затвердженому за відповідними властивостями.
3. Розпочато дослідження стабільності відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) для принаймні двох дослідно-промислових або промислових серій із задовільними результатами стабільності протягом трьох місяців або більше. Однак, якщо запропонований пакувальний матеріал — більш стійкий, ніж затверджений (наприклад, матеріал блістера має більшу товщину), дані щодо стабільності за три місяці ще є недоступними. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці (за необхідності).
2. Відповідні дані щодо нової упаковки (порівняльні дані про проникність, наприклад для O2, CO2, вологи).
3. Підтвердження відсутності (за необхідності) будь-якої взаємодії між лікарським засобом та пакувальним матеріалом (наприклад, не було перенесення компонентів запропонованого пакувального матеріалу до лікарського засобу та навпаки), включаючи підтвердження того, що пакувальний матеріал відповідає чинним фармакопейним вимогам щодо пакувальних матеріалів або законодавству ЄС щодо безпеки взаємодії пластикових пакувальних матеріалів та упаковок з харчовими продуктами.
4. Заява, що дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням номерів серій) та (за необхідності) що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності, а також наявні дані не вказували на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності, одразу після завершення досліджень дані будуть подані до Центру (із запропонованими заходами).
5. Результати дослідження стабільності, які було проведено відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання), за відповідними параметрами принаймні для двох дослідно-промислових або промислових серій лікарського засобу із задовільними показниками щодо стабільності принаймні за три місяці. При цьому мають бути надані гарантії того, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності дані будуть подані до Центру (із запропонованими заходами).
6. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій (за необхідності).
7. Зразки нової системи упаковка/укупорка (за необхідності).
Примітка. Будь-яка зміна, що призводить до нової лікарської форми, потребує подання заяви про державну реєстрацію лікарського засобу, форма якої наведена у додатку 1 до Порядку.
| 2.2.5.2. Зміна параметрів специфікацій та/або допустимих меж первинної упаковки готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж, визначених у специфікації | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфікації новим показником з відповідним методом випробування | 1, 2, 5 | 1, 2, 3, 4, 6 | IА |
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 5 | ІА |
| г) додавання або заміна показника за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 6 | ІБ |
Умови
1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду параметрів специфікації (наприклад, проведеною під час подання заяви про реєстрацію або внесення змін типу II).
2. Зміна, не обумовлена непередбаченими обставинами у процесі виробництва.
3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.
4. Методи випробування залишаються незмінними або такі зміни є незначними.
5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій.
3. Опис нового методу випробування та дані з його валідації (за необхідності).
4. Результати аналізу для двох серій первинної упаковки за усіма показниками, зазначеними у специфікації.
5. Обґрунтування того, що показник є незначним.
6. Обґрунтування нового показника якості та допустимих меж.
| 2.2.5.3. Зміна у методах випробування первинної упаковки готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробувань | 1, 2, 3 | 1, 2 | IА |
| б) інші зміни у методах випробувань (включаючи заміну або додавання) | 1, 3, 4 | 1, 2 | IА |
| в) вилучення методу випробування, якщо вже затверджений альтернативний | 5 | 1 | ІА |
Умови
1. Дослідження з валідації були проведені відповідно до чинних фармакопейних вимог або настанов та керівних принципів з валідації (чинне видання) та результати проведених досліджень підтверджують, що нова методика — ідентична затвердженій.
2. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу, але тип колонки або методика — незмінні).
3. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.
4. Субстанція/готовий лікарський засіб не є речовиною/лікарським засобом біологічного/імунологічного походження.
5. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні зміни типу ІА.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи опис методу випробування та звіт з його валідації.
2. Порівняльні дані валідації або, якщо обумовлено, порівняльні результати аналізу, які підтверджують ідентичність результатів, отриманих за затвердженим та новим методами випробувань. Це не стосується випадків додавання нового методу випробування.
| 2.2.5.4. Зміна форми або розміру контейнера чи закупорювального засобу (первинної упаковки) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) нестерильні лікарські засоби | 1, 2, 3 | 1, 2, 4 | IА |
| б) зміна форми або розміру основної частини пакувального матеріалу (вторинної упаковки), що може мати значний вплив на доставку, застосування, безпеку та стабільність готового лікарського засобу | IІ | ||
| в) стерильні лікарські засоби | 1, 2, 3, 4 | ІБ |
Умови
1. Відсутні якісні або кількісні зміни складу пакувального матеріалу.
2. Зміна не стосується основної частини пакувального матеріалу, яка впливає на доставку, застосування, безпеку та стабільність готового лікарського засобу.
3. У разі зміни вільного простору над лікарським засобом або зміни у співвідношенні поверхня/об’єм дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) принаймні для двох дослідно-промислових (трьох для лікарських засобів біологічного/імунологічного походження) або промислових серій лікарського засобу із задовільними показниками щодо стабільності принаймні за три місяці (шість місяців для лікарських засобів біологічного/імунологічного походження). При цьому мають бути надані гарантії того, що дослідження будуть завершені, і одразу після завершення досліджень у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності отримані дані будуть подані до Центру (із запропонованими заходами).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи опис, детальне зображення та склад контейнера або закупорювального матеріалу, оновлені коротку характеристику лікарського засобу та інструкцію для медичного застосування (за необхідності).
2. Зразок нового пакування (за необхідності).
3. Дослідження з ревалідації проводилися для стерильних лікарських засобів, які пройшли кінцеву стерилізацію. Слід зазначити номери серій лікарського засобу, що використовувались в дослідженнях (за необхідності).
4. У разі зміни вільного простору над лікарським засобом або зміни співвідношення поверхня/об’єм має бути надане підтвердження того, що дослідження стабільності розпочато відповідно до вимог настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням номерів серій лікарського засобу) та (за необхідності) що на дату подання заяви про внесення змін типу ІА або ІБ у розпорядженні заявника було мінімум, який вимагається, даних щодо стабільності, а також наявні дані не вказували на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності, одразу після завершення досліджень дані будуть подані до Центру (із запропонованими заходами).
| 2.2.5.5. Зміна розміру упаковки готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна кількості одиниць (наприклад, таблеток, ампул та ін.) в упаковці: | |||
| зміна у діапазоні затверджених розмірів упаковки | 1, 2 | 1, 3 | IА |
| зміна поза діапазоном затверджених розмірів упаковки | 1, 2, 3 | IБ | |
| б) вилучення упаковки певного розміру | 3 | 1, 2 | ІА |
| в) зміна маси/об’єму вмісту контейнера багатодозового стерильного лікарського засобу (або однодозового часткового використання) та багатодозового лікарського засобу біологічного/імунологічного походження для парентерального застосування | IІ | ||
| г) зміна маси/об’єму вмісту контейнера багатодозового лікарського засобу для непарентеральних застосувань (або однодозового часткового використання) | 1, 2, 3 | ІБ |
Умови
1. Новий розмір упаковки повинен відповідати дозуванню і тривалості лікування відповідно до затвердженої короткої характеристики лікарського засобу.
2. Первинний пакувальний матеріал не змінився.
3. Незмінені форми випуску лікарського засобу повинні відповідати інструкції з дозування та тривалості лікування, як вказано в короткій характеристиці лікарського засобу.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи оновлену коротку характеристику лікарського засобу (за необхідності).
2. Обґрунтування нового/незміненого розміру упаковки та підтвердження, що новий/незмінений розмір упаковки відповідає схемі дозування та тривалості лікування, затвердженим у короткій характеристиці лікарського засобу.
3. Заява, що дослідження стабільності будуть проводитись відповідно до настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (чинне видання), якщо показники стабільності будуть змінені. Дані мають бути подані до Центру лише у разі невідповідності специфікаціям (з відповідною пропозицією).
Примітка до підпунктів «в» та «г» пункту 2.2.5.5. Будь-які зміни у силі дії лікарського засобу потребують подання заяви про реєстрацію лікарського засобу.
| 2.2.5.6. Зміна будь-якої частини матеріалу первинної упаковки, що не контактує з готовим лікарським засобом (наприклад, колір кришечок з контролем першого відкриття, колір кодових кілець на ампулах, контейнера для голок (різні види пластмаси) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна, яка впливає на коротку характеристику лікарського засобу | 1 | 1 | IА |
| б) зміна, яка не впливає на коротку характеристику лікарського засобу | 1 | 1 | IА |
Умови
1. Зміна не стосується тієї частини пакувального матеріалу, яка б могла вплинути на доставку, застосування, безпеку та стабільність готового лікарського засобу.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи оновлену коротку характеристику лікарського засобу (за необхідності).
| 2.2.5.7. Зміна постачальника пакувальних матеріалів або комплектуючих (якщо зазначено в досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) вилучення постачальника | 1 | 1 | IА |
| б) заміна або додавання постачальника | 1, 2, 3, 4 | 1, 2, 3 | IА |
| в) будь-яка зміна постачальника спейсерів для дозованих інгаляторів | ІІ |
Умови
1. Жодних вилучень у компонентах упаковки або комплектуючих не відбулося.
2. Кількісний та якісний склад пакувального матеріалу/комплектуючих та проектні специфікації не змінилися.
3. Специфікації та методи контролю якості — принаймні ідентичні.
4. Метод та умови стерилізації залишаються незмінними (за необхідності).
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Для комплектуючих для лікарських засобів — підтвердження CE-маркування (сертифікат СЕ).
3. Порівняльна таблиця затвердженої та запропонованої специфікацій, якщо необхідно.
Примітка. СЕ-маркування (CE-marking) — скорочення від Conformit Europeane (Європейська відповідність) — особливий знак, який наноситься на виріб і засвідчує, що даний виріб відповідає основним вимогам Європейських Директив, а також те, що він пройшов процедуру оцінки відповідності Директивам. Маркування «СЕ» вказує на те, що виріб є безпечним для здоров’я людини, а також для навколишнього середовища.
2.2.6. Стабільність
| 2.2.6.1. Зміна у термінах придатності або умовах зберігання готового лікарського засобу: | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зменшення терміну придатності готового лікарського засобу: | |||
| для торговельної упаковки | 1 | 1, 2, 3 | IА |
| після першого розкриття | 1 | 1, 2, 3 | IА |
| після розчинення або відновлення | 1 | 1, 2, 3 | IА |
| б) збільшення терміну придатності готового лікарського засобу: | |||
| для торговельної упаковки (підтверджується даними стабільності реального часу) | 1, 2, 3 | IБ | |
| після першого розкриття (підтверджується даними стабільності реального часу) | 1, 2, 3 | IБ | |
| після розчинення або відновлення (підтверджується даними стабільності реального часу) | 1, 2, 3 | IБ | |
| збільшення терміну придатності на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності до вимог настанови щодо дослідження стабільності лікарських засобів або керівних принципів ICH щодо дослідження стабільності лікарських засобів* | ІІ | ||
| збільшення терміну придатності лікарського засобу біологічного/імунологічного походження на основі результатів досліджень стабільності, проведених відповідно до затвердженого протоколу стабільності | 1, 2, 3 | IБ | |
| в) зміна в умовах зберігання лікарського засобу біологічного походження, якщо дослідження стабільності були проведені не у відповідності до затвердженого протоколу стабільності | ІІ | ||
| г) зміна в умовах зберігання готового лікарського засобу або після розчинення/відновлення | 1, 2, 3 | IБ |
Умови
1. Зміна, не обумовлена непередбаченими обставинами у процесі виробництва або пересторогами щодо стабільності.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє повинні містити результати досліджень стабільності у реальному часі (що включають повний термін придатності), проведених відповідно до настанови щодо дослідження стабільності лікарських засобів або керівних принципів ICH щодо дослідження стабільності лікарських засобів (чинне видання) принаймні на двох дослідно-промислових серіях1 готового лікарського засобу у затвердженій упаковці та/або після першого розкриття, або відновлення (за необхідності). За потреби мають бути включені результати мікробіологічних досліджень.
2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці.
3. Копія затвердженої специфікації наприкінці терміну придатності готового лікарського засобу та, якщо необхідно, специфікації на лікарський засіб після розчинення/відновлення або першого розкриття.
*Екстраполяція не застосовується до лікарських засобів біологічного/імунологічного походження.
____________________________________
1 Із зобов’язанням підтвердити термін придатності на промисловій серії.
2.2.7. Проектний простір
| 2.2.7.1. Введення нового проектного простору або розширення затвердженого для готового лікарського засобу (за винятком лікарських засобів біологічного походження) щодо | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) одного або більше елементів (блок, частина) виробничого процесу готового лікарського засобу, включаючи контроль у процесі виробництва та/або методи випробувань | 1, 2, 3 | II | |
| б) методів випробувань для допоміжних речовин/проміжних продуктів та/або готового лікарського засобу | 1, 2, 3 | II |
Документація
1. Результати досліджень розробки лікарського засобу або виробничого процесу (включаючи дослідження оцінки ризику та багатомірні дослідження, якщо необхідно), які демонструють, що функціональна взаємодія властивостей матеріалу та параметрів процесу, які можуть впливати на критичні характеристики якості готового лікарського засобу, була досягнута.
2. Опис проектного простору у формі таблиці, включаючи змінні властивості матеріалу та параметри процесу (якщо необхідно) та їх запропоновані діапазони.
3. Зміни до відповідних розділів матеріалів реєстраційного досьє.
| 2.2.7.2. Внесення змін після затвердження протоколу управління змінами для готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | II |
Документація
1. Детальний опис запропонованої зміни.
2. Протокол управління змінами для готового лікарського засобу.
| 2.2.7.3. Вилучення затвердженого протоколу управління змінами для готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| 1 | 1 | IА |
Умови
1.Вилучення не спричинено непередбаченими обставинами або результатами випробувань, що виходять за межі специфікацій під час внесення зміни, описаної у протоколі.
Документація
1. Обґрунтування запропонованого вилучення.
2.3. Сертифікат відповідності Європейській фармакопеї/Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії
| 2.3.1. Подання нового або оновленого сертифіката відповідності Європейській фармакопеї | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| Для АФІ або діючої речовини/для вихідного матеріалу/реагенту/проміжного продукту, що використовуються у виробництві АФІ або діючої речовини/для допоміжної речовини | |||
| а) сертифікат відповідності Європейській фармакопеї | |||
| новий сертифікат відповідності від діючого виробника | 1, 2, 3, 4, 5, 8 | 1, 2, 3, 4, 5 | IА |
| оновлений сертифікат відповідності від діючого виробника | 1, 2, 3, 4, 8 | 1, 2, 3, 4, 5 | IА |
| новий сертифікат відповідності від нового виробника (заміна або доповнення) | 1, 2, 3, 4, 5, 8 | 1, 2, 3, 4, 5 | IА |
| б) сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії для АФІ або діючої речовини /вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини | |||
| новий сертифікат відповідності для АФІ або діючої речовини від нового або діючого виробника | 3, 6 | 1, 2, 3, 4, 5 | IА |
| новий сертифікат відповідності для вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини від нового або діючого виробника | 3, 6 | 1, 2, 3, 4, 5 | IА |
| оновлений сертифікат відповідності від діючого виробника | 1, 2, 3, 4, 5 | IА |
Умови
1. Специфікації при випуску та наприкінці терміну придатності готового лікарського засобу залишаються незмінними.
2. Додаткові (до Європейської фармакопеї) специфікації щодо домішок (за винятком залишкових розчинників, за умови, що вони відповідають керівним принципам ІСН) та відповідні вимоги до лікарського засобу (наприклад, профілі розміру часток, поліморфна форма) залишаються незмінними, за виключенням звуження (за необхідності).
3. Виробничий процес АФІ або діючої речовини,вихідного матеріалу/реагенту/проміжного продукту не включає використання матеріалу людського або тваринного походження, для якого проводиться оцінка даних щодо вірусної безпеки.
4. Тільки для АФІ або діючої речовини необхідно проводити дослідження безпосередньо перед використанням, якщо період повторного випробування не включений до сертифіката відповідності Європейській фармакопеї або якщо дані щодо періодичності повторного випробування ще не представлені в досьє.
5. АФІ або діюча речовина/вихідний матеріал/реагент/проміжний продукт/допоміжна речовина — нестерильні.
6. Для рослинних субстанцій: шлях виробництва, фізична форма, екстрагент та співвідношення лікарський засіб/екстрагент повинні залишатися незмінними.
Документація
1. Копія затвердженого (оновленого) сертифіката відповідності Європейській фармакопеї.
2. У випадку додавання виробничої дільниці у заяві слід чітко визначати затвердженого та запропонованого виробників, як зазначено у пункті 2.5 заяви про державну реєстрацію лікарського засобу, форма якої наведена у додатку 1 до Порядку.
3. Зміни до відповідних розділів матеріалів реєстраційного досьє.
4. Якщо необхідно, документ, в якому представлена інформація про будь-які матеріали, що підлягають оцінці вірусної безпеки відповідно до керівництва з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами, включаючи ті, що використовуються при виробництві АФІ або діючої речовини/допоміжної речовини. Інформація повинна містити таке: найменування виробника, види та тканини тварин, з яких було отримано матеріал, країна-постачальник тваринної сировини, її використання та попередній дозвіл.
5. Для АФІ або діючої речовини — заява від уповноваженої особи (УО) кожного з виробників, перелічених у заяві на внесення змін, якщо АФІ або діюча речовина використовується як вихідний матеріал, та заява УО кожного з виробників, відповідальних за випуск серії, перелічених у заяві. Ці заяви повинні підтверджувати, що виробник(и) діючої речовини, зазначений(і) у заяві, діє(ють) відповідно до вимог належної виробничої практики для вихідних матеріалів (чинне видання). Узагальнена заява може бути прийнятною за певних умов (примітка до підпункту 2.2.2.1). Виробництво проміжних продуктів також вимагає заяви УО, оскільки будь-які зміни до сертифікатів на діючу речовину та проміжні продукти — пов’язані. Заява УО надається лише у тому випадку, якщо порівняно з діючим сертифікатом є зміна у затвердженому переліку виробничих дільниць.
| 2.3.2. Зміни, пов’язані з необхідністю приведення у відповідність до монографії Державної фармакопеї України або Європейської фармакопеї | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна у специфікації нефармакопейної АФІ або діючої речовини для приведення у відповідність до вимог Державної фармакопеї України або Європейської фармакопеї | |||
| АФІ або діюча речовина | 1, 2, 3, 4, 5 | 1, 2, 3, 4, 5 | IА |
| допоміжна речовина/вихідний матеріал для виробництва АФІ або діючої речовини | 1, 2, 4 | 1, 2, 3, 4, 5 | IА |
| б) зміна у специфікації, пов’язана зі змінами в Державній фармакопеї України або Європейській фармакопеї | 1, 2, 4, 5 | 1, 2, 3, 4 | IА |
| в) зміна у специфікації, пов’язана із заміною вимог монографії Державної фармакопеї України на вимоги монографії Європейської фармакопеї | 1, 4, 5 | 1, 2, 3, 4 | IА |
Умови
1. Зміна вноситься виключно для приведення у відповідність до вимог Державної фармакопеї України або Європейської фармакопеї.
2. Додаткові специфікації до Державної фармакопеї України або Європейської фармакопеї залишаються незмінними (наприклад, профіль розміру часток, поліморфна форма або, наприклад, біопроби, агреганти).
3. Жодних значних змін у якісних та кількісних показниках профілю домішок не відбулося, якщо специфікації не звужені.
4. Додаткова валідація нового або незміненого фармакопейного методу не потрібна.
5. Для рослинних субстанцій: метод виробництва, фізична форма, екстрагент та співвідношення лікарський засіб/екстрагент повинні залишатися незмінними.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє.
2. Порівняльна таблиця вимог затвердженої та оновленої специфікацій.
3. Дані аналізу двох промислових серій АФІ або діючої речовини за всіма показниками оновленої специфікації.
4. Дані, які підтверджують придатність монографії для контролю АФІ або діючої речовини, наприклад, порівняння потенційних домішок із зазначеними у примітці до монографії.
5. Дані аналізів (у формі таблиць порівняння) двох промислових серій готового лікарського засобу, що містять діючу речовину, яка відповідає затвердженій та оновленій специфікаціям, а також порівняльні дані профілю розчинення принаймні однієї дослідно-промислової серії готового лікарського засобу. Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.
Примітка. Немає необхідності вносити зміни, пов’язані зі змінами у Державній фармакопеї України або Європейській фармакопеї, у випадку, якщо відповідність вимогам оновленої монографії виконується протягом шести місяців з дати її публікації та зроблено посилання на поточне видання Фармакопеї у досьє на затверджений лікарський засіб.
2.4. Медичні пристрої
| 2.4.1. Зміна пристроїв для вимірювання дози або введення лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) додавання або заміна пристрою, який не є невід’ємною частиною первинної упаковки | |||
| пристрій, який має СЕ-маркування | 1, 2, 3 | 1, 2, 4 | IА |
| спейсер для дозованих інгаляторів | ІІ | ||
| б) вилучення пристрою | 4, 5 | 1, 5 | IА |
| в) додавання або заміна пристрою, який є невід’ємною частиною первинної упаковки | IІ |
Умови
1. Запропонований пристрій для вимірювання дози повинен точно видавати необхідну дозу препарату згідно із затвердженим дозуванням. Необхідно надати результати відповідних досліджень.
2. Новий пристрій повинен бути сумісним з лікарським засобом.
3. Зміни не повинні призвести до суттєвих виправлень у короткій характеристиці лікарського засобу, інструкції для медичного застосування та маркуванні.
4. Лікарський засіб, як і раніше, демонструє відтворюваність дози.
Документація
1. Зміни до відповідних розділів матеріалів реєстраційного досьє, включаючи опис, детальне зображення та склад матеріалу пристрою, та постачальника (за необхідності), оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці (за необхідності).
2. Підтвердження СЕ-маркування (сертифікат СЕ).
3. Дані, які підтверджують безпеку, точність дозування та сумісність матеріалу пристрою та лікарського засобу.
4. Зразки нового пристрою (за потреби).
5. Обґрунтування виключення пристрою.
Примітка до підпункту «в» пункту 2.4.1. Будь-яка зміна, що призводить до нової лікарської форми, потребує подання заяви про державну реєстрацію лікарського засобу.
2.5. Зміни до реєстраційного посвідчення внаслідок інших регуляторних процедур
2.5.1. ПМФ/ВАЗФ (мастер-файл на плазму/мастер-файл на вакцинний антиген)
| 2.5.1.1. Включення нового, оновленого або зміненого мастер-файла на плазму у реєстраційне досьє на лікарський засіб (процедура 2-го етапу для мастер-файла на плазму) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) первинне включення нового мастер-файла на плазму, що впливає на властивості готового лікарського засобу | II | ||
| б) первинне включення нового мастер-файла на плазму, що не впливає на властивості готового лікарського засобу | 1, 2, 3, 4 | IБ | |
| в) включення оновленого/зміненого мастер-файла на плазму, якщо зміни впливають на властивості готового лікарського засобу | 1, 2, 3, 4 | IБ | |
| г) включення оновленого/зміненого мастер-файла на плазму, якщо зміни не впливають на властивості готового лікарського засобу | 1 | 1, 2, 3, 4 | IA |
Умови
1. Оновлений або змінений мастер-файл на плазму (далі — ПМФ)отримав сертифікат.
Документація
1. Заява, що сертифікат на ПМФ та звіт про проведену оцінку, видані уповноваженим органом, цілком придатні для реєстрації лікарського засобу; власник ПМФ надав сертифікат, звіт про оцінку та мастер-файл на плазму заявнику (у випадку, якщо заявник не є власником ПМФ), сертифікат та звіт про оцінку замінює документацію на попередній ПМФ для даного реєстраційного посвідчення.
2. Сертифікат на ПМФ та звіт про проведену оцінку, видані уповноваженим органом.
3. Експертний висновок дає стислий опис усіх змін, які внесені до сертифікованого ПМФ, та оцінює їх потенціальний вплив на готовий лікарський засіб, включаючи оцінки специфічного ризику.
4. Форма заяви на внесення змін повинна чітко визначати «затверджений» та «запропонований» сертифікат на ПМФ (номер сертифіката) у матеріалах реєстраційного досьє на готовий лікарський засіб. Заява на внесення зміни має чітко перелічувати також всі інші ПМФ, на які посилається заявник, навіть якщо вони не є предметом заяви (за необхідності).
| 2.5.1.2. Включення нового, оновленого або зміненого мастер-файла на вакцинний антиген у матеріали реєстраційного досьє на готовий лікарський засіб (процедура 2-го етапу для ВАЗФ) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) первинне включення нового мастер-файла на вакцинний антиген | II | ||
| б) включення оновленого або зміненого мастер-файла на вакцинний антиген, коли зміни впливають на властивості готового лікарського засобу | 1, 2, 3, 4 | IБ | |
| в) включення оновленого або зміненого мастер-файла на вакцинний антиген, коли зміни не впливають на властивості готового лікарського засобу | 1 | 1, 2, 3, 4 | IA |
Умови
1. Оновлений або змінений мастер-файл на вакцинний антиген отримав сертифікат відповідності.
Документація
1. Заява, що сертифікат відповідності та звіт про поведену оцінку ВАЗФ, видані уповноваженим органом, цілком придатні для реєстрації медичного імунобіологічного препарату; власник ВАЗФ надав сертифікат відповідності та звіт про проведену оцінку ВАЗФ, видані уповноваженим органом, заявнику (коли заявник не є власником ВАЗФ), сертифікат та звіт про оцінку заміщають документацію попереднього ВАЗФ для даного реєстраційного посвідчення.
2. Сертифікат відповідності та звіт про оцінку, видані уповноваженим органом.
3. Експертний висновок дає стислий опис усіх змін, внесених до сертифікованого ВАЗФ, та оцінює їх потенціальний вплив на готові лікарські засоби, включаючи оцінку специфічного ризику.
4. Форма заяви на внесення змін повинна чітко визначати «затверджений» та «запропонований» сертифікат відповідності на ВАЗФ (номер сертифіката) у реєстраційному досьє. Форма заяви на внесення зміни має чітко перелічувати також всі інші ВАЗФ, на які посилається заявник, навіть якщо вони не є предметом заяви (за необхідності).
2.5.2. Протокол управління змінами
| 2.5.2.1. Перегляд досьє з якості для внесення змін на вимогу компетентного органу після оцінки протоколу управління змінами | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) внесення зміни не вимагає будь-яких додаткових супровідних даних | 1 | 1, 2, 4 | IА |
| б) внесення зміни вимагає додаткових супровідних даних | 1, 2, 3, 4 | IB | |
| в) внесення зміни для лікарського засобу біологічного/імунологічного походження | 1, 2, 3, 4, 5 | IB |
Умови
1. Запропонована зміна введена цілком відповідно до затвердженого протоколу управління змінами, який вимагає негайного повідомлення після затвердження.
Документація
1. Посилання на затверджений протокол управління змінами.
2. Заява про те, що зміна відповідає затвердженому протоколу управління змінами, результати дослідження відповідають критеріям прийнятності, зазначеним у протоколі. Додатково заява про те, що оцінка порівнянності не вимагається для лікарських засобів біологічного/імунологічного походження.
3. Результати досліджень, що проводилися згідно із затвердженим протоколом управління змінами.
4. Зміни до відповідних розділів матеріалів реєстраційного досьє.
5. Копія затверджених специфікацій на АФІ або діючу речовину або готовий лікарський засіб.
III. ЗМІНИ ЩОДО БЕЗПЕКИ, ЕФЕКТИВНОСТІ, ФАРМАКОНАГЛЯДУ
3.1. Лікарські засоби
| 3.1.1. Зміна у короткій характеристиці лікарського засобу, інструкції для медичного застосування або маркуванні упаковок | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| а) потребує негайного внесення, але не пізніше 30 днів | 1, 2, 3 | ІА | |
| б) потребує внесення відповідно до інформації референтного/оригінального лікарського засобу без надання жодних нових додаткових даних заявником | 1, 2, 3 | ІБ | |
| в) потребує внесення відповідно до посилань на нові додаткові дані, надані заявником | 1, 3 | ІІ |
Документація
1. До супровідного листа заяви на внесення зміни додати обґрунтування внесення змін, включаючи оновлені: коротку характеристику лікарського засобу, інструкцію для медичного застосування та пропозиції до маркування на упаковці.
2. Заява про те, що запропоновані коротка характеристика лікарського засобу, інструкція для медичного застосування та пропозиції до маркування на упаковці — ідентичні такій інформації на референтний/оригінальний лікарський засіб.
3. Оновлена інформація про лікарський засіб.
| 3.1.2. Зміна у короткій характеристиці лікарського засобу, інструкції для медичного застосування, маркуванні упаковок генеричних/комбінованих/біоподібних лікарських засобів після оцінки тієї ж зміни щодо референтного/оригінального лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| а) впровадження змін(и), щодо яких(ої) заявник не надає жодних нових додаткових даних | 1, 2 | IБ | |
| б) впровадження змін(и), які (яку) необхідно надалі обґрунтувати новими додатковими даними, які має надати заявник (наприклад, порівнянність) | II |
Документація
1. До супровідного листа заяви на внесення зміни додати рішення МОЗ щодо зміни у короткій характеристиці лікарського засобу, інструкції для медичного застосування, маркуванні упаковок на лікарський засіб (за необхідності).
2. Оновлена інформація про лікарський засіб.
| 3.1.3. Внесення змін(и) на вимогу МОЗ згідно з рекомендаціями національних компетентних органів, міжнародних організацій, регуляторних агенцій інших країн після оцінки негайного обмеження щодо безпеки, маркування, характерного для певної фармакологічної та/або фармакотерапевтичної групи, регулярно оновлюваного звіту з безпеки, плану управління ризиками, контрольних заходів/спеціальних зобов’язань, змін для відображення короткої характеристики лікарського засобу та інструкції для медичного застосування | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| a) впровадження узгоджених змін у формулюванні, щодо яких заявник не надає жодних нових додаткових даних | 1, 2 | IБ | |
| б) впровадження змін, які вимагають подальшого обґрунтування новими додатковими даними, що надаються заявником | ІІ |
Документація
1. До супровідного листа заяви на внесення зміни додати рішення МОЗ щодо необхідності внесення змін та відповідний звіт з оцінки (за необхідності).
2. Оновлена інформація про лікарський засіб.
Примітка. Заявникам після отримання нової інформації, яка може призвести до змін у реєстраційному посвідченні, слід одразу надати таку інформацію та відповідну заяву про внесення змін у матеріали реєстраційного досьє і не чекати оцінки цих даних компетентними органами відповідно до процедури, описаної вище.
| 3.1.4. Зміни, що пов’язані зі значними змінами у короткій характеристиці лікарського засобу, інструкції для медичного застосування згідно з новими даними з якості, доклінічними, клінічними даними та даними фармаконагляду | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| ІІ | |||
| 3.1.5. Зміна у правовому статусі лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| а) для генеричних/комбінованих/біоподібних лікарських засобів після зміни затвердженого правового статусу референтного/оригінального лікарського засобу | 1, 2 | ІБ | |
| б) усі інші зміни правового статусу | ІІ |
Документація
1. До супровідного листа заяви на внесення зміни додати обґрунтування внесення змін з наданням документації, яка підтверджує зміну правового статусу лікарського засобу.
2. Оновлена інформація про лікарський засіб.
| 3.1.6. Зміна(и) до терапевтичних показань | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| а) додавання нового терапевтичного показання або зміни затвердженого показання | ІІ | ||
| б) вилучення терапевтичного показання | ІБ |
Примітка. Якщо додавання або зміна терапевтичного показання має місце в контексті застосування результату процедури посилання або змін до інформації на генеричний/ комбінований/біоподібний лікарський засіб після оцінки тієї ж зміни для референтного/оригінального лікарського засобу, застосовуються відповідно зміни 3.1.1 та 3.1.2.
| 3.1.7. Вилучення | Умови, які необхідно виконати | Документи, які мають бути представлені | Тип змін |
| а) лікарська форма | 1, 2 | ІБ | |
| б) сила дії | 1, 2 | ІБ |
Документація
1. Заява, що незмінені форми випуску лікарського засобу відповідають інструкції з дозування та тривалості лікування, як вказано в короткій характеристиці лікарського засобу.
2. Переглянута інформація про лікарський засіб.
Примітка. У випадках, якщо на дану лікарську форму або силу дії отримано реєстраційне посвідчення, яке відрізняється від реєстраційного посвідчення на інші лікарські форми або сили дії, строк дії реєстраційного посвідчення на попередню лікарську форму буде припинений.
| 3.1.8. Введення нової системи фармаконагляду | Умови, які необхідно виконати | Документи, які мають бути представлені | Тип змін |
| а) яка не пройшла оцінки відповідним національним компетентним органом/Європейською медичною агенцією (ЕМА) для іншого продукту того ж заявника | ІІ | ||
| б) яка пройшла оцінки відповідним національним компетентним органом/іншими регуляторними агенціями для іншого лікарського засобу того ж заявника* | 1 | IБ |
Документація
1. Новий детальний опис системи фармаконагляду.
*Стосується випадків, коли прийнятність вже оціненої системи фармаконагляду буде оцінюватися для нового реєстраційного посвідчення.
| 3.1.9. Зміни до існуючої системи фармаконагляду, як зазначено в описі системи фармаконагляду | Умови, які необхідно виконати | Документи, які мають бути представлені | Тип змін |
| а) зміна уповноваженої особи, відповідальної за фармаконагляд | 1 | 1 | IА |
| б) зміна в контактних даних уповноваженої особи, відповідальної за фармаконагляд | 1 | 2 | ІА |
| в) зміна до процедури підтримки уповноваженої особи, відповідальної за фармаконагляд | 1 | 2 | ІА |
| г) зміна до бази даних з безпеки (наприклад, введення нової бази даних з безпеки, включаючи перенесення зібраних даних з безпеки та/або аналіз та повідомлення до нової системи) | 1, 2, 3 | 2 | ІА |
| ґ) зміни в контрактних домовленостях з іншими фізичними або юридичними особами, що залучені до виконання вимог з фармаконагляду та описані в системі фармаконагляду, а саме: якщо складається договір субпідряду про подання електронних повідомлень про індивідуальний випадок, пов’язаний з безпекою лікарського засобу; про ведення головних баз даних; про відстеження сигналу або про підготовку регулярно оновлюваного звіту з безпеки | 1 | 2 | ІА |
| д) виключення тем, що увійшли у письмову процедуру(и), яка(і) описує(ють) діяльність з фармаконагляду | 1 | 2 | ІА |
| е) зміна дільниці, що проводить діяльність з фармаконагляду | 1 | 2 | ІА |
| є) інші зміни до системи фармаконагляду, які не впливають на функціонування системи фармаконагляду (наприклад, зміни розташування основного архіву, адміністративні зміни, оновлення скорочень, зміни щодо назви функцій/процедур) | 1 | 2 | ІА |
Умови
1. Система фармаконагляду залишається незмінною.
2. Система бази даних має бути валідована.
3. Перенесення даних з інших систем баз даних було валідовано.
4. Такі самі зміни до системи фармаконагляду введені для всіх лікарських засобів того самого заявника (така сама кінцева версія опису системи фармаконагляду).
Документація
1. Остання версія опису системи фармаконагляду, включаючи:
а) резюме нової уповноваженої особи, відповідальної за фармаконагляд;
б) доказ реєстрації уповноваженої особи, відповідальної за фармаконагляд в базі ЕМА Eudravigilance, у разі наявності (якщо контактні дані уповноваженої особи, відповідальної за фармаконагляд, не були включені до опису системи фармаконагляду, подання переглянутої версії опису системи фармаконагляду не вимагається, повинна подаватися лише форма заяви/повідомлення) та;
в) нове положення про заявника та уповноважену особу, відповідальну за фармаконагляд, щодо їх наявності та засоби повідомлення про побічні реакції, які підписані новою уповноваженою особою, відповідальною за фармаконагляд, та заявником, і відображення будь-яких наступних змін, наприклад, до організаційної схеми.
2. Остання версія опису системи фармаконагляду та/або остання версія додатка стосовно лікарського засобу, якщо необхідно.
3. Посилання на заяву/процедуру та лікарський засіб, до яких було внесено зміни.
Примітка до підпункту «є» пункту 3.1.9. Оцінка поданого опису системи фармаконагляду як частини нової заяви на отримання реєстраційного посвідчення/розширення/зміну може призвести до необхідності внесення змін у цьому описі системи фармаконагляду на вимогу національного компетентного органу. У цьому випадку такі самі зміни можуть бути внесені до опису системи фармаконагляду в інших реєстраційних посвідченнях того самого заявника шляхом подання загальної зміни типу ІА.
IV. Мастер-файл на плазму (далі — ПМФ)/мастер-файл на вакцинний антиген (далі — ВАЗФ)
| 4.1. Зміна найменування та/або місцезнаходження власника ВАЗФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1 | IА |
Умови
1. Власником ВАЗФ повинна залишатися одна й та сама юридична особа або фізична особа — підприємець.
Документація
1. Офіційний документ відповідного компетентного уповноваженого органу, в якому зазначено нове найменування та/або нове місцезнаходження власника ВАЗФ.
| 4.2. Зміна найменування та/або місцезнаходження власника ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1 | IА |
Умови
1. Власником ПМФ повинна залишатися одна й та сама юридична особа або фізична особа — підприємець.
Документація
1. Офіційний документ відповідного компетентного уповноваженого органу, в якому зазначено нове найменування та/або нове місцезнаходження власника ПМФ.
| 4.3. Зміна або передача власності на ПМФ від затвердженого власника новому власнику ПМФ (а саме, іншому суб’єкту господарювання) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2, 3, 4, 5, 6 | IА |
Документація
1. Документ, який містить персональні дані (найменування та місцезнаходження) затвердженого власника ПМФ, а також персональні дані особи, якій передано право користування ПМФ, разом із запропонованою датою підписання передачі права користування обома сторонами.
2. Копія останньої сторінки сертифіката на ПМФ (сертифікат відповідності, виданий компетентним уповноваженим органом).
3. Обґрунтування утворення нового власника (витяг з торгового (комерційного) реєстру, його переклад на англійську та українську мови), підписане обома сторонами.
4. Підтвердження передачі повного комплекту матеріалів ПМФ з дати його первинної оцінки правонаступнику, підписане обома сторонами.
5. Доручення, оформлене на контактну особу, відповідальну за роботу з компетентними органами та власником ПМФ, підписане новим власником ПМФ.
6. Лист-підтвердження нового власника про виконання всіх зобов’язань, які залишились не виконаними попереднім власником ПМФ (якщо такі є).
| 4.4. Зміна найменування та/або місцезнаходження установи (закладу) відбору крові/плазми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | 1, 2, 3 | IА |
Умови
1. Така установа (заклад) повинна залишатися тією самою юридичною особою.
2. Така зміна є адміністративною (наприклад, об’єднання, передача повноважень); зміна найменування установи (закладу) відбору крові/плазми, за умови, що установи (заклади) будуть залишатись тими самими.
Документація
1. Підписана заява про те, що така зміна не спричиняє змін у системі якості установ (закладів) крові.
2. Заява про те, що перелік установ (закладів) крові залишається незмінним.
3. Оновлені відповідні розділи ПМФ та додатки до нього.
| 4.5. Заміна або додавання нової установи (закладу) відбору крові/плазми, внесеної у ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2, 3 | IБ |
Документація
1. Епідеміологічні дані щодо вірусних маркерів, пов’язані з установами (закладами) відбору крові/плазми, за останні 3 роки. Для нових установ (закладів) крові або у випадку, коли такі дані ще не доступні, надається заява про те, що епідеміологічні дані будуть представлені під час наступного щорічного їх оновлення.
2. Підтвердження, що установа (заклад) крові на даний момент працює в тих самих умовах, що зазначені у стандартному контракті, укладеному між установою (закладом) крові та власником ПМФ.
3. Оновлені відповідні розділи ПМФ та додатки до нього.
| 4.6. Вилучення або зміна статусу (робоча/неробоча) установи (закладу), яка займається відбором крові/плазми або аналізом зразків донорської крові та пулів плазми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | 1 | IА |
Умови
1. Підстави для вилучення або зміни статусу установи (закладу) не повинні бути пов’язані з питаннями належної виробничої практики.
2. Такі установи (заклади) мають дотримуватись вимог законодавства у разі зміни статусу.
Документація
1. Оновлені відповідні розділи ПМФ та додатки до нього.
| 4.7. Додавання нової установи (закладу) забору крові, яка не внесена у ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| IІ | |||
| 4.8. Заміна або додавання установи (закладу), яка здійснює аналіз зразків донорської крові та/або пулів плазми, у складі установи (закладу) крові, внесеної у ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | IБ |
Документація
1. Заява про те, що аналіз здійснюється за тією самою стандартною операційною процедурою та/або затвердженими методами.
2. Оновлені відповідні розділи ПМФ та додатки до нього.
| 4.9. Додавання нової установи (закладу), яка здійснює аналіз зразків донорської крові та/або пулів плазми, не внесеної у ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| IІ | |||
| 4.10. Заміна або додавання нової установи (закладу) крові, в якій здійснюється зберігання плазми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | IБ |
Документація
1. Заява про те, що установа (заклад) крові, в якій здійснюється зберігання плазми, здійснюється за тією самою стандартною операційною процедурою, яка вже була затверджена.
2. Оновлені відповідні розділи ПМФ та додатки до нього.
| 4.11. Вилучення установи (закладу) крові, в якій здійснюється зберігання плазми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни | |
| 1 | 1 | IА | ||
| Умови | ||||
| 1. Підстави для вилучення установи (закладу) не повинні бути пов’язані з питаннями належної виробничої практики. | ||||
| Документація | ||||
| 1. Оновлені відповідні розділи ПМФ та додатки до нього. | ||||
| 4.12. Заміна або додавання суб’єкта господарювання, який здійснює транспортування плазми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни | |
| 1 | IБ | |||
Документація
1. Оновлені відповідні розділи ПМФ та додатки до нього, що містять перелік усіх установ (закладів) крові, які користуються послугами цього суб’єкта господарювання, резюме щодо системи на місцях для забезпечення належних умов транспортування (час, температура, дотримання вимог належної виробничої практики), а також підтвердження того, що умови транспортування будуть валідовані.
| 4.13. Вилучення суб’єкта господарювання, який здійснює транспортування плазми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1 | IА |
Умови
1. Підстави для вилучення суб’єкта господарювання не повинні бути пов’язані з питаннями належної виробничої практики.
Документація
1. Оновлені відповідні розділи ПМФ та додатки до нього.
| 4.14. Додавання тестового набору, який має СЕ-маркування, для тестування окремих зразків донорської крові в якості нового тестового набору або заміни зазначеного у ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2 | IА |
Умови
1. Новий тестовий набір має СЕ-маркування.
Документація
1. Перелік суб’єктів господарювання, де проводиться тестування з використанням тестового набору.
2. Оновлені відповідні розділи ПМФ та додатки до нього, включаючи оновлену інформацію щодо тестування відповідно до керівництв, що містять вимоги до ПМФ.
| 4.15. Додавання тестового набору, який не має СЕ-маркування, для тестування окремих зразків донорської крові в якості нового тестового набору або заміни зазначеного у ПМФ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) новий тестовий набір, який не був затверджений у ПМФ для тестування окремих зразків донорської крові в іншій установі крові | ІІ | ||
| б) новий тестовий набір, який був затверджений у ПМФ для тестування окремих зразків донорської крові в іншій установі крові | 1, 2 | IА |
Документація
1. Перелік суб’єктів господарювання, де проводиться тестування з використанням тестового набору, а також перелік організацій, де даний набір буде використовуватися.
2. Оновлені відповідні розділи ПМФ та додатки до нього, включаючи оновлену інформацію щодо тестування відповідно до керівництв, що містять вимоги до ПМФ.
| 4.16. Зміна набору/методу, які використовуються для тестування пулів плазми (антитіла або антигени, або NAT-тестування (технологія ампліфікації нуклеїнових кислот)) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| ІІ | |||
| 4.17. Введення або продовження строку інвентаризаційної процедури для зразків донорської крові | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1 | IА |
Умови
1. Інвентаризаційна процедура проводиться за більш строгими вимогами.
Документація
1. Оновлені відповідні розділи ПМФ, включаючи обґрунтування щодо введення або продовження строку інвентаризаційної процедури, установи, де застосовується процедура, а також зміни у процедурі прийняття рішень, у тому числі нові умови.
| 4.18. Вилучення або скорочення строку інвентаризаційної процедури для зразків донорської крові | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | IБ |
Документація
1. Оновлені відповідні розділи ПМФ.
| 4.19. Заміна або додавання контейнерів для крові (наприклад, пляшки, пакети) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) нові контейнери для крові мають СЕ-маркування | 1, 2 | 1 | IА |
| б) нові контейнери для крові не мають СЕ-маркування | ІІ |
Умови
1. Контейнер має СЕ-маркування.
2. Критерії щодо якості крові залишаються незмінними для контейнера.
Документація
1. Оновлені відповідні розділи ПМФ та додатки до нього, включаючи назву контейнера, найменування виробника, специфікацію розчину антикоагулянта, підтвердження СЕ-маркування, а також назву установи крові, де використовується даний контейнер.
| 4.20. Зміни у зберіганні/транспортуванні | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) умови зберігання та/або транспортування | 1 | 1 | IА |
| б) максимальний строк зберігання для плазми | 1, 2 | 1 | ІА |
Умови
1. Зміни повинні посилити умови зберігання та відповідати положенням монографії Європейської фармакопеї «Плазма людини для фракціонування».
2. Максимальний строк зберігання — коротший за попередній.
Документація
1. Оновлені відповідні розділи ПМФ та додатки до нього, включаючи детальний опис нових умов зберігання, дані з валідації умов зберігання/транспортування та найменування установи крові, де впроваджується зміна (за необхідності).
| 4.21. Впровадження тестування на вірусні маркери за умови, що таке впровадження буде мати суттєвий вплив на оцінку вірусної безпеки | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| ІІ | |||
| 4.22. Зміни у приготуванні пулів плазми (наприклад, метод приготування, розмір пулу, зберігання зразків пулів плазми) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | IБ |
Документація
1. Оновлені відповідні розділи ПМФ.
| 4.23. Зміни у заходах, які будуть вжиті, якщо ретроспективно буде встановлено, що зразки донорської крові необхідно виключити з процесу (ретроспективні дослідження) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| IІ |
Додаток 12
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
ЗАЯВА
про внесення змін до реєстраційних матеріалів
на лікарський засіб
(у тому числі медичний
імунобіологічний препарат)
Дата надходження заяви
«___» ____________ 20____ року № ______________
Цим я заявляю, що:
немає інших змін, крім тих, що вказані у даній заяві (виняток: інші зміни, що подаються паралельно та вказані у графі «Інша(і) заява(и)» цього додатка;
зміна(и) не спричинить(ять) негативного впливу на якість, ефективність та безпеку лікарського засобу;
усі умови (згідно з додатком 11 Порядку), які стосуються змін(и), виконано;
необхідні документи, що стосуються змін(и), подано;
Заявник гарантує достовірність інформації, що міститься у матеріалах реєстраційного досьє, а також підтверджує, що всі наявні дані стосовно якості, безпеки та ефективності лікарського засобу представлено в реєстраційному досьє;
усі внески сплачено/буде сплачено відповідно до вимог чинного законодавства.
Зміни будуть введені:
з наступного виробничого циклу/наступного випуску
дата __________________________________________
| Від імені заявника | |
|
____________________________________ (підпис) _____________________________________ (ім’я та прізвище) |
|
| М. П. |
_____________________________________ (посада) |
У разі ненадання матеріалів реєстраційного досьє протягом трьох місяців з дати подання заяви або листа з обґрунтуванням строку у розгляді даної заяви буде відмовлено. Надалі така заява може бути подана до МОЗ в установленому порядку.
Тип змін
Тип ІА
Тип ІБ
безпека
термінові обмеження, пов’язані із безпекою
якість
інші
| Назва лікарського засобу | Найменування та місцезнаходження заявника (місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця) |
| Діюча(і) речовина(и) | Прізвище, ім’я представника заявника (уповноважена особа, що виступає від імені заявника)Місцезнаходження юридичної особи або місце проживання фізичної особи — підприємця |
| Лікарська форма та сила дії | ТелефонФаксе-mail |
| Номер реєстраційного посвідчення | Посилання заявника |
Зміни типів ІА та ІБ (нижче позначте необхідне).
У разі змін типу II із заяви вилучають перелік змін типів IА, ІБ, наведений нижче.
У разі змін типів IА, ІБ вилучають ті зміни типів IА, ІБ, на які не поширюється заява.
У разі змін, які потребують нової реєстрації, вилучають ті зміни типів IА, ІБ та II, на які не розповсюджується заява.
| І. АДМІНІСТРАТИВНІ ЗМІНИ | ||||||||
| Назва зміни | Тип зміни | |||||||
| 1.1. Зміна найменування та/або місцезнаходження заявника (власника реєстраційного посвідчення) | IА | IБ* | ||||||
| 1.2. Зміна торговельної назви лікарського засобу | IБ | |||||||
| 1.3. Зміна назви АФІ або діючої речовини | IА | IБ* | ||||||
| 1.4. Зміна найменування та/або місцезнаходження виробника (включаючи за необхідності місце проведення контролю якості) або постачальника АФІ або діючої речовини/вихідного матеріалу/реагенту/проміжного продукту, що застосовуються у виробництві АФІ або діючої речовини (за відсутності сертифіката відповідності Європейській фармакопеї у затвердженому реєстраційному досьє) | IА | IБ* | ||||||
| 1.5. Зміна найменування та/або місцезнаходження виробника готового лікарського засобу, включаючи місце проведення контролю якості | ||||||||
| а) виробнича дільниця випуску серій | IА | IБ* | ||||||
| б) усі інші дільниці | IА | IБ* | ||||||
| 1.6. Зміна коду АТХ | IА | IБ* | ||||||
| 1.7. Вилучення виробничої дільниці (включаючи дільниці для АФІ або діючої речовини, проміжного продукту або готового лікарського засобу, дільниці для проведення пакування, виробника, відповідального за випуск серій, місце проведення контролю серії) або постачальника вихідного матеріалу, реагенту або допоміжної речовини (якщо зазначено у реєстраційному досьє) | IА | IБ* | ||||||
| ІІ. ЗМІНИ ЩОДО ЯКОСТІ | ||||||||
| 2.1. АФІ або діюча речовина | ||||||||
| 2.1.1. Виробництво | ||||||||
| 2.1.1.1. Зміна виробника вихідного/проміжного продукту/реагенту, що використовується у виробничому процесі АФІ або діючої речовини, або зміна виробника (включаючи, де необхідно, місце проведення контролю якості) АФІ або діючої речовини (за відсутності сертифіката відповідності Європейській фармакопеї у матеріалах реєстраційного досьє) | ||||||||
| а) запропонована додаткова виробнича дільниця є дільницею одного виробника | IА | IБ* | ||||||
| б) введення нового виробника АФІ або діючої речовини з наданням матеріалів реєстраційного досьє (мастер-файла) на діючу речовину | II | |||||||
| в) запропонований виробник використовує спосіб синтезу, що суттєво відрізняється від попереднього, або умови виробництва, які потенційно можуть змінити важливі характеристики якості АФІ або діючої речовини, такі як якісний та/або кількісний профіль домішок, що потребує кваліфікації, або фізико-хімічні властивості АФІ або діючої речовини, що впливають на біодоступність | II | |||||||
| г) новий виробник вихідного продукту, для якого вимагається попередня оцінка вірусної безпеки та/або ризику передачі збудників губчатої енцефалопатії | II | |||||||
| ґ) зміна у АФІ або діючої речовини біологічного походження або вихідному матеріалі/реагенті/проміжному продукті, що використовується для виробництва лікарського засобу біологічного походження | II | |||||||
| д) зміни у методах контролю якості АФІ або діючої речовини або додавання дільниці для проведення контролю серії/випробування | IА | IБ* | ||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.1.1.2. Зміни в процесі виробництва АФІ або діючої речовини | ||||||||
| а) незначна зміна у процесі виробництва АФІ або діючої речовини | IА | IБ* | ||||||
| б) значна зміна у процесі виробництва АФІ або діючої речовини, що може мати істотний вплив на якість, безпеку або ефективність лікарського засобу | II | |||||||
| в) зміна стосується АФІ або діючої речовини біологічного/імунологічного походження або використання хімічних субстанцій у виробництві лікарського засобу біологічного/імунологічного походження і не відноситься до протоколу виробництва | II | |||||||
| г) зміна у лікарському засобі рослинного походження, що стосується однієї з таких характеристик: джерело походження сировини, спосіб виробництва або виготовлення | II | |||||||
| ґ) незначна зміна у закритій частині матеріалів реєстраційного досьє (мастер-файла) на АФІ або діючу речовину | IБ | |||||||
| д) інші зміни | IА | IБ | II | |||||
| 2.1.1.3. Зміна розміру серії (включаючи діапазони) АФІ або діючої речовини або проміжного продукту | ||||||||
| а) збільшення до 10 разів порівняно із затвердженим розміром серії | IА | IБ* | ||||||
| б) зменшення обсягу виробництва | IА | IБ* | ||||||
| в) зміна розміру серії, що потребує доведення подібності АФІ або діючої речовини біологічного/імунологічного походження порівняно із затвердженою | II | |||||||
| г) збільшення у понад 10 разів порівняно із затвердженим розміром серії | IБ | |||||||
| ґ) розмір серії АФІ або діючої речовини біологічного/імунологічного походження збільшився/зменшився без зміни параметрів процесу (наприклад, дублювання лінії) | IБ | |||||||
| д) інші зміни | IА | IБ | II | |||||
| 2.1.1.4. Зміни випробувань або допустимих меж, що встановлені у специфікаціях, у процесі виробництва АФІ або діючої речовини | ||||||||
| а) звуження допустимих меж | IА | IБ* | ||||||
| б) додавання нового випробування та допустимих меж | IА | IБ* | ||||||
| в) вилучення несуттєвого випробування | IА | IБ* | ||||||
| г) розширення затверджених допустимих меж для показників, які можуть істотно вплинути на якість АФІ або діючої речовини | II | |||||||
| ґ) вилучення випробування, що може мати істотний влив на загальну якість АФІ або діючої речовини | II | |||||||
| д) додавання або заміна випробування за результатами досліджень з безпеки або якості | IБ | |||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.1.1.5. Зміни у АФІ або діючій речовині для сезонних, передпандемічних або пандемічних вакцин для профілактики грипу | ||||||||
| а) заміна штаму(ів) у сезонних, передпандемічних або пандемічних вакцинах для профілактики грипу | II | |||||||
| 2.1.2. Контроль АФІ або діючої речовини | ||||||||
| 2.1.2.1. Зміна у параметрах специфікацій та/або допустимих меж, визначених у специфікаціях на АФІ або діючу речовину, або вихідний/проміжний продукт/реагент, що використовуються у процесі виробництва АФІ або діючої речовини | ||||||||
| а) звуження допустимих меж, визначених у специфікації на лікарські засоби, що підлягають випуску серії | IA | ІБ* | ||||||
| б) звуження допустимих меж, визначених у специфікації | IA | ІБ* | ||||||
| в) доповнення специфікації новим показником якості та відповідним методом випробування | IA | ІБ* | ||||||
| г) вилучення незначного показника якості (наприклад, вилучення застарілого показника) | IА | ІБ* | ||||||
| ґ) вилучення параметра специфікації, який може мати суттєвий влив на якість АФІ або діючої речовини та/або готового лікарського засобу | II | |||||||
| д) розширення допустимих меж, визначених у специфікації на АФІ або діючу речовину | II | |||||||
| е) розширення допустимих меж, визначених у специфікаціях на вихідні матеріали/проміжні продукти, які мають істотний вплив на якість АФІ або діючої речовини та/або готового лікарського засобу | II | |||||||
| є) доповнення або заміна параметра специфікації за результатами досліджень з безпеки або якості (за винятком АФІ або діючої речовини біологічного або імунологічного походження) | IБ | |||||||
| ж) інші зміни | IА | IБ | II | |||||
| 2.1.2.2. Зміна у методах випробування АФІ або діючої речовини або вихідного/проміжного продукту/реагенту, що використовується у процесі виробництва АФІ або діючої речовини | ||||||||
| а) незначні зміни у затверджених методах випробування | IА | ІБ* | ||||||
| б) видалення методу випробування для АФІ або діючої речовини /реагенту/проміжного продукту, якщо альтернативний метод вже затверджений | IА | ІБ* | ||||||
| в) інші зміни в методах випробування (включаючи заміну або доповнення) для реагенту, що не спричиняє істотного впливу на якість АФІ або діючої речовини | IA | ІБ* | ||||||
| г) зміна у біологічному/імунобіологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент для АФІ або діючої речовини біологічного походження, наприклад, пептидні карти, глік-карти тощо | II | |||||||
| ґ) інші зміни у методах випробування (включаючи заміну або доповнення) АФІ або діючої речовини або вихідного/проміжного продукту | IБ | |||||||
| д) інші зміни | IА | IБ | II | |||||
| 2.1.3. Система упаковка/укупорка | ||||||||
| 2.1.3.1. Зміна у безпосередній упаковці АФІ або діючої речовини | ||||||||
| а) якісні та/або кількісні зміни складу | IА | ІБ* | ||||||
| б) якісні та/або кількісні зміни складу для стерильних та незаморожених АФІ або діючих речовин біологічного/імунологічного походження | II | |||||||
| в) рідких АФІ або діючих речовин (нестерильних) | IБ | |||||||
| г) інші зміни | IА | IБ | II | |||||
| 2.1.3.2. Зміни у параметрах специфікацій та/або допустимих меж, зазначених у специфікаціях, для первинної упаковки АФІ або діючої речовини | ||||||||
| а) звуження допустимих меж, зазначених у специфікаціях | IА | ІБ* | ||||||
| б) доповнення специфікації новим показником та відповідним методом випробування | IА | ІБ* | ||||||
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | IА | ІБ* | ||||||
| г) доповнення або заміна показника за результатами досліджень з безпеки або якості | IБ | |||||||
| ґ) інші зміни | IА | IБ | II | |||||
| 2.1.3.3. Зміна в методах випробування первинної упаковки АФІ або діючої речовини | ||||||||
| а) незначні зміни у затверджених методах випробування | IА | ІБ* | ||||||
| б) інші зміни в методах випробування (включаючи заміну або доповнення) | IА | ІБ* | ||||||
| в) вилучення методу випробування, якщо альтернативний метод випробування вже затверджено | IА | ІБ* | ||||||
| 2.1.4. Стабільність | ||||||||
| 2.1.4.1. Зміна періоду повторних випробувань/терміну придатності або умов зберігання АФІ або діючої речовини (за наявності у матеріалах реєстраційного досьє сертифіката відповідності Європейській фармакопеї, що включає період повторного випробування) | ||||||||
| а) період повторного випробування/термін придатності | ||||||||
| зменшення | ІА | ІБ* | ||||||
| збільшення періоду повторного випробування на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності до настанови щодо випробування стабільності або керівних принципів ІСН щодо випробування стабільності (крім АФІ або діючих речовин біологічного/імунологічного походження) | ІІ | |||||||
| збільшення терміну придатності АФІ або діючої речовини біологічного/імунологічного походження на основі результатів досліджень, виконаних не у відповідності до затвердженого протоколу стабільності | ІІ | |||||||
| збільшення або введення періоду повторного випробування/терміну придатності на основі результатів досліджень у реальному часі | ІБ | |||||||
| б) умови зберігання | ||||||||
| обмеження умов зберігання | ІА | ІБ* | ||||||
| зміна умов зберігання АФІ або діючої речовини біологічного/імунологічного походження, якщо дослідження стабільності ще не проводились відповідно до затвердженого протоколу стабільності | II | |||||||
| зміна умов зберігання АФІ або діючої речовини | ІБ | |||||||
| в) інші зміни | IА | IБ | II | |||||
| 2.1.5. Проектний простір | ||||||||
| 2.1.5.1. Введення нового проектного простору або розширення затвердженого для АФІ або діючої речовини щодо | ||||||||
| а) одного елементу (блок, частина) виробничого процесу АФІ або діючої речовини, включаючи контроль у процесі виробництва та/або методи випробування | II | |||||||
| б) методів випробувань для вихідних матеріалів/реагентів/проміжних продуктів та/або АФІ або діючої речовини | II | |||||||
| 2.1.5.2. Внесення змін після затвердження протоколу управління АФІ або діючої речовини | II | |||||||
| 2.1.5.3. Вилучення затвердженого протоколу управління змінами для АФІ або діючої речовини | IА | ІБ* | ||||||
| 2.2. Готовий лікарський засіб | ||||||||
| 2.2.1. Опис та склад | ||||||||
| 2.2.1.1. Зміна або додавання штампів, потовщень або інших маркувань, уключаючи заміну або додавання фарб для маркування лікарського засобу | ||||||||
| а) зміни штампів, потовщень або інших маркувань | IА | ІБ* | ||||||
| б) зміна риски, призначеної для поділу таблетки на рівні дози | IБ | |||||||
| в) інші зміни | IА | IБ | II | |||||
| 2.2.1.2. Зміна форми або розмірів лікарської форми | ||||||||
| а) таблетки з негайним вивільненням, капсули, супозиторії та песарії | IА | ІБ* | ||||||
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим або пролонгованим вивільненням та ділимі таблетки, призначені для поділу на рівні дози | IБ | |||||||
| в) інші зміни | IА | IБ | II | |||||
| 2.2.1.3. Зміна у складі (допоміжних речовинах) готового лікарського засобу | ||||||||
| а) смакові добавки або барвники | ||||||||
| додавання, вилучення або заміна | IА | ІБ* | ||||||
| збільшення або зменшення | IА | ІБ* | ||||||
| б) інші допоміжні речовини | II | |||||||
| будь-яка незначна зміна кількісного складу допоміжних речовин у готовому лікарському засобі | IА | ІБ* | ||||||
| якісні або кількісні зміни щодо однієї або декількох допоміжних речовин, які можуть значно вплинути на безпеку, якість або ефективність готового лікарського засобу | II | |||||||
| зміна у лікарському засобі біологічного/імунологічного походження | II | |||||||
| будь-яка нова допоміжна речовина, що включає використання матеріалів людського або тваринного походження, для яких вимагається оцінка даних з вірусної безпеки або ризику передачі збудників губчатої енцефалопатії | II | |||||||
| зміна, яка підтверджується дослідженнями з еквівалентності | II | |||||||
| заміна однієї допоміжної речовини на іншу з тими самими функціональними характеристиками та на тому самому рівні | IБ | |||||||
| в) інші зміни | IА | IБ | II | |||||
| 2.2.1.4. Зміна маси покриття лікарських форм для перорального застосування або зміна маси оболонки капсул | ||||||||
| а) тверді лікарські форми для перорального застосування | IА | ІБ* | ||||||
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим вивільненням або пролонгованої дії, для яких покриття є вирішальним чинником механізму вивільнення | II | |||||||
| в) інші зміни | IА | IБ | II | |||||
| 2.2.1.5. Зміна концентрації в окремій дозі багатодозового лікарського засобу для парентерального застосування, коли кількість діючої речовини на одиницю дози (тобто сила дії) не змінюється | II | |||||||
| 2.2.1.6. Вилучення контейнера з розчинником з упаковки | IБ | |||||||
| 2.2.2. Виробництво | ||||||||
| 2.2.2.1. Заміна або введення додаткової дільниці виробництва для частини або всього виробничого процесу готового лікарського засобу | ||||||||
| а) дільниця для вторинного пакування | IА | ІБ* | ||||||
| б) дільниця для первинного пакування | IА | ІБ* | ||||||
| в) дільниця, на якій проводяться будь-які виробничі стадії, за винятком випуску серій, проведення контролю якості та вторинного пакування, для лікарських засобів біологічного/імунологічного походження | II | |||||||
| г) дільниця, яка вимагає проведення первинної перевірки виробництва або перевірки виробництва конкретного готового лікарського засобу | II | |||||||
| ґ) дільниця, на якій проводяться будь-які виробничі стадії, за винятком випуску серій, контролю якості, первинного та вторинного пакування, для нестерильних лікарських засобів | IБ | |||||||
| д) дільниця, на якій проводяться будь-які виробничі стадії, за винятком випуску серії, контролю якості та вторинного пакування для стерильних лікарських засобів, що вироблені з використанням асептичного методу, за винятком лікарських засобів біологічного/імунологічного походження | IБ | |||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.2.2.2. Зміни, що стосуються випуску серії та контролю якості готового лікарського засобу | ||||||||
| а) заміна або додавання дільниці, на якій здійснюється контроль/випробування серії | IА | ІБ* | ||||||
| б) заміна або додавання виробника, відповідального за випуск серії | ||||||||
| не включаючи контроль/випробування серії | IА | ІБ* | ||||||
| включаючи контроль/випробування серії | IА | ІБ* | ||||||
| включаючи контроль/випробування серії для лікарського засобу біологічного/імунологічного походження та один з методів аналізу, що застосовується на дільниці, є біологічним/імунологічним/імунохімічним методом | II | |||||||
| 2.2.2.3. Зміни у процесі виробництва готового лікарського засобу | ||||||||
| а) незначна зміна у процесі виробництва твердої лікарської форми для перорального застосування з негайним вивільненням або розчинів для перорального застосування | ІА | ІБ* | ||||||
| б) зміна у процесі виробництва, яка може мати істотний влив на якість, безпеку та ефективність лікарського засобу | II | |||||||
| в) лікарський засіб є лікарським засобом біологічного/імунологічного походження та зміна вимагає проведення порівняльних досліджень | ІІ | |||||||
| г) ведення нестандартного методу кінцевої стерилізації | ІІ | |||||||
| ґ) введення або збільшення припустимого надлишку діючої речовини | ІІ | |||||||
| д) незначна зміна у процесі виробництва водної суспензії для перорального застосування | IБ | |||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.2.2.4. Зміна розміру серії (включаючи діапазон розміру серії) готового лікарського засобу | ||||||||
| а) збільшення до 10 разів порівняно із затвердженим розміром серії | IА | ІБ* | ||||||
| б) зменшення до 10 разів порівняно із затвердженим розміром серії | IА | ІБ* | ||||||
| в) зміна вимагає оцінки порівнянності (проведення порівняльних досліджень) лікарського засобу біологічного/імунологічного походження | ІІ | |||||||
| г) зміна стосується всіх інших лікарських форм сукупного (комплексного) виробничого процесу | ІІ | |||||||
| ґ) збільшення розміру серії більш ніж у 10 разів порівняно із затвердженим розміром для твердих лікарських форм з негайним вивільненням | ІБ | |||||||
| д) масштаб для лікарського засобу біологічного/імунологічного походження збільшився/зменшився без зміни виробничого процесу (наприклад, дублювання лінії) | ІБ | |||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.2.2.5. Зміни випробувань або допустимих меж, встановлених у специфікаціях, під час виробництва готового лікарського засобу | ||||||||
| а) звуження допустимих меж | IА | ІБ* | ||||||
| б) доповнення нового методу випробування та допустимих меж | IА | ІБ* | ||||||
| в) вилучення несуттєвого випробування | IА | ІБ* | ||||||
| г) вилучення випробування у процесі виробництва, яке може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | |||||||
| ґ) розширення затверджених допустимих меж для показників, які можуть мати істотний вплив на загальну якість готового лікарського засобу | ІІ | |||||||
| д) доповнення або заміна випробування за результатами досліджень з безпеки або якості | ІБ | |||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.2.3. Контроль допоміжних речовин | ||||||||
| 2.2.3.1. Зміна параметрів специфікацій та/або допустимих меж для допоміжної речовини | ||||||||
| а) звуження допустимих меж | ІА | ІБ* | ||||||
| б) доповнення специфікації новим показником з відповідним методом випробування | ІА | ІБ* | ||||||
| в) вилучення із специфікації незначного показника (наприклад, вилучення застарілого показника) | ІА | ІБ* | ||||||
| г) зміна знаходиться поза затвердженими допустимими межами специфікацій | ІІ | |||||||
| ґ) вилучення із специфікації показника, який може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | |||||||
| д) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за винятком лікарських засобів біологічного та імунологічного походження) | ІБ | |||||||
| е) інші зміни | IА | IБ | II | |||||
| 2.2.3.2. Зміна у методах випробування допоміжної речовини | ||||||||
| а) незначні зміни у затверджених методах випробувань | IА | ІБ* | ||||||
| б) вилучення методу випробування, якщо альтернативний метод випробування вже затверджено | ІА | ІБ* | ||||||
| в) заміна біологічного/імунологічного/імунохімічного методу випробування або методу, у якому використовується біологічний реагент | ІІ | |||||||
| г) інші зміни у методах випробування (включаючи заміну або додавання) | IБ | |||||||
| 2.2.3.3. Заміна джерела одержання допоміжної речовини або реактиву, що становить ризик передачі збудників губчатої енцефалопатії | ||||||||
| а) матеріалу, що становить ризик передачі збудників губчатої енцефалопатії, на матеріал рослинного або синтетичного походження | ||||||||
| для допоміжних речовин або реактивів, які не використовуються у виробництві активної субстанції біологічного/імунологічного походження або лікарського засобу біологічного/імунологічного походження | IА | ІБ* | ||||||
| для допоміжних речовин або реактивів, які використовуються у виробництві активної субстанції біологічного/імунологічного походження або лікарського засобу біологічного/імунологічного походження | IБ | |||||||
| б) заміна або додавання речовини, що становить ризик передачі збудників губчатої енцефалопатії, або заміна речовини, що становить ризик передачі збудників губчатої енцефалопатії, на іншу речовину, що становить ризик передачі збудників губчатої енцефалопатії, для якої немає сертифіката відповідності Європейській фармакопеї щодо губчатої енцефалопатії | II | |||||||
| 2.2.3.4. Зміни в методі синтезу або регенерації нефармакопейної допоміжної речовини (за наявності в досьє) | ||||||||
| а) незначні зміни у методі синтезу або регенерації | IА | ІБ* | ||||||
| б) зміни у специфікації або зміна фізико-хімічних властивостей допоміжної речовини, що може мати вплив на якість готового лікарського засобу | ІІ | |||||||
| в) допоміжна речовина є речовиною біологічного/імунологічного походження | ІІ | |||||||
| г) інші зміни | IА | IБ | II | |||||
| 2.2.4. Контроль готового лікарського засобу | ||||||||
| 2.2.4.1. Зміна параметрів специфікацій та/або допустимих меж готового лікарського засобу | ||||||||
| а) звуження допустимих меж | IА | ІБ* | ||||||
| б) звуження допустимих меж для лікарського засобу, що підлягає офіційному випуску серії | IА | ІБ* | ||||||
| в) доповнення специфікації новим показником з відповідним методом випробування | IА | ІБ* | ||||||
| г) вилучення незначного показника (наприклад, вилучення застарілого показника) | IА | ІБ* | ||||||
| ґ) зміна знаходиться поза затвердженими допустимими межами | ІІ | |||||||
| д) вилучення показника, який може мати істотний вплив на якість готового лікарського засобу | ІІ | |||||||
| е) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за винятком лікарських засобів біологічного/імунологічного походження) | ІБ | |||||||
| є) інші зміни | IА | IБ | II | |||||
| 2.2.4.2. Зміна у методах випробування готового лікарського засобу | ||||||||
| а) незначна зміна у затверджених методах випробування | IА | ІБ* | ||||||
| б) вилучення методу випробування, якщо вже затверджено альтернативний метод | IА | ІБ* | ||||||
| в) зміна у біологічному/імунологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент | ІІ | |||||||
| г) інші зміни у методах випробувань (включаючи заміну або доповнення) | IБ | |||||||
| 2.2.4.3. Зміни, які стосуються виробничого процесу у реальному часі або випуску за параметрами для готового лікарського засобу | ІІ | |||||||
| 2.2.5. Система упаковка/укупорка | ||||||||
| 2.2.5.1. Зміна у первинній упаковці готового лікарського засобу | ||||||||
| а) якісний та кількісний склад | ||||||||
| тверді лікарські форми | IА | ІБ* | ||||||
| м’які та нестерильні рідкі лікарські форми | IБ | |||||||
| стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | |||||||
| зміна стосується зниження ступеня захисту оновленої упаковки, якщо наявні відповідні зміни в умовах зберігання та/або скорочення терміну придатності | ІІ | |||||||
| б) тип контейнера | ||||||||
| тверді, м’які та нестерильні рідкі лікарські форми | ІБ | |||||||
| стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | |||||||
| в) інші зміни | IА | IБ | II | |||||
| 2.2.5.2. Зміна параметрів специфікацій та/або допустимих меж первинної упаковки готового лікарського засобу | ||||||||
| а) звуження допустимих меж, визначених у специфікації | IА | ІБ* | ||||||
| б) доповнення специфікації новим показником з відповідним методом випробування | IА | ІБ* | ||||||
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | ІА | ІБ* | ||||||
| г) додавання або заміна показника за результатами досліджень з безпеки або якості | ІБ | |||||||
| ґ) інші зміни | IА | IБ | II | |||||
| 2.2.5.3. Зміна у методах випробування первинної упаковки готового лікарського засобу | ||||||||
| а) незначні зміни у затверджених методах випробувань | IА | ІБ* | ||||||
| б) інші зміни у методах випробувань (включаючи заміну або додавання) | IА | ІБ* | ||||||
| в) вилучення методу випробування, якщо вже затверджено альтернативний | ІА | ІБ* | ||||||
| 2.2.5.4. Зміна форми або розміру контейнера чи закупорювального засобу (первинної упаковки) | ||||||||
| а) нестерильні лікарські засоби | IА | ІБ* | ||||||
| б) зміна форми або розміру основної частини пакувального матеріалу (вторинної упаковки), що може мати значний вплив на доставку, застосування, безпеку та стабільність готового лікарського засобу | IІ | |||||||
| в) стерильні лікарські засоби | ІБ | |||||||
| 2.2.5.5. Зміна розміру упаковки готового лікарського засобу | ||||||||
| а) зміна кількості одиниць (наприклад, таблеток, ампул та ін.) в упаковці: | ||||||||
| зміна у діапазоні затверджених розмірів упаковки | IА | ІБ* | ||||||
| зміна поза діапазоном затверджених розмірів упаковки | IБ | |||||||
| б) вилучення упаковки певного розміру | ІА | ІБ* | ||||||
| в) зміна маси/об’єму вмісту контейнера багатодозового стерильного лікарського засобу (або однодозового часткового використання) та багатодозового лікарського засобу біологічного/імунологічного походження для парентерального застосування | IІ | |||||||
| г) зміна маси/об’єму вмісту контейнера багатодозового лікарського засобу для непарентеральних застосувань (або однодозового часткового використання) | ІБ | |||||||
| ґ) інші зміни | IА | IБ | II | |||||
| 2.2.5.6. Зміна будь-якої частини матеріалу первинної упаковки, що не контактує з готовим лікарським засобом (наприклад, колір кришечок з контролем першого відкриття, колір кодових кілець на ампулах, контейнера для голок (різні види пластмаси) | ||||||||
| а) зміна, яка впливає на коротку характеристику лікарського засобу | IА | ІБ* | ||||||
| б) зміна, яка не впливає на коротку характеристику лікарського засобу | IА | ІБ* | ||||||
| 2.2.5.7. Зміна постачальника пакувальних матеріалів або комплектуючих (якщо зазначено в досьє) | ||||||||
| а) вилучення постачальника | IА | ІБ* | ||||||
| б) заміна або додавання постачальника | IА | ІБ* | ||||||
| в) будь-яка зміна постачальника спейсерів для дозованих інгаляторів | ІІ | |||||||
| 2.2.6. Стабільність | ||||||||
| 2.2.6.1. Зміна у термінах придатності або умовах зберігання готового лікарського засобу: | ||||||||
| а) зменшення терміну придатності готового лікарського засобу: | ||||||||
| для торговельної упаковки | IА | ІБ* | ||||||
| після першого розкриття | IА | ІБ* | ||||||
| після розчинення або відновлення | IА | ІБ* | ||||||
| б) збільшення терміну придатності готового лікарського засобу: | ||||||||
| для торговельної упаковки (підтверджується даними стабільності реального часу) | IБ | |||||||
| після першого розкриття (підтверджується даними стабільності реального часу) | IБ | |||||||
| після розчинення або відновлення (підтверджується даними стабільності реального часу) | IБ | |||||||
| збільшення терміну придатності на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності з вимогами настанови щодо дослідження стабільності лікарських засобів або керівних принципів ICH щодо дослідження стабільності лікарських засобів | ІІ | |||||||
| збільшення терміну придатності лікарського засобу біологічного/імунологічного походження на основі результатів досліджень стабільності, проведених відповідно до затвердженого протоколу стабільності | IБ | |||||||
| в) зміна в умовах зберігання лікарського засобу біологічного походження, якщо дослідження стабільності були проведені не у відповідності до затвердженого протоколу стабільності | ІІ | |||||||
| г) зміна в умовах зберігання готового лікарського засобу або після розчинення/відновлення | IБ | |||||||
| ґ) інші зміни | IА | IБ | II | |||||
| 2.2.7. Проектний простір | ||||||||
| 2.2.7.1. Введення нового проектного простору або розширення затвердженого для готового лікарського засобу (за винятком лікарських засобів біологічного походження) щодо | ||||||||
| а) одного або більше елементів (блок, частина) виробничого процесу готового лікарського засобу, включаючи контроль у процесі виробництва та/або методи випробувань | II | |||||||
| б) методів випробувань для допоміжних речовин/проміжних продуктів та/або готового лікарського засобу | II | |||||||
| 2.2.7.2. Внесення змін після затвердження протоколу управління змінами для готового лікарського засобу | II | |||||||
| 2.2.7.3. Вилучення затвердженого протоколу управління змінами для готового лікарського засобу | IА | ІБ* | ||||||
| 2.3. Сертифікат відповідності Європейській фармакопеї/Сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії | ||||||||
| 2.3.1. Подання нового або оновленого сертифіката відповідності Європейській фармакопеї | ||||||||
| Для АФІ або діючої речовини/для вихідного матеріалу/реагенту/проміжного продукту, що використовуються у виробництві АФІ або діючої речовини/для допоміжної речовини | ||||||||
| а) сертифікат відповідності Європейській фармакопеї | ||||||||
| новий сертифікат відповідності від діючого виробника | IА | ІБ* | ||||||
| оновлений сертифікат відповідності від діючого виробника | IА | ІБ* | ||||||
| новий сертифікат відповідності від нового виробника (заміна або доповнення) | IА | ІБ* | ||||||
| б) сертифікат відповідності Європейській фармакопеї щодо губчатої енцефалопатії для АФІ або діючої речовини /вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини | ||||||||
| новий сертифікат відповідності для АФІ або діючої речовини від нового або діючого виробника | IА | ІБ* | ||||||
| новий сертифікат відповідності для вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини від нового або діючого виробника | IА | ІБ* | ||||||
| оновлений сертифікат відповідності від діючого виробника | IА | ІБ* | ||||||
| 2.3.2. Зміни, пов’язані з необхідністю приведення у відповідність до монографії Державної фармакопеї України або Європейської фармакопеї | ||||||||
| а) зміна у специфікації нефармакопейної АФІ або діючої речовини для приведення у відповідність до вимог Державної фармакопеї України або Європейської фармакопеї | ||||||||
| АФІ або діюча речовина | IА | ІБ* | ||||||
| допоміжна речовина/вихідний матеріал для виробництва АФІ або діючої речовини | IА | ІБ* | ||||||
| б) зміна у специфікаціях, пов’язана зі змінами в Державній фармакопеї України або Європейській фармакопеї | IА | ІБ* | ||||||
| в) зміна у специфікаціях, пов’язана із заміною вимог монографії Державної фармакопеї України на вимоги монографії Європейської фармакопеї | IА | ІБ* | ||||||
| 2.4. Медичні пристрої | ||||||||
| 2.4.1. Зміна пристроїв для вимірювання дози або введення лікарського засобу | ||||||||
| а) додавання або заміна пристрою, який не є невід’ємною частиною первинної упаковки | ||||||||
| пристрій, який має СЕ-маркування | IА | ІБ* | ||||||
| спейсер для дозованих інгаляторів | ІІ | |||||||
| б) вилучення пристрою | IА | ІБ* | ||||||
| в) додавання або заміна пристрою, який є невід’ємною частиною первинної упаковки | IІ | |||||||
| 2.5. Зміни до реєстраційного посвідчення внаслідок інших регуляторних процедур | ||||||||
| 2.5.1. ПМФ/ВАЗФ (мастер-файл на плазму/мастер-файл на вакцинний антиген) | ||||||||
| 2.5.1.1. Включення нового, оновленого або зміненого мастер-файла на плазму у матеріали реєстраційного досьє на лікарський засіб (процедура 2-го етапу для мастер-файла на плазму) | ||||||||
| а) первинне включення нового мастер-файла на плазму, що впливає на властивості готового лікарського засобу | II | |||||||
| б) первинне включення нового мастер-файла на плазму, що не впливає на властивості готового лікарського засобу | IБ | |||||||
| в) включення оновленого/зміненого мастер-файла на плазму, якщо зміни впливають на властивості готового лікарського засобу | IБ | |||||||
| г) включення оновленого/зміненого мастер-файла на плазму, якщо зміни не впливають на властивості готового лікарського засобу | IA | ІБ* | ||||||
| 2.5.1.2. Включення нового, оновленого або зміненого мастер-файла на вакцинний антиген у матеріали реєстраційного досьє на готовий лікарський засіб (процедура 2-го етапу для ВАЗФ) | ||||||||
| а) первинне включення нового мастер-файла на вакцинний антиген | II | |||||||
| б) включення оновленого або зміненого мастер-файла на вакцинний антиген, коли зміни впливають на властивості готового лікарського засобу | IБ | |||||||
| в) включення оновленого або зміненого мастер-файла на вакцинний антиген, коли зміни не впливають на властивості готового лікарського засобу | IA | ІБ* | ||||||
| 2.5.2. Протокол управління змінами | ||||||||
| 2.5.2.1. Перегляд досьє з якості для внесення змін на вимогу компетентного органу після оцінки протоколу управління змінами | ||||||||
| а) внесення зміни не вимагає будь-яких додаткових супровідних даних | IА | ІБ* | ||||||
| б) внесення зміни вимагає додаткових супровідних даних | IБ | |||||||
| в) внесення зміни для лікарського засобу біологічного/імунологічного походження | IБ | |||||||
| ІІІ. ЗМІНИ ЩОДО БЕЗПЕКИ, ЕФЕКТИВНОСТІ, ФАРМАКОНАГЛЯДУ | ||||||||
| 3.1. Лікарські засоби | ||||||||
| 3.1.1. Зміна у короткій характеристиці лікарського засобу, інструкції для медичного застосування або маркуванні упаковок | ||||||||
| а) потребує негайного внесення, але не пізніше 30 днів | ІА | ІБ* | ||||||
| б) потребує внесення відповідно до інформації референтного/оригінального лікарського засобу без надання жодних нових додаткових даних заявником | ІБ | |||||||
| в) потребує внесення відповідно до посилань на нові додаткові дані, надані заявником | ІІ | |||||||
| 3.1.2. Зміна у короткій характеристиці лікарського засобу, інструкції для медичного застосування, маркуванні упаковок генеричних/комбінованих/біоподібних лікарських засобів після оцінки тієї ж зміни щодо референтного/оригінального лікарського засобу | ||||||||
| а) впровадження змін(и), щодо яких(ої) заявник не надає жодних нових додаткових даних | IБ | |||||||
| б) впровадження змін(и), які(у) необхідно надалі обґрунтувати новими додатковими даними, які має надати заявник (наприклад, порівнянність) | II | |||||||
| 3.1.3. Внесення змін(и) на вимогу МОЗ згідно з рекомендаціями національних компетентних органів, міжнародних організацій, регуляторних агенцій інших країн після оцінки негайного обмеження щодо безпеки, маркування, характерного для певної фармакологічної та/або фармакотерапевтичної групи, регулярно оновлюваного звіту з безпеки, плану управління ризиками, контрольних заходів/спеціальних зобов’язань, змін для відображення короткої характеристики лікарського засобу та інструкції для медичного застосування | ||||||||
| a) впровадження узгоджених змін у формулюванні, щодо яких заявник не надає жодних нових додаткових даних | IБ | |||||||
| б) впровадження змін, які вимагають подальшого обґрунтування новими додатковими даними, що надаються заявником | ІІ | |||||||
| 3.1.4. Зміни, що пов’язані зі значними змінами у короткій характеристиці лікарського засобу, інструкції для медичного застосування згідно з новими даними з якості, доклінічними, клінічними даними та даними фармаконагляду | ІІ | |||||||
| 3.1.5. Зміна у правовому статусі лікарського засобу | ||||||||
| а) для генеричних/комбінованих/біоподібних лікарських засобів після зміни затвердженого правового статусу референтного/оригінального лікарського засобу | ІБ | |||||||
| б) усі інші зміни правового статусу | ІІ | |||||||
| 3.1.6. Зміна(и) до терапевтичних показань | ||||||||
| а) додавання нового терапевтичного показання або зміни затвердженого показання | ІІ | |||||||
| б) вилучення терапевтичного показання | ІБ | |||||||
| 3.1.7. Вилучення | ||||||||
| а) лікарська форма | ІБ | |||||||
| б) сила дії | ІБ | |||||||
| 3.1.8. Введення нової системи фармаконагляду | ||||||||
| а) яка не пройшла оцінки відповідним національним компетентним органом/Європейською медичною агенцією (ЕМА) для іншого продукту того ж заявника | ІІ | |||||||
| б) яка пройшла оцінку відповідним національним компетентним органом/іншими регуляторними агенціями для іншого лікарського засобу того ж заявника | IБ | |||||||
| 3.1.9. Зміни до існуючої системи фармаконагляду, як зазначено в описі системи фармаконагляду | ||||||||
| а) зміна уповноваженої особи, відповідальної за фармаконагляд | IА | ІБ* | ||||||
| б) зміна в контактних даних уповноваженої особи, відповідальної за фармаконагляд | ІА | ІБ* | ||||||
| в) зміна до процедури підтримки уповноваженої особи, відповідальної за фармаконагляд | ІА | ІБ* | ||||||
| г) зміна до бази даних з безпеки (наприклад, введення нової бази даних з безпеки, включаючи перенесення зібраних даних з безпеки та/або аналіз та повідомлення до нової системи) | ІА | ІБ* | ||||||
| ґ) зміни в контрактних домовленостях з іншими фізичними або юридичними особами, що залучені до виконання вимог з фармаконагляду та описані в системі фармаконагляду, а саме: якщо складається договір субпідряду про подання електронних повідомлень про індивідуальний випадок, пов’язаний з безпекою лікарського засобу; про ведення головних баз даних; про відстеження сигналу або про підготовку регулярно оновлюваного звіту з безпеки | ІА | ІБ* | ||||||
| д) виключення тем, що увійшли у письмову процедуру(и), яка(і) описує(ють) діяльність з фармаконагляду | ІА | ІБ* | ||||||
| е) зміна дільниці, що проводить діяльність з фармаконагляду | ІА | ІБ* | ||||||
| є) інші зміни до системи фармаконагляду, які не впливають на функціонування системи фармаконагляду (наприклад, зміни розташування основного архіву, адміністративні зміни, оновлення скорочень, зміни щодо назви функцій/процедур) | ІА | ІБ* | ||||||
| ж) інші зміни | IА | IБ | II | |||||
| IV. Мастер-файл на плазму (далі — ПМФ)/мастер-файл на вакцинний антиген (далі — ВАЗФ) | ||||||||
| 4.1. Зміна найменування та/або місцезнаходження власника ВАЗФ | ІА | ІБ* | ||||||
| 4.2. Зміна найменування та/або місцезнаходження власника ПМФ | ІА | ІБ* | ||||||
| 4.3. Зміна або передача власності на ПМФ від затвердженого власника новому власнику ПМФ (а саме, іншому суб’єкту господарювання) | ІА | ІБ* | ||||||
| 4.4. Зміна найменування та/або місцезнаходження установи (закладу) відбору крові/плазми | IА | ІБ* | ||||||
| 4.5. Заміна або додавання нової установи (закладу) відбору крові/плазми, внесеної у ПМФ | IБ | |||||||
| 4.6. Вилучення або зміна статусу (робоча/неробоча) установи (закладу), яка займається відбором крові/плазми або аналізом зразків донорської крові та пулів плазми | IА | ІБ* | ||||||
| 4.7. Додавання нової установи (закладу) забору крові, яка не внесена у ПМФ | IІ | |||||||
| 4.8. Заміна або додавання установи (закладу), яка здійснює аналіз зразків донорської крові та/або пулів плазми, у складі установи (закладу) крові, внесеної у ПМФ | IБ | |||||||
| 4.9. Додавання нової установи (закладу), яка здійснює аналіз зразків донорської крові та/або пулів плазми, не внесеної у ПМФ | IІ | |||||||
| 4.10. Заміна або додавання нової установи (закладу) крові, в якій здійснюється зберігання плазми | IБ | |||||||
| 4.11. Вилучення установи (закладу) крові, в якій здійснюється зберігання плазми | IА | ІБ* | ||||||
| 4.12. Заміна або додавання суб’єкта господарювання, який здійснює транспортування плазми | IБ | |||||||
| 4.13. Вилучення суб’єкта господарювання, який здійснює транспортування плазми | IА | ІБ* | ||||||
| 4.14. Додавання тестового набору, який має СЕ-маркування, для тестування окремих зразків донорської крові в якості нового тестового набору або заміни зазначеного у ПМФ | IА | ІБ* | ||||||
| 4.15. Додавання тестового набору, який не має СЕ-маркування, для тестування окремих зразків донорської крові в якості нового тестового набору або заміни зазначеного у ПМФ | ||||||||
| а) новий тестовий набір, який не був затверджений у ПМФ для тестування окремих зразків донорської крові в іншій установі крові | ІІ | |||||||
| б) новий тестовий набір, який був затверджений у ПМФ для тестування окремих зразків донорської крові в іншій установі крові | IА | ІБ* | ||||||
| 4.16. Зміна набору/методу, які використовуються для тестування пулів плазми (антитіла або антигени, або NAT-тестування (технологія ампліфікації нуклеїнових кислот) | ІІ | |||||||
| 4.17. Введення або продовження строку інвентаризаційної процедури для зразків донорської крові | IА | ІБ* | ||||||
| 4.18. Вилучення або скорочення строку інвентаризаційної процедури для зразків донорської крові | IБ | |||||||
| 4.19. Заміна або додавання контейнерів для крові (наприклад, пляшки, пакети) | ||||||||
| а) нові контейнери для крові мають СЕ-маркування | IА | ІБ* | ||||||
| б) нові контейнери для крові не мають СЕ-маркування | ІІ | |||||||
| 4.20. Зміни у зберіганні/транспортуванні | ||||||||
| а) умови зберігання та/або транспортування | IА | ІБ* | ||||||
| б) максимальний строк зберігання для плазми | ІА | ІБ* | ||||||
| 4.21. Впровадження тестування на вірусні маркери за умови, що таке впровадження буде мати суттєвий вплив на оцінку вірусної безпеки | ІІ | |||||||
| 4.22. Зміни у приготуванні пулів плазми (наприклад, метод приготування, розмір пулу, зберігання зразків пулів плазми) | IБ | |||||||
| 4.23. Зміни у заходах, які будуть вжиті, якщо ретроспективно буде встановлено, що зразки донорської крові необхідно виключити з процесу (ретроспективні дослідження) | IІ | |||||||
*Якщо одна з умов не виконується і цю зміну неможливо віднести до змін ІІ типу.
Для документів, що подавалися на реєстрацію у форматі чотирьох частин (Додаток 9 до Порядку), надаються:
| Зміни у частині I реєстраційного досьє | Звіт експерта | ||
| Зміни у частині II реєстраційного досьє | Оновлення | ||
| Зміни у частині III реєстраційного досьє | Доповнення | ||
| Зміни у частині IV реєстраційного досьє |
Для документів, що подавалися на реєстрацію у форматі загального технічного документа (Додаток 5 до Порядку), надаються:
| Зміни у модулі 1 реєстраційного досьє | Короткий огляд | ||
| Зміни у модулі 2 реєстраційного досьє | Резюме | ||
| Зміни у модулі 3 реєстраційного досьє | Оновлення | ||
| Зміни у модулі 4 реєстраційного досьє | Доповнення | ||
| Зміни у модулі 5 реєстраційного досьє | |||
|
Інша(і) заява(и) (дайте коротке обґрунтування запропонованим змінам або іншим змінам, заявленим паралельно, або поновленню заяви, або розширенню заяви) |
|||
| Зміст (перерахуйте всі зміни в стислій формі) | |||
|
Передумови для внесення змін та обґрунтування послідовних змін (якщо необхідно) (надайте коротке пояснення передумовам для пропонованих змін, а також обґрунтування у разі послідовних змін) |
|||
| Діюча редакція* | Пропонована редакція* |
*Укажіть точну діючу та пропоновану редакцію або специфікацію; у разі змін, які стосуються маркування первинної, вторинної (за наявності) упаковок та інструкції для медичного застосування лікарського засобу, підкресліть або виділіть змінені слова, указані в таблиці вище, або подайте окремим додатком.
Додайте (за необхідності) пропоновані тексти інформації про лікарський засіб (коротка характеристика лікарського засобу, інструкція для медичного застосування, маркування первинної та вторинної (за наявності) упаковок) та інші матеріали, які обґрунтовують внесення змін.
Додаток 13
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
ЗМІНИ ВНЕСЕНО
Наказ Міністерства
охорони здоров’я України
_____________ № _________
Заявник, країна:
Виробник, країна:
ЗМІНИ
до інструкції для медичного застосування лікарського засобу/лікарського засобу (медичного імунобіологічного препарату)
НАЗВА ЛІКАРСЬКОГО ЗАСОБУ
лікарська форма, дозування, упаковка
| Попередня редакція | Нова редакція |
| Розділ «…» | Розділ «…» |
Уповноважена особа,
що виступає від імені заявника Печатка, підпис
Додаток 14
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
СТРУКТУРА
короткої характеристики лікарського засобу/лікарського засобу (медичного імунобіологічного препарату)
1. Назва лікарського засобу, дозування, лікарська форма.
2. Якісний і кількісний склад.
3. Лікарська форма.
4. Клінічна інформація:
4.1. Терапевтичні показання.
4.2. Дози та спосіб застосування.
4.3. Діти.
4.4. Протипоказання.
4.5. Особливі застереження та запобіжні заходи при застосуванні.
4.6. Взаємодія з іншими лікарськими засобами та інші види взаємодій.
4.7. Застосування під час вагітності та годування груддю.
4.8. Вплив на здатність керувати транспортними засобами або працювати з іншими автоматизованими системами.
4.9. Побічні реакції.
4.10. Передозування.
5. Фармакологічні властивості. Фармакотерапевтична група. Код АТХ:
5.1. Фармакодинамічні властивості.
5.2. Фармакокінетичні властивості.
5.3. Доклінічні дані з безпеки.
6. Фармацевтична інформація:
6.1. Допоміжні речовини.
6.2. Основні випадки несумісності.
6.3. Термін придатності.
6.4. Особливі запобіжні заходи при зберіганні.
6.5. Тип та вміст первинної упаковки.
6.6. Спеціальні заходи безпеки при поводженні з невикористаним лікарським засобом або відходами лікарського засобу (у разі необхідності).
7. Власник реєстраційного посвідчення.
Виробник лікарського засобу.
8. Номер реєстраційного посвідчення.
9. Дата першої реєстрації лікарського засобу.
10. Дата останнього перегляду.
Додаток 15
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські
засоби, що подаються на державну
реєстрацію (перереєстрацію), а також
експертизи матеріалів про внесення
змін до реєстраційних матеріалів
протягом дії реєстраційного посвідчення
Генеральному директору
Державного експертного центру МОЗ
_____________________________________
ЛИСТ
Заявник (представник заявника) _____________________, в особі _________________________,
(найменування) (прізвище та ініціали)
гарантує, що при наданні на державну реєстрацію такого лікарського засобу:
____________________________________________________________________________________________,
(назва лікарського засобу із зазначенням лікарської форми)
на який надаються матеріали реєстраційного досьє, мною додержано вимоги частини чотирнадцятої статті 9 Закону України «Про лікарські засоби», а саме: права третьої сторони, захищені патентом або передані за ліцензією, не порушуються у зв’язку із реєстрацією вказаного лікарського засобу.
Мені відомо, що у державній реєстрації даного лікарського засобу може бути відмовлено у разі, коли внаслідок такої реєстрації будуть порушені захищені патентом чинні майнові права інтелектуальної власності, у тому числі при виробництві, використанні, продажу лікарських засобів.
Дата ______________________ _________________________________
(прізвище та ініціали)
М. П.
Додаток 16
до Порядку проведення експертизи
реєстраційних матеріалів на лікарські засоби,
що подаються на державну реєстрацію
(перереєстрацію), а також експертизи
матеріалів про внесення змін до реєстраційних
матеріалів протягом дії реєстраційного
посвідчення
Перелік допоміжних речовин, які мають бути обов’язково зазначені на упаковці, та інформація щодо впливу цих речовин залежно від шляху введення та вмісту допоміжної речовини, що має бути наведена в інструкції для медичного застосування лікарського засобу
| Назва допоміжної речовини | Код речовини | Шлях введення | Граничний вміст | Інформація в інструкції для медичного застосування |
|---|---|---|---|---|
|
Азобарвники: тартразин жовтий захід FCF азорубін, кармоїзин амарант понсо 4R (пунцовий 4R), кошеніль червона А діамантовий чорний BN, чорний PN |
E 102 E 110 E 122 E 123 E 124 E 151 |
Пероральний | Нульовий | Може спричиняти алергічні реакції |
| Апротинін | Зовнішньо | Нульовий | Може спричиняти гіперчутливість та тяжкі алергічні реакції | |
| Аспартам | E 951 | Пероральний | Нульовий | Є похідним фенілаланіну, що являє небезпеку для хворих на фенілкетонурію |
| Бензалконію хлорид | Для застосування в офтальмології | Нульовий | Може спричиняти подразнення. Необхідно уникати контакту з м’якими контактними лінзами (зняти контактні лінзи перед нанесенням препарату та встановити через 15 хв. після застосування). Знебарвлює м’які контактні лінзи | |
| Зовнішньо | Спричиняє подразнення, може спричинити шкірні реакції | |||
| Інгаляційний | 10 мкг/доза | Може спричинити бронхоспазм | ||
| Бергамотова оліяБергаптен | Зовнішньо | Нульовий | Може підвищувати чутливість до ультрафіолетового випромінювання (природного та штучного) | |
| Бронопол | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) | |
| Бутилгідрокситолуол | E 321 | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) або подразнення очей та слизових оболонок |
| Бутилгідроксіанізол | E 320 | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) або подразнення очей та слизових оболонок |
| Галактоза | Парентеральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | |
| Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | ||
|
Пероральний Парентеральний |
5 г | |||
| Гепарин (як допоміжна речовина) | Парентеральний | Нульовий | Може спричинити алергічні реакції та вплинути на систему згортання крові. Пацієнтам з наявністю в анамнезі гепарин-індукованих алергічних реакцій слід уникати застосування лікарських засобів, що містять гепарин | |
| Гліцерин (гліцерол) | Пероральний | 10 г/доза | Може спричинити головний біль, подразнення ШКТ та діарею | |
| Ректальний | 1 г | Може чинити м’яку послаблюючу дію | ||
| Глюкоза | Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | |
|
Пероральний Парентеральний |
5 г | Містить … г глюкози на дозу. З обережністю застосовують хворим на цукровий діабет | ||
| Рідкі лікарські форми для перорального застосування, пастилки, льодяники, жувальні таблетки | Нульовий | Може бути шкідливим для зубів | ||
| Диметилсульфоксид | Зовнішньо | Нульовий | Може спричинити подразнення шкіри | |
| Етанол (спирт) | Пероральний та парентеральний | Менше 100 мг/доза | Цей лікарський засіб містить невелику кількість етанолу (алкоголю), менше 100 мг/дозу | |
| 100 мг — 3 г/доза | Цей лікарський засіб містить … об.% етанолу (алкоголю), тобто … мг/дозу, що еквівалентно … мл пива, … мл вина у дозі.Шкідливий для пацієнтів, хворих на алкоголізм. Слід бути обережним при застосуванні вагітним та жінкам, які годують груддю, дітям та пацієнтам із захворюваннями печінки та хворим на епілепсію | |||
| Пероральний та парентеральний | 3 г/доза | Цей лікарський засіб містить … об.% етанолу (алкоголю), тобто… мг/дозу, що еквівалентно … мл пива, … мл вина у дозі.Шкідливий для пацієнтів, хворих на алкоголізм. Слід бути обережним при застосуванні вагітним та жінкам, які годують груддю, дітям та пацієнтам із захворюваннями печінки та хворим на епілепсію.Кількість алкоголю у цьому лікарському засобі може впливати на дію інших препаратів.Кількість алкоголю у цьому лікарському засобі може впливати на здатність керувати транспортними засобами або працювати з механізмами | ||
| Калію сполуки | Парентеральний | Менше1 ммоль/доза | Цей лікарський засіб містить менше 1 ммоль (39 мг)/дозу калію, тобто практично вільний від калію | |
|
Парентеральний Пероральний |
1 ммоль/доза | Цей лікарський засіб містить … ммоль (або … мг)/дозу калію. Слід бути обережним при застосуванні пацієнтам зі зниженою функцією нирок або таким, хто застосовує калій-контрольовану дієту | ||
| Парентеральний (внутрішньо-венний) | 30 ммоль/л | Може спричинити біль у місці введення | ||
|
Кислота бензойна та бензоати: кислота бензойна натрію бензоат калію бензоат |
E 210 E 211 E 212 |
Зовнішньо | Нульовий | Помірно подразнює шкіру, очі та слизові оболонки |
| Парентеральний | Нульовий | Може підвищувати ризик виникнення жовтяниці у новонароджених | ||
| Кислота сорбінова та її солі | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад контактний дерматит) | |
| Крохмаль пшеничний | Пероральний | Нульовий | Можна застосовувати хворим на целіакію. Пацієнти з алергією на пшеницю (відмінну від целіакії) не повинні застосовувати цей лікарський засіб | |
| Ксиліт | Пероральний | 10 г | Може чинити послаблюючу дію. Енергетична цінність 1 г ксиліту — 2,4 ккал | |
| Лактит | E 966 | Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб |
| 10 г | Може чинити м’яку послаблюючу дію. Енергетична цінність 1 г лактиту — 2,1 ккал | |||
| Лактоза | Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | |
| 5 г | Містить … г лактози (…/2 г глюкози та …/2 г галактози) на дозу. З обережністю застосовують хворим на цукровий діабет | |||
| Ланолін | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) | |
| ЛатексНатуральний каучук | Будь-який | Нульовий | Упаковка цього лікарського засобу містить латексну гуму. Може спричиняти тяжкі алергічні реакції | |
|
Мальтит Ізомальтит Мальтит рідкий (сироп глюкози гідрогенізований) |
E 965 Е 953 |
Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб |
| 10 г | Може чинити м’яку послаблюючу дію. Енергетична цінність 1 г мальтиту (або ізомальтиту) — 2,3 ккал | |||
| Маніт | E 421 | Пероральний | 10 г | Може чинити м’яку послаблюючу дію |
| Натрію сполуки | Парентеральний | Менше1 ммоль/доза | Цей лікарський засіб містить менше 1 ммоль (23 мг)/дозу натрію, тобто практично вільний від натрію | |
|
Пероральний Парентеральний |
1 ммоль/доза | Цей лікарський засіб містить … ммоль (або … мг)/дозу натрію. Слід бути обережним при застосуванні пацієнтам, які застосовують натрій-контрольовану дієту | ||
| Олія арахісова | Будь-який | Нульовий | Лікарський засіб містить арахісову олію. Якщо у Вас є алергія на арахіс або сою, не вживайте цей лікарський засіб | |
| Олія кунжутна | Будь-який | Нульовий | Рідко може спричиняти тяжкі алергічні реакції | |
| Олія рицинова поліетоксильована та олія рицинова поліетоксильована, гідрогенізована | Парентеральний | Нульовий | Може спричинити тяжкі алергічні реакції | |
| Пероральний | Нульовий | Може спричинити розлад шлунка та діарею | ||
| Зовнішньо | Нульовий | Може спричинити шкірні реакції | ||
| Олія соєва та олія соєва гідрогенізована | Будь-який | Нульовий | Лікарський засіб містить соєву олію. Якщо у Вас є алергія на арахіс або сою, не вживайте цей лікарський засіб | |
|
Органічні сполуки ртуті, наприклад: тіомерсал фенілртуті нітрат фенілртуті ацетат фенілртуті борат |
Для застосування в офтальмології | Нульовий | Може спричинити алергічні реакції | |
| Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад контактний дерматит) та знебарвлення шкіри | ||
| Парентеральний | Нульовий | Цей лікарський засіб містить тіомерсал як консервант та може спричинити у Вас/Вашої дитини алергічну реакцію. Повідомте лікаря, якщо у Вас/Вашої дитини були/виникли алергічні реакції | ||
|
Парагідроксибензоати та їх ефіри: етилпарагідроксибензоат пропілпарагідроксибензоат натрію пропілпарагідрокси-бензоат метилпарагідроксибензоат натрію метилпарагідрокси-бензоат |
E 214 E 216 E 217 E 218 E 219 |
Пероральний Для застосування в офтальмології Зовнішньо |
Нульовий | Може спричинити алергічні реакції (можливо, уповільнені) |
|
Парентеральний Інгаляційний |
Нульовий | Може спричинити алергічні реакції (можливо, уповільнені) та в окремих випадках бронхоспазм | ||
| Перуанський бальзам | Зовнішньо | Нульовий | Може спричинити шкірні реакції | |
| Пропіленгліколь та ефіри | Зовнішньо | Нульовий | Може спричинити подразнення шкіри | |
|
Пероральний Парентеральний |
400 мг/кг (для дорослих)200 мг/кг (для дітей) | Може спричинити симптоми, схожі з такими, що виникають при вживанні алкоголю | ||
| Сорбіт | E 420 |
Пероральний Парентеральний |
Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб |
| Пероральний | 10 г | Може чинити м’яку послаблюючу дію. Енергетична цінність 1 г сорбіту — 2,6 ккал | ||
| Спирт бензиловий | Парентеральний | Менше90 мг/кг/добу | Не застосовують недоношеним дітям та новонародженим.Може спричинити токсичні та алергічні реакції у немовлят та дітей віком до 3 років | |
| 90 мг/кг/добу | Не застосовують недоношеним дітям та новонародженим.Через ризик фатальних токсичних реакцій не застосовувати немовлятам та дітям віком до 3 років | |||
| Спирт стеариловий | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) | |
| Спирт цетиловий | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) | |
| Спирт цетостеариловий | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад, контактний дерматит) | |
|
Сульфіти, включаючи метабісульфіти, наприклад: сірки діоксид натрію сульфіт натрію гідросульфіт натрію метабісульфіт калію метабісульфіт калію гідросульфіт |
E 220 E 221 E 222 E 223 E 224 E 228 |
Пероральний Парентеральний Респіраторний |
Нульовий | Рідко може спричиняти реакції гіперчутливості та бронхоспазм |
| Фенілаланін | Будь-який | Нульовий | Цей лікарський містить фенілаланін.Може бути небезпечним для хворих на фенілкетонурію | |
| Формальдегід | Зовнішньо | Нульовий | Може спричинити місцеві шкірні реакції (наприклад контактний дерматит) | |
| Пероральний | Нульовий | Може спричинити розлад шлунка та діарею | ||
| Фруктоза |
Пероральний Парентеральний |
Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | |
| 5 г | Містить … г фруктози на дозу. З обережністю застосовують хворим на цукровий діабет | |||
| Рідкі лікарські форми для перорального застосування, пастилки, льодяники, жувальні таблетки | Нульовий | Може бути шкідливим для зубів | ||
| Хлоркрезол |
Зовнішньо Парентеральний |
Нульовий | Може спричиняти алергічні реакції | |
| Цукор інвертний | Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | |
| 5 г | Містить … г суміші фруктози та глюкози на дозу. З обережністю застосовують хворим на цукровий діабет | |||
| Рідкі лікарські форми для перорального застосування, пастилки, льодяники, жувальні таблетки | Нульовий | Може бути шкідливим для зубів | ||
| Цукроза (сахароза) | Пероральний | Нульовий | Якщо у Вас встановлена непереносимість деяких цукрів, проконсультуйтеся з лікарем, перш ніж приймати цей лікарський засіб | |
| 5 г | Містить … г цукрози на дозу. З обережністю застосовують хворим на цукровий діабет | |||
| Рідкі лікарські форми для перорального застосування, пастилки, льодяники, жувальні таблетки | Нульовий | Може бути шкідливим для зубів |
________________
Термін «нульовий» означає будь-який вміст речовини, починаючи з мінімального.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим