 На теперішній час нові лікарські засоби, незалежно від того, у якій країні вони розроблені, оцінюються на підставі результатів досліджень, проведених із дотриманням уніфікованих вимог, у тому числі й правил належної клінічної практики (Good Clinical Practice — GCP). Для виконання сучасних вимог GCP необхідна співпраця усіх зацікавлених сторін — промисловості, дослідників, державних органів, відповідальних за реєстрацію лікарських препаратів й контроль за їх обігом, комісій з питань етики. Дотримання вимог GCP — необхідна умова для належного проведення клінічних випробувань у більшості розвинених країн світу.
На теперішній час нові лікарські засоби, незалежно від того, у якій країні вони розроблені, оцінюються на підставі результатів досліджень, проведених із дотриманням уніфікованих вимог, у тому числі й правил належної клінічної практики (Good Clinical Practice — GCP). Для виконання сучасних вимог GCP необхідна співпраця усіх зацікавлених сторін — промисловості, дослідників, державних органів, відповідальних за реєстрацію лікарських препаратів й контроль за їх обігом, комісій з питань етики. Дотримання вимог GCP — необхідна умова для належного проведення клінічних випробувань у більшості розвинених країн світу.
Протягом останніх років в Україні з метою поступової гармонізації з міжнародними стандартами здійснювалися розробка та впровадження регламентуючих документів щодо удосконалення процедури експертизи матеріалів та проведення клінічних випробувань. Так, у 2009 р. наказом МОЗ України від 16.02.2009 р. № 95 була затверджена настанова з клінічних досліджень — Лікарські засоби. Належна клінічна практика. Настанова 42-7.0:2008. Цей документ розроблений на підставі Настанови з Належної клінічної практики (Настанова СРСР/ICH/135/95 (Е6) «Note for Guidance on Good Clinical Practice», 1997 р.).
З метою удосконалення процедури клінічних випробувань в Україні були внесені відповідні зміни до нормативно-правової бази. Зокрема, наказами МОЗ України від 06.05.2014 р. № 304 (зареєстровано в Міністерстві юстиції України 07.07.2014 р. за № 739/25516), від 18.12.2014 р. № 966 (зареєстровано в Міністерстві юстиції України 17.01.2015 р. за № 62/26507) та від 01.10.2015 р. № 639 (зареєстровано в Міністерстві юстиції України 08.12.2015 р. за № 1520/27965) змінено Порядок проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань, затверджений наказом МОЗ України від 23.09.2009 р. № 690, зареєстрований в Міністерстві юстиції України 29.10.2009 р. за № 1010/17026 (далі — Порядок), розроблений з урахуванням вимог Директиви Європейського Парламенту та Ради 2001/20/ЕС.
З метою реалізації положень Податкового кодексу України (застосування 7% ставки податку на додану вартість (ПДВ) під час ввезення незареєстрованих лікарських засобів на митну територію України) змінами до Порядку передбачене затвердження клінічного випробування/суттєвої поправки наказами МОЗ. Заяву про проведення клінічного випробування лікарських засобів/про внесення суттєвої поправки до протоколу клінічного випробування подається до Центру адміністративних послуг МОЗ України — «Єдине вікно». Після затвердження накази оприлюднюються на сайті МОЗ. Ця процедура наблизила створення єдиного відкритого реєстру клінічних випробувань в Україні та зробила процес їх проведення прозорішим.
ІНФОРМАЦІЯ ЩОДО КЛІНІЧНИХ ВИПРОБУВАНЬ ЗА 2010 — І КВ. 2017 Р.
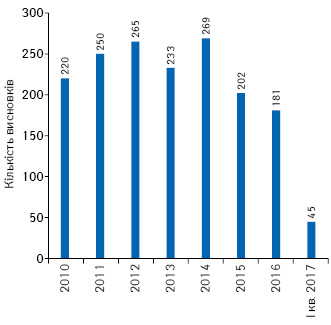
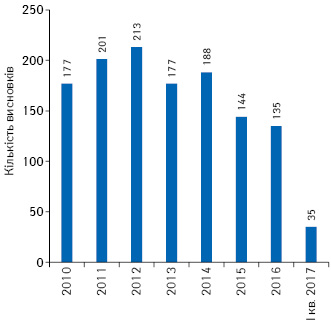
За 2010 — І кв. 2017 р. заявники отримали 1665 позитивних висновків щодо можливості проведення клінічних випробувань лікарських засобів як вітчизняних, так і зарубіжних (рис. 1), з них надано 1270 позитивних висновків щодо можливості проведення міжнародних багатоцентрових клінічних випробувань (рис. 2).
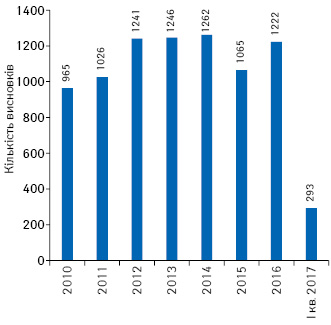
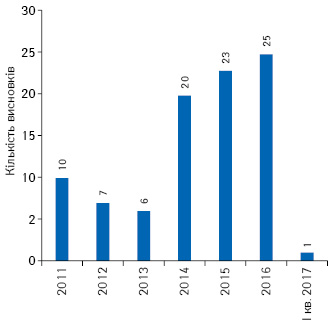
Під час проведення клінічних випробувань заявник може внести суттєві поправки до протоколу клінічних випробувань, які також розглядаються в установленому порядку. З 2010 до І кв. 2017 р. після проведеної експертизи надано 8320 позитивних висновків щодо суттєвих поправок до міжнародних багатоцентрових клінічних випробувань (рис. 3).
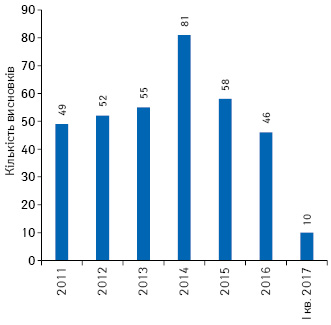
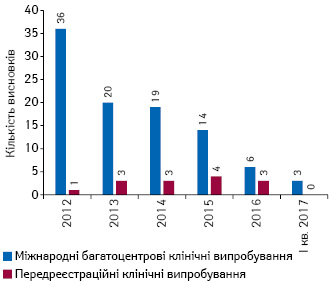
За останні 6 років було надано 390 позитивних висновків щодо можливості проведення передреєстраційних клінічних випробувань в Україні (рис. 4), з них за останні 3 роки надано 36 позитивних висновків щодо проведення досліджень з біоеквівалентності (рис. 5).
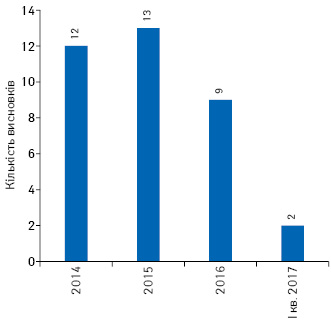
У період 2011 — І кв. 2017 р. сумарно кількість суттєвих поправок до матеріалів передреєстраційних клінічних випробувань становила 92 (рис. 6).
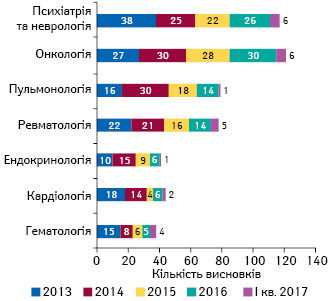
Аналіз матеріалів клінічних випробувань за останні роки свідчить, що частіше проводяться клінічні випробування лікарських засобів для лікування онкологічних, психіатричних, неврологічних, пульмонологічних, ревматологічних захворювань (рис. 7). Це пов’язано з тим, що такі захворювання найчастіше стають причиною інвалідизації людини та погіршення якості її життя.
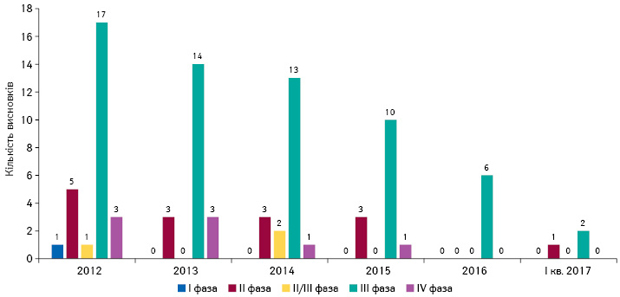
Під час аналізу матеріалів клінічних випробувань визначено, що найчастіше проводяться клінічні випробування ІІ–III фази за участю пацієнтів із захворюваннями, для лікування яких розробляється досліджуваний лікарський засіб (рис. 8).
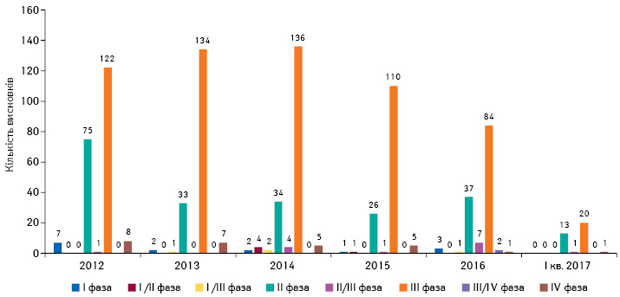
Саме ці фази клінічних випробувань потребують участі значної кількості пацієнтів з відповідною патологією. Участь України в багатоцентрових клінічних випробуваннях, які одночасно проводяться у декількох країнах світу, має велике значення для пацієнтів, лікарів та країни в цілому. Для пацієнтів це можливість отримати безкоштовно інноваційні лікарські засоби, для лікарів — знайомство з новими технологіями, для країни — можливість подальшої реєстрації препаратів з доведеними ефективністю та безпекою.
Експертиза матеріалів клінічних випробувань з метою подальшого складання висновку щодо проведення клінічного випробування/суттєвої поправки проводиться співробітниками ДП «Державний експертний центр МОЗ України» із залученням фахівців консультативно-експертних груп відповідного профілю, з дотриманням вищезазначених нормативно-правових документів та відповідно до посадових інструкцій. Крім того, етичну оцінку матеріалів клінічних випробувань здійснюють комісії з питань етики при лікувально-профілактичних закладах, у яких вони проводяться.
За оцінками експертів, потенціал України щодо можливості проведення клінічних випробувань використовується лише на 10–15%, незважаючи на імплементацію міжнародних вимог та наявність висококваліфікованих лікарів у різних напрямках медицини.
КЛІНІЧНІ ВИПРОБУВАННЯ ЗА УЧАСТЮ ДІТЕЙ
У світі, у тому числі й в Україні, наразі існує нагальна потреба в розробці нових і більш детального вивчення вже існуючих лікарських засобів для застосування в педіатрії. У частині проведення клінічних випробувань за участю дітей законодавство України на сьогодні більш регламентоване та суворіше, ніж у країнах ЄС, що свідчить про високий рівень відповідальності у сфері клінічних випробувань, які проводяться із залученням дітей. Це обумовлено тим, що діти мають фізіологічні та психологічні особливості, пов’язані з розвитком організму, недосконалістю ферментативних і рецепторних систем, які визначають характер фармакокінетики та фармакодинаміки лікарського засобу.
Крім того, існує ряд захворювань, які зустрічаються тільки в дитячому віці. Наявні відмінності між дитячою та дорослою популяцією, а також між дітьми різних вікових груп є основними чинниками, які визначають необхідність проведення клінічних випробувань лікарських засобів у педіатрії, з урахуванням фізіологічних особливостей і віку дитини.
В Україні за період 2012 — І кв. 2017 р. надано 98 позитивних висновків щодо проведення багатоцентрових клінічних випробувань лікарських засобів, призначених для застосування у педіатрії, та 14 позитивних висновків щодо передреєстраційних клінічних випробувань (рис. 9).
У педіатрії найчастіше проводяться клінічні випробування ІІІ фази за участю дітей різних вікових категорій (рис. 10).
КЛІНІЧНИЙ АУДИТ
Важливою складовою у забезпеченні якості клінічних випробувань є клінічний аудит, який проводять співробітники відділу клінічного аудиту Державного експертного центру. Клінічний аудит клінічного випробування — це процедура офіційної перевірки Державним експертним центром документів, приміщень, устаткування та обладнання, записів, системи якості та інших ресурсів, які мають відношення до клінічних випробувань та можуть бути розташовані на клінічних базах, у приміщеннях спонсора, контрактної дослідницької організації та інших місцях, які задіяні у процесі клінічного випробування. Вимоги до проведення клінічного аудиту визначені в Розділі ХІІІ Поряду.
Попередній план клінічного аудиту оприлюднюється на офіційному сайті Державного експертного центру та оновлюється щоквартально. У плані зазначаються дата та місце проведення клінічного аудиту.
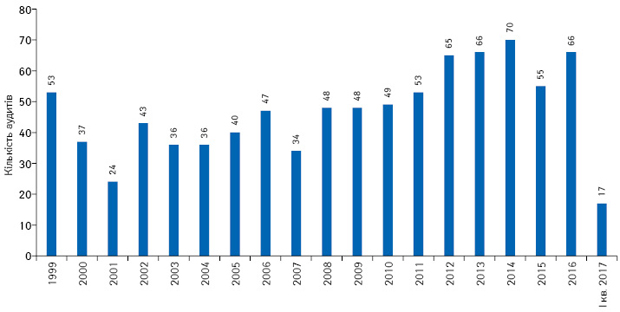
Державний експертний центр проводить клінічні аудити (інспекції) клінічних випробувань з 1999 р. (рис. 11)
Основні завдання клінічного аудиту клінічного випробування — це перевірка:
- відповідності проведення клінічного випробування затвердженому протоколу й діючим нормативно-правовим актам;
- можливості проведення клінічного випробування в місці проведення випробування, включення достатньої кількості досліджуваних відповідно до профілю клінічного випробування, наявності необхідного устаткування, функціонування лабораторії, первинної медичної документації — медичних карт стаціонарних/амбулаторних хворих, даних лабораторних та інструментальних методів обстеження;
- захисту прав пацієнтів — дотримання процедури отримання інформованої згоди, наявність письмової інформованої згоди на участь у клінічному випробуванні (датована й підписана);
- достовірності й відповідності записів в індивідуальних реєстраційних формах даним первинної документації;
- наявності документації про одержання, зберігання, видачу, розподіл й повернення досліджуваного лікарського засобу;
- реєстрації побічних ефектів та їх лікування тощо.
Клінічний аудит клінічного випробування проводиться не раніше ніж через 14 календарних днів після надсилання попереднього повідомлення та узгодження з відповідальним дослідником та/або заявником клінічного випробування дати початку та тривалості перевірки, її мети, переліку документів та приміщень, що перевірятимуться.
За результатами клінічного аудиту клінічного випробування складається акт, у якому за наявності зазначаються зауваження (несуттєві, суттєві, критичні, що визначені в Розділі ХІІІ Поряду) та терміни їх усунення. Державний експертний центр надсилає акт про перевірку спонсору та відповідальному досліднику клінічного випробування.
За період з 2012 до І кв. 2017 р. співробітники Державного експертного центру взяли участь (на території України) у 5 інспекційних перевірках клінічних випробувань лікарських засобів, що проводилися представниками Управління з контролю за харчовими продуктами та лікарськими засобами США (Food and Drug Administration — FDA), 2 інспекційних перевірках клінічних випробувань лікарського засобу, що проводилися представниками Європейського агентства з лікарських засобів (European Medicines Agency — ЕМА), та 1 інспекційній перевірці клінічного випробування лікарського засобу, що проводилась представниками регуляторного органу Японії — Pharmaceuticals and Medical Devices Agency (PMDA).
Інспектори ЕМА, FDA та PMDA визнали систему клінічних випробувань в Україні достатньо розвиненою. Це означає, що в нашій країні клінічні випробування проводяться на відповідному рівні з дотриманням вимог нормативної бази України та міжнародних стандартів.
У 2014–2016 рр. співробітники Державного експертного центру взяли участь у робочих нарадах (семінарах) GCP-інспекторів ЕМА. У 2015 р. співробітники відділу клінічного аудиту отримали сертифікати після успішного проходження навчання, організованого ЕМА: «GCP Inspector’s basic training course», ЕМА.
З метою роз’яснення основних положень Порядку та міжнародних вимог щодо проведення клінічних випробувань Державний експертний центр регулярно організовує навчальні семінари. Так, у 2015 р. проведено 10 навчальних семінарів для 815 лікарів-дослідників, а в 2016 р. — 17 семінарів, у яких взяли участь 977 слухачів.
У 2015 р. Державний експертний центр виступив організатором V Науково-практичної конференції з міжнародною участю «Клінічні випробування лікарських засобів в Україні». Загальна кількість учасників заходу становила 600 осіб.
На сьогодні в Україні створена нормативна база, яка дозволяє проводити клінічні випробування відповідно до міжнародних стандартів. У той же час стратегія інтеграції України в ЄС вимагає проведення подальших заходів щодо гармонізації нормативно-правової базі у сфері обігу лікарських засобів з вимогами стандартів та директив ЄС.
Л.Я. Янкова, С.С. Распутняк,
за даними роботи Департаменту експертизи матеріалів доклінічних та клінічних випробувань Державного експертного центру МОЗ України
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим