ПОВІДОМЛЕННЯ
про оприлюднення проекту наказу Міністерства охорони здоров’я України
Про внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики
Проект наказу Міністерства охорони здоров’я України «Про внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики» (далі — проект наказу) розроблений з метою забезпечення належного державного контролю якості лікарських засобів в державі, а також гармонізації законодавства України із законодавством ЄС.
Наказ було розроблено та оприлюднено для громадського обговорення на офіційних сайтах Міністерства охорони здоров’я України (www.moz.gov.ua) та Державної служби України з лікарських засобів та контролю за наркотиками ([email protected]) до 13.09.2019 року.
За результатами громадського обговорення було проведено узгоджувальну нараду, доопрацьовано проект наказу.
Наразі доопрацьований проект наказу та повідомлення про оприлюднення проекту наказу розміщено на сайті Міністерства охорони здоров’я України (www.moz.gov.ua) та на сайті Державної служби України з лікарських засобів та контролю за наркотиками (www.dls.gov.ua).
Зауваження та пропозиції просимо надсилати до 02.12.2019 року до:
– Міністерства охорони здоров’я України за адресою: м. Київ, 01601, вул. Грушевського, 7, e-mail: [email protected]; Фармацевтичний директорат e-mail: [email protected];
– Державної служби України з лікарських засобів та контролю за наркотиками за адресою: м. Київ, 03115, просп. Перемоги, 120-А, e-mail: [email protected]; [email protected].
ПОЯСНЮВАЛЬНА ЗАПИСКА
до проекту наказу Міністерства охорони здоров’я України
«Про внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики»
1. Резюме
Метою затвердження проекту наказу «Про внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики» є забезпечення належного контролю якості лікарських засобів й умов їх виробництва, а також гармонізація законодавства України із законодавством Європейського Союзу.
2. Проблема, яка потребує роз’яснення
З 16 вересня 2014 року Європейський парламент ратифікував Угоду про асоціацію між Україною та Європейським Союзом синхронно з Верховною Радою України. 01 вересня 2017 року після тривалого процесу ратифікації Угода про асоціацію між Україною та ЄС набула чинності у повному обсязі. Угода за своїм обсягом і тематичним охопленням стала найбільшим міжнародно-правовим документом за всю історію України та найбільшим міжнародним договором з третьою країною, укладеним Європейським Союзом. Вона стала яскравою демонстрацією якісно нового формату відносин між Україною та ЄС на принципах «політичної асоціації та економічної інтеграції». Держлікслужба відповідно до покладених на неї завдань, зокрема здійснює державний контроль за дотриманням вимог законодавства щодо забезпечення якості та безпеки лікарських засобів на всіх етапах обігу, в тому числі контроль за виробництвом на відповідність умов вимогам європейського стандарту — належної виробничої практики.
Відтак, Україна йде шляхом євроінтеграції, а приведення нормативно-правових актів України до європейських норм є обов’язковим.
Внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики (GMP) (далі — Порядок) передбачає гармонізацію термінології з нормами збірки процедур Європейського Співтовариства з проведення інспекцій та обміну інформацією від 03.10.2014 № EMA/572454/2014 Rev 17 (Compilation of Community Procedures on Inspections and Exchange of Information) (далі — Компіляційна Процедура), а саме: визначення понять «критичне порушення», «суттєве порушення» та «несуттєве порушення».
Також Компіляційною Процедурою передбачена методологія проведення оцінки ризиків щодо розрахунку строку наступної інспекції виробництва лікарського засобу на відповідність вимогам GMP, а саме розрахунок здійснюється на основі аналізу внутрішнього ризику, пов’язаного з виробничою дільницею, складністю виробничої дільниці, її процесів і продукції та критичності лікарських засобів, що виробляється виробничою дільницею, класифікацією виявлених порушень. Зміни, що вносяться новою редакцією Порядку передбачають актуалізацію вимог українського законодавства щодо розрахунку строку наступної інспекції.
Компіляційною Процедурою ЄС передбачена видача сертифіката GMP лише за наявності документальних підтверджень усунення критичних та суттєвих порушень. Водночас змінами, передбаченими новою редакцією Порядку, пропонується інтегрований, але спрощений підхід до цієї норми: видача сертифіката GMP після надання прийнятного плану коригувальних дій (без документальних підтверджень, надання яких передбачене пізніше згідно з термінами, зазначеними в плані коригувальних дій).
Відповідно до вимог Директиви 2001/83/ЄС Європейського Парламенту та Ради від 06 листопада 2001 року щодо Кодексу Співтовариства стосовно лікарських засобів, призначених для застосування людиною; Компіляційної Процедури та інших документів ЄС інспектування на відповідність умов виробництва лікарських засобів вимогам GMP здійснюється регуляторним органом країни-члена ЄС або країни, яка має угоду з ЄС про взаємне визнання результатів інспектування на відповідність умов виробництва лікарських засобів вимогам GMP (далі — угода про взаємне визнання з ЄС), лише за наявності заяви на реєстрацію лікарського засобу в країні ЄС або в країні, яка має угоду про взаємне визнання з ЄС (заява на отримання marketing authorization).
В подальшому регуляторний орган, що видав сертифікат GMP, здійснює наглядові інспектування, які стосуються виробництва конкретних лікарських засобів, що були зареєстровані після проведення інспектування на території ЄС або країни, яка має угоду про взаємне визнання з ЄС. Діючим Порядком передбачене документальне визнання сертифікатів GMP, виданих регуляторними органами країн-членів PIC/S (в тому числі ЄС), без проведення інспектування. Водночас останнім часом частина лікарських засобів, які подаються на процедуру визнання сертифіката GMP в Україні, не були предметом інспектування з боку жодного регуляторного органу країн-членів PIC/S або ЄС та не зареєстровані на території країн PIC/S та ЄС.
Тобто жоден регуляторний орган не несе відповідальності за лікарські засоби, які не були предметом їх перевірки, та немає достовірної інформації щодо відповідності їх виробництва вимогам належної виробничої практики та вимогам реєстраційного досьє.
Пунктом 2, 3 розділу І діючим Порядком визначено, що процедура підтвердження — це підтвердження відповідності умов виробництва лікарських засобів чинним в Україні вимогам GMP запроваджено з метою доведення, що лікарські засоби постійно виробляються і контролюються згідно зі стандартами якості, які відповідають їх призначенню, а також відповідно до вимог реєстраційного досьє, досьє досліджуваного лікарського засобу для клінічних випробувань або специфікації на цю продукцію.
Процес визнання надав можливість забезпечити пацієнтів України лікарськими засобами європейської якості за спрощеною процедурою, водночас, для не добропорядних виробників ця процедура на сьогодні надає можливість ввозити лікарські засоби не встановленої відповідності, так звані «ліки сірого виробництва».
В Україні не повинно бути подвійної сертифікації, але в Україну повинні потрапляти лише ті лікарські засоби, які дозволені в ЄС або країнах, які мають угоди про взаємне визнання з ЄС, та мають в цих країнах реєстрацію.
Враховуючи вищезазначене, в новій редакції Порядку впроваджено норму щодо подання в комплекті документів при підтвердженні відповідності умов виробництва лікарських засобів чинним в Україні вимогам GMP для виробників, виробничі потужності яких розташовані в третіх країнах та які подають заяви на визнання (без проведення інспектування з боку України) копій реєстраційних посвідчень (marketing authorization), виданих компетентним органом країни ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною.
Проектом змін, передбачених новою редакцією Порядку, переглянута процедура визнання сертифікатів GMP (без проведення інспектування з боку України), які видані іншими регуляторними органами (процедура підтвердження відповідності умов виробництва лікарських засобів чинним в Україні вимогам GMP) з позиції, по-перше, євроінтеграції та майбутнього підписання угоди про взаємне визнання результатів інспектування на відповідність умов виробництва лікарських засобів вимогам GMP між Україною та ЄС, по-друге, розуміння всіма учасниками фармацевтичного ринку що таке PIC/S.
Підписання угоди про взаємне визнання між Україною та ЄС не можливе без ідентичності процедур в Україні вимогам ЄС щодо ліцензування виробництва та імпорту лікарських засобів, впровадження вимог стандарту GMP, ідентичність процедур інспектування та сертифікації лікарських засобів та, головне, в цілому ідентичність допуску на ринки України та ЄС лікарських засобів з інших країн.
Процедура Визнання в діючому Порядку зовсім не забезпечує та не гарантує прозорої відповідності, як вже зазначено, і допуск на ринок лікарських засобів в Україну не є ідентичним на сьогодні з ЄС. Терміново потребує приведення у відповідність норм діючого Порядку до Компіляційної Процедури в частині звуження країн визнання. Відповідно до Компіляційної Процедури країнами визнання можуть були лише країни ЄС та країни, з якими ЄС має угоди про взаємне визнання.
З 2011 року Україна, в особі Держлікслужби, є членом міжнародної Системи фармацевтичних інспекцій, PIC/S. PIC/S — це неофіційний договір про співпрацю в сфері GMP. Цілі та завдання PIC/S не полягають в процедурі визнання, PIC/S спрямована на гармонізацію процедур інспектування у всьому світі шляхом розробки загальних стандартів у сфері GMP та наданням можливостей для навчання інспекторам.
Цілі PIC/S досягаються шляхом розробки та впровадження стандартів GMP та керівних документів; підготовки компетентних органів, зокрема інспекторів GMP; оцінці (та переоцінці) інспекторатів та сприяння співпраці та роботі в системі для компетентних органів та міжнародних організацій. На сьогодні PIC/S нараховує 52 регуляторні з усього світу (Європи, Африки, Америки, Азії та Австралії).
Міжнародна організація постійно збільшує кількість членів, таким чином просуваючи вимоги GMP у всьому світі. Водночас, взаємного визнання між членами PIC/S немає, кожна країна має внутрішні нормативно-правові акти, які регулюють відповідні сфери. Наразі, заяви на вступ до PIC/S подано: Вірменія, Бразилія, Болгарія. Також на сьогодні вже розглядаються пре-заяви на вступ до PIC/S: Бангладеш, Пакистан, Російська Федерація, Йорданія, Саудівська Аравія.
Враховуючи вищезазначене, змінами, передбаченими в новій редакції Порядку, передбачено відповідність до Компіляційної Процедури в частині визначення країн визнання результатів інспектування на відповідність умов виробництва лікарських засобів вимогам GMP, а саме це країни — члени ЄС або країни, які мають угоду про взаємне визнання з ЄС. З метою розвитку фармацевтичного сектору галузі охорони здоров’я України до країн визнання внесено країни, які будуть мати угоду про взаємне визнання з Україною.
01 січня 2019 року прийнято в дію Настанову PIC/S PI 040-1 з класифікації невідповідностей GMP, у зв’язку з чим Держлікслужба замінює Додаток 10 до Порядку.
Внесення змін та викладення в новій редакції Порядку забезпечить відповідність процедури проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики вимогам ЄС.
3. Суть проекту акта
Після впровадження проекту акта очікується:
посилення контролю за якістю лікарських засобів;
недопущення виробництва лікарських засобів невідповідної якості;
створення умов, що гарантують надходження на національний ринок України тільки якісних, ефективних та безпечних лікарських засобів, що вироблені відповідно до вимог стандартів і є запорукою захисту здоров’я і життя громадян та безпеки держави в цілому.
4. Вплив на бюджет
Реалізація проекту акта не потребує додаткових матеріальних та інших витрат з Державного бюджету України.
5. Позиція заінтересованих сторін
Від реалізації проекту акта очікується позитивний вплив на:
забезпечення подальшої гармонізації національної нормативно-правової бази у сфері обігу лікарських засобів із міжнародним та європейським законодавством;
створення належних умов для ведення підприємницької діяльності на фармацевтичному ринку України, що позитивно вплине на процеси детінізації економіки у цій сфері;
випуск в обіг лікарських засобів, які відповідають встановленим вимогам;
дотримання законних вимог посадових осіб центрального органу виконавчої влади, що реалізує державну політику у сфері контролю якості та безпеки лікарських засобів;
забезпечення населення України якісними, безпечними та ефективними лікарськими засобами.
Проект акта не стосується розвитку адміністративно-територіальних одиниць та соціально-трудової сфери, тому не потребує погодження з уповноваженими представниками від всеукраїнських профспілок, їх об’єднань та всеукраїнськими об’єднаннями організацій роботодавців.
Проект акта не стосується сфери наукової та науково-технічної діяльності. Реалізація проекту акта не впливає на ринок праці.
Прогноз впливу реалізації акта на ключові інтереси заінтересованих сторін додається.
6. Прогноз впливу
Проект акта впливає на ринкове середовище, суб’єктів господарської діяльності з виробництва лікарських засобів та забезпечення прав та інтересів населення.
7. Позиція заінтересованих органів
Проект акта потребує погодження з Державною регуляторною службою України, Уповноваженим Верховної Ради України з прав людини та потребує проведення правової експертизи Міністерством юстиції України.
8. Ризики та обмеження
У проекті акта відсутні положення, які порушують принцип забезпечення рівних прав та можливостей жінок і чоловіків та положення, що мають ознаки дискримінації.
Проект акта не містить норм, що зачіпають права і свободи, гарантовані Конвенцією про захист прав і основоположних свобод.
У проекті акта відсутні правила і процедури, які можуть містити ризики вчинення корупційних правопорушень та правопорушень, пов’язаних з корупцією.
9. Підстава розроблення проекту акта
Відповідно до Положення про Державну службу України з лікарських засобів та контролю за наркотиками, затвердженого постановою Кабінету Міністрів України від 12 серпня 2015 року № 647, Державна служба України з лікарських засобів та контролю за наркотиками (далі — Держлікслужба) є центральним органом виконавчої влади, діяльність якого спрямовується і координується Кабінетом Міністрів України через Міністра охорони здоров’я, який реалізує державну політику у сферах контролю якості та безпеки лікарських засобів, у тому числі медичних імунобіологічних препаратів, медичної техніки і виробів медичного призначення, та обігу наркотичних засобів, психотропних речовин і прекурсорів, протидії їх незаконному обігу.
Згідно зі статтею 14 Закону України «Про лікарські засоби» контроль за якістю лікарських засобів та умовами їх виробництва здійснюється центральним органом виконавчої влади, що реалізує державну політику у сфері контролю якості та безпеки лікарських засобів.
Відповідно до статті 15 Закону України «Про лікарські засоби» посадові особи центрального органу виконавчої влади, що реалізує державну політику у сфері контролю якості та безпеки лікарських засобів в межах компетенції, визначеної законодавством, мають право, зокрема перевіряти додержання вимог законодавства щодо якості лікарських засобів, правил здійснення належних практик (виробничої, дистриб’юторської, зберігання, аптечної) на всіх етапах обігу, а також під час їх виробництва, зберігання, транспортування та реалізації суб’єктами господарської діяльності, утилізації та знищення.
ПРОГНОЗ ВПЛИВУ
реалізації проекту наказу Міністерства охорони здоров’я України
«Про внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики»
1. Суть проекту акта: посилення контролю за якістю лікарських засобів; недопущення виробництва лікарських засобів невідповідної якості; створення умов, що гарантують надходження на національний ринок України тільки якісних, ефективних та безпечних лікарських засобів, які вироблені відповідно до вимог міжнародного стандарту з належної виробничої практики, що є запорукою захисту здоров’я і життя громадян та безпеки держави в цілому.
2. Вплив на ключові інтереси усіх заінтересованих сторін
| Заінтересована сторона | Ключовий інтерес | Очікуваний (позитивний чи негативний) вплив на ключовий інтерес із зазначенням передбачуваної динаміки змін основних показників (у числовому або якісному вимірі) | Пояснення (чому саме реалізація акта призведе до очікуваного впливу | |
|---|---|---|---|---|
| короткостроковий вплив (до року) | середньостроковий вплив (більше року) | |||
| Суб’єкти господарювання | Удосконалення процедури отримання Сертифіката та Висновку щодо підтвердження відповідності умов виробництва лікарського засобу вимогам належної виробничої практики | – | + | Проект акта надасть можливість суб’єктам господарської діяльності з виробництва лікарських засобів отримувати адміністративні послуги на належному рівні. |
| Громадяни | Забезпечення права на отримання якісних, ефективних та безпечних лікарських засобів | – | + | Проект акта надасть можливість отримувати тільки якісні, ефективні та безпечні лікарські засоби, що вироблені відповідно до вимог стандартів, і є запорукою захисту здоров’я і життя громадян |
Проект оприлюднено
на сайті МОЗ України
04.11.2019 р.
МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ
НАКАЗ
Про внесення змін до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики
Відповідно до статті 15 Закону України «Про лікарські засоби», пункту 5 Порядку здійснення державного контролю якості лікарських засобів, що ввозяться в Україну, затвердженого постановою Кабінету Міністрів України від 14 вересня 2005 року № 902,
НАКАЗУЮ:
1. Внести зміни до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики, затвердженого наказом Міністерства охорони здоров’я України від 27 грудня 2012 року № 1130, зареєстрованого у Міністерстві юстиції України 21 січня 2013 року за № 133/22665 (у редакції наказу Міністерства охорони здоров’я України від 22 липня 2015 року № 452), виклавши його в новій редакції, що додається.
2. Фармацевтичному директорату (Комаріда О.О.) в установленому порядку забезпечити подання цього наказу на державну реєстрацію до Міністерства юстиції України.
3. Цей наказ набирає чинності з дня його офіційного опублікування.
4. Контроль за виконанням цього наказу залишаю за собою.
ЗАТВЕРДЖЕНО
наказом Міністерства охорони здоров’я України
______ № ___
ПОРЯДОК
проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики
І. Загальні положення
1. Цей Порядок розроблено відповідно до Закону України «Про лікарські засоби», з урахуванням вимог Директиви 2001/83/ЄС Європейського Парламенту та Ради від 06 листопада 2001 року щодо Кодексу Співтовариства стосовно лікарських засобів, призначених для застосування людиною, Директиви Комісії 2003/94/ЄС від 08 листопада 2003 року, яка встановлює принципи і правила належної виробничої практики щодо лікарських засобів, призначених для застосування людиною, та досліджуваних лікарських засобів, призначених для застосування людиною, Збірника процедур Співтовариства з питань інспекцій та обміну інформацією (EMA/572454/2014, 17 редакція) та рекомендацій документів міжнародної Системи співробітництва фармацевтичних інспекцій (PIC/S) РІ 002-3 Вимоги до системи якості фармацевтичних інспекторатів, PI 040-1 Настанови PIC/S з класифікації невідповідностей GMP.
2. У цьому Порядку терміни вживаються у таких значеннях:
активний фармацевтичний інгредієнт (лікарська речовина, діюча речовина, субстанція) (далі — АФІ або діюча речовина) — будь-яка речовина чи суміш речовин, що призначена для використання у виробництві лікарського засобу і під час цього використання стає його активним інгредієнтом. Такі речовини мають фармакологічну чи іншу безпосередню дію на організм людини, у складі готових форм лікарських засобів їх застосовують для лікування, діагностики чи профілактики захворювання, для зміни стану, структур або фізіологічних функцій організму, для догляду, обробки та полегшення симптомів;
атестована лабораторія — лабораторія з контролю якості та безпеки лікарських засобів, яка пройшла галузеву атестацію Держлікслужбою;
виробник лікарських засобів — суб’єкт господарювання, який здійснює хоча б один із етапів виробництва лікарських засобів та має ліцензію (дозвіл) на виробництво лікарських засобів (якщо останнє передбачено національним законодавством країни, на території якої знаходяться виробничі потужності виробника);
виробництво лікарських засобів — діяльність, пов’язана із серійним випуском лікарських засобів, яка включає всі або хоча б одну з операцій щодо технологічного процесу, контролю якості, видачі дозволу на випуск (сертифікації) серії, а також закупівлі матеріалів і продукції, зберігання, оптової торгівлі (дистрибуції) лікарських засобів власного виробництва;
висновок щодо підтвердження відповідності умов виробництва лікарського засобу вимогам належної виробничої практики (далі — Висновок) — документ, виданий Держлікслужбою, який засвідчує, що за результатами проведеної спеціалізованої експертизи поданих документів офіційний документ щодо відповідності виробництва лікарських засобів вимогам GMP, виданий уповноваженим органом країни — члена ЄС або країни, що має угоду про взаємне визнання з ЄС або з Україною, вважається таким, що підтверджує відповідність умов виробництва лікарських засобів чинним в Україні вимогам належної виробничої практики;
Заявник — суб’єкт господарювання — власник реєстраційного посвідчення (торгової ліцензії) та/або ліцензії на виробництво лікарських засобів, який подає до Держлікслужби особисто або через уповноважену особу (представника), що знаходиться в Україні, заяву на видачу сертифіката відповідності умов виробництва лікарських засобів вимогам належної виробничої практики або заяву на видачу висновку щодо підтвердження відповідності умов виробництва лікарського засобу вимогам належної виробничої практики (далі — Заява) та який відповідає за достовірність наданої інформації та документів;
зразок (проба) від серії — частина від серії, відібрана таким чином і в такій
кількості, що є репрезентативною для всієї серії;
інспектор — посадова особа Держлікслужби та/або фахівець, який залучається нею, має вищу освіту за однією з таких спеціальностей: фармація, технологія фармацевтичних препаратів, хімія, хімічна технологія, біологія, біотехнологія, має досвід роботи у виробництві лікарських засобів, контролі якості, управлінні (забезпеченні) якістю або створенні лікарських засобів та має підтвердження компетентності з питань належної виробничої практики;
інспектування — процедура оцінки відповідності фармацевтичної системи якості суб’єкта господарювання та фактичного стану наявних умов виробництва лікарських засобів та умов контролю якості чинним в Україні вимогам належної виробничої практики за місцем провадження діяльності (місцезнаходженням виробничих потужностей, у тому числі зон контролю якості та зон зберігання за контрактом (договором));
критичне порушення — порушення, що спричиняє або веде до високої ймовірності виробництва лікарського засобу, який шкідливий для людини або або тварини, або такого, що може призвести до появи шкідливих залишків у харчовій продукції тваринного походження;
лабораторний аналіз — аналіз зразків лікарських засобів на відповідність показників якості лікарських засобів вимогам специфікації якості методів контролю якості лікарських засобів або загальним вимогам до лікарських засобів, встановленим Державною фармакопеєю України, в атестованих лабораторіях;
лікарський засіб критичного рівня ризику — лікарський засіб, для якого виконується будь-яка з таких умов: вузький терапевтичний індекс; висока токсичність; стерильний продукт; біологічний лікарський препарат або складний виробничий процес, проте лікарські засоби низького рівня ризику не можуть розглядатися як критичні, навіть якщо для їх виробництва застосовуються складні виробничі процеси;
лікарський засіб високого рівня ризику — лікарський засіб, що може становити ризик для здоров’я людини навіть при малих кількостях внаслідок перехресної контамінації продуктами, які включають, але не обмежуються пеніцилінами, певними цитотоксинами та біологічними лікарськими засобами; лікарські засоби низького рівня ризику — лікарські засоби, такі як: місцевого застосування для терапії проти акне; лікарські засоби проти лупи; антисептичні очищувачі шкіри; протигрибкові лікарські засоби для ніг; продукція по догляду за шкірою, яка містить лікарські засоби; засоби захисту від сонячних опіків; льодяники від болю в горлі або кашлю, лікарські засоби, аналогічні їм, а також які не відносяться до стерильних або рецептурних лікарських засобів;
належна виробнича практика (Good Manufacturing Practice, GMP) — частина управління якістю, яка гарантує, що лікарські засоби постійно виробляються і контролюються відповідно до стандартів якості, які відповідають їх призначенню, а також відповідно до вимог реєстраційного досьє, досьє досліджуваного лікарського засобу для клінічних випробувань або специфікації на цю продукцію;
несуттєве порушення — порушення вимог GMP, яке не належить до критичних або суттєвих порушень (така класифікація присвоюється, якщо невідповідність оцінена як така або якщо не вистачає інформації для оцінки невідповідності як критичної чи суттєвої);
офіційний документ щодо відповідності умов виробництва лікарських засобів вимогам GMP — сертифікат відповідності умов виробництва лікарських засобів вимогам GMP, виданий уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною (відомості з офіційного електронного реєстру — для FDA США), або ліцензія на виробництво лікарських засобів (якщо уповноваженим органом країни — члена ЄС або країни, що має угоду про взаємне визнання з ЄС або з Україною не передбачена видача сертифіката відповідності умов виробництва лікарських засобів вимогам GMP);
підтвердження відповідності умов виробництва лікарських засобів вимогам GMP — процедура підтвердження Держлікслужбою відповідності умов виробництва лікарських засобів вимогам GMP шляхом видачі сертифіката відповідності умов виробництва лікарських засобів вимогам GMP або висновку щодо підтвердження відповідності виробництва лікарського засобу вимогам GMP;
представник Заявника (уповноважена особа, що знаходиться в Україні та виступає від імені Заявника) — юридична або фізична особа, яка діє на підставі відповідного доручення (довіреності), у якому Заявником надано право представляти його інтереси в Україні при проведенні процедур підтвердження відповідності умов виробництва лікарських засобів вимогам GMP;
прекваліфікація лікарського засобу — стандартизована процедура/програма ВООЗ, яка проводиться з метою оцінки якості, безпечності та ефективності лікарського засобу;
препарат обмеженого застосування (препарат-сирота) — лікарський засіб, що призначений для діагностики, профілактики чи лікування рідкісного захворювання, тобто захворювання, що загрожує життю чи призводить до втрати працездатності (зазвичай не більше 5 осіб з кожних 10 000 жителів на дату подання заяви про державну реєстрацію);
продукція «in bulk» — будь-який лікарський засіб, призначений для виробництва готового лікарського засобу, який пройшов усі стадії технологічного процесу, крім стадії фасування та/або кінцевого пакування і маркування;
сертифікат відповідності умов виробництва лікарських засобів вимогам GMP (далі — Сертифікат) — документ, виданий Держлікслужбою за результатами інспектування, який засвідчує відповідність умов виробництва лікарських засобів чинним в Україні вимогам GMP;
складний виробничий процес — процес, для якого навіть незначні відхилення в контрольних параметрах можуть призвести до отримання неоднорідного продукту або продукту, який не відповідає вимогам специфікації (наприклад, процеси гомогенізації або грануляції для твердих лікарських форм з низькою дозою, препарати пролонгованої або відкладеної дії, стерильні продукти);
спеціалізована експертиза — експертиза поданих документів на відповідність вимогам законодавства, у тому числі вимогам цього Порядку, вимогам GMP, аналіз їх повноти і достовірності наданої інформації, а також перевірка комплектності документів;
суттєве порушення — порушення, яке не є критичним, але призвело або може призвести до виробництва лікарського засобу, який не відповідає вимогам реєстраційного досьє на цей лікарський засіб, або при виробництві лікарського засобу не додержуються вимоги належної виробничої практики, гармонізованої із законодавством ЄС, або полягає в невідповідній процедурі випуску серій чи неналежному виконанні уповноваженою особою суб’єкта господарювання своїх обов’язків, або є комбінацією декількох несуттєвих порушень, кожне з яких власне не може класифікуватися як суттєве порушення, але разом вони мають бути класифіковані та відображені у звіті як суттєве порушення;
угода про взаємне визнання — угода про взаємне визнання результатів інспектування на відповідність умов виробництва лікарських засобів вимогам GMP;
фармацевтична система якості — система управління, що спрямовує та контролює діяльність суб’єкта господарювання щодо якості.
3. Підтвердження відповідності умов виробництва лікарських засобів чинним в Україні вимогам GMP запроваджено з метою доведення, що лікарські засоби постійно виробляються і контролюються згідно зі стандартами якості, які відповідають їх призначенню, а також відповідно до вимог реєстраційного досьє, досьє досліджуваного лікарського засобу для клінічних випробувань або специфікації на цю продукцію. Підтвердження відповідності умов виробництва лікарських засобів, у тому числі активних фармацевтичних інгредієнтів (субстанцій), вимогам GMP здійснюється на добровільних засадах за бажанням заявника.
4. Підтвердження відповідності умов виробництва лікарських засобів вимогам GMP для резидентів та нерезидентів здійснюється Держлікслужбою шляхом проведення експертизи документів, поданих відповідно до цього Порядку, та інспектування виробництва у випадках, передбачених цим Порядком.
5. Відповідно до цього Порядку проводиться підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики на виробничих дільницях згідно із відповідною заявою:
на видачу сертифіката відповідності умов виробництва лікарських засобів вимогам належної виробничої практики згідно з додатком 1 до цього Порядку (далі — Заява на видачу Сертифіката);
на видачу висновку щодо підтвердження відповідності умов виробництва лікарських засобів вимогам GMP згідно з додатком 2 до цього Порядку (далі — Заява на видачу Висновку).
6. Згідно з цим Порядком Держлікслужба видає документи, що підтверджують відповідність умов виробництва лікарських засобів вимогам GMP:
сертифікат відповідності умов виробництва лікарських засобів вимогам належної виробничої практики за формою згідно з додатком 3 до цього Порядку;
висновок щодо підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики за формою згідно з додатком 4 до цього Порядку.
ІІ. Етапи підтвердження відповідності умов виробництва лікарських засобів вимогам GMP
1. Процедура підтвердження відповідності умов виробництва лікарських засобів вимогам GMP включає такі етапи:
подання до Держлікслужби Заяви на видачу Сертифіката або Заяви на видачу Висновку та комплекту документів, передбаченого пунктами 2, 3, 8 цього розділу;
перевірку та опрацювання поданої Заяви на видачу Сертифіката або Заяви на видачу Висновку та комплекту документів (спеціалізована експертиза);
інспектування виробництва лікарських засобів за місцем провадження діяльності, в тому числі лабораторій, що здійснюють контроль якості за контрактом (договором), складських зон за контрактом (договором), у випадках, передбачених цим Порядком;
прийняття рішення щодо видачі документа про відповідність умов виробництва лікарських засобів вимогам GMP або вмотивованого висновку про відмову у його видачі;
внесення змін до виданого документа про відповідність виробництва лікарських засобів вимогам GMP у випадках, передбачених пунктом 12 розділу V цього Порядку.
2. Для підтвердження відповідності умов виробництва лікарських засобів вимогам GMP Заявник (представник Заявника) подає до Держлікслужби Заяву на видачу Сертифіката або Заяву на видачу Висновку, до яких додаються такі документи:
1) копія заяви про державну реєстрацію лікарського засобу, що була подана до МОЗ України, засвідчена підписом і печаткою (за наявності) Заявника або його представника, — для лікарських засобів, що подаються на державну реєстрацію, або при внесенні відповідних змін, що стосуються зміни виробника або його виробничих потужностей, або внесення додаткового виробника до реєстраційних матеріалів зареєстрованих в Україні лікарських засобів, або зміни назви лікарського засобу, або зміни Заявника;
2) засвідчена в установленому порядку копія офіційного документа, який дає право на здійснення господарської діяльності з виробництва лікарських засобів та виданий відповідним державним органом країни, де розташоване виробництво, на виробничу дільницю, що вказана в Заяві на видачу Сертифіката (Заяві на видачу Висновку), та засвідчений в установленому порядку переклад українською та англійською мовами (для нерезидентів). Якщо згідно із законодавством країни-виробника ліцензія на виробництво існує лише в електронному вигляді, мають бути надані роздруківка із посиланням на відповідний офіційний сайт, засвідчена підписом та/або (за наявності) печаткою Заявника (представника Заявника), та переклад українською мовою.
Якщо внесено до бази даних Eudra GMP або електронного реєстру FDA США, Заявник (представник Заявника) надає засвідчену Заявником/представником Заявника роздруківку з бази даних Eudra GMP або електронного реєстру FDA США із засвідченим перекладом на українську мову;
3) засвідчена в установленому порядку копія офіційного документа, виданого державним органом країни, де розташовано виробництво, про відповідність вимогам GMP виробничої дільниці, що вказана в Заяві (за наявності); засвідчені в установленому порядку копії додатків до цього документа з переліком лікарських засобів (за наявності) та засвідчені в установленому порядку переклади цих документів українською та англійською мовами.
Якщо внесено до бази даних Eudra GMP або електронного реєстру FDA США, Заявник (представник Заявника) надає засвідчену Заявником/представником Заявника роздруківку з бази даних Eudra GMP або електронного реєстру FDA США із засвідченим перекладом на українську мову;
4) засвідчена в установленому порядку копія офіційного документа щодо відповідності умов виробництва лікарських засобів вимогам GMP, виданого уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною.
Якщо внесено до бази даних EudraGMP або електронного реєстру FDA США, Заявник (представник Заявника) надає роздруківку з цієї бази даних та переклад українською мовою, у тому числі із додатками (за наявності). Ці документи повинні бути засвідчені Заявником (представником Заявника);
5) копія досьє виробничої дільниці (Site Master File) та його переклад на українську та/або англійську мову, засвідчені Заявником (представником Заявника) (для резидентів та нерезидентів, за винятком виробників, виробничі потужності яких знаходяться на території країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною);
6) засвідчена Заявником (представником Заявника) копія звіту, складеного за результатами останньої перевірки, проведеної уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною (для нерезидентів, за винятком виробників, виробничі потужності яких знаходяться на території країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або Україною). При цьому перевірка повинна бути проведена не раніше ніж за 3 роки до подання Заяви;
7) інформація щодо останньої перевірки, проведеної за процедурою прекваліфікації ВООЗ (за наявності). При цьому перевірка повинна бути проведена не раніше ніж за 3 роки до подання Заяви на видачу Сертифіката або Висновку (якщо немає інших поданих Заявником або представником Заявника відповідним чином оформлених копій офіційних документів ВООЗ);
8) довідка про якість продукції, що виробляється, згідно з додатком 5 до цього Порядку;
9) довідка про результати перевірок виробничої дільниці, проведених органами державного контролю, за формою згідно з додатком 6 до цього Порядку, що зазначена в Заяві на видачу Сертифіката (Заяві на видачу Висновку);
10) Загальний перелік номенклатури продукції за формою згідно з додатком 7 до цього Порядку (за формою 1 — у разі подання Заяви на видачу Висновку; за формою 2 — у разі подання Заяви на видачу Сертифіката), в якому наводиться перелік лікарських засобів, які зареєстровані в Україні та/або знаходяться в процесі реєстрації/внесення змін і виробництво яких вже здійснюється або планується здійснювати на виробничій дільниці, що вказана в Заяві на видачу Висновку (Заяві на видачу Сертифіката). Такий перелік подається в письмовій формі та на електронному носії інформації (файл Excel). У разі коли Заявником є власник ліцензії на виробництво для виробничої дільниці, зазначеної у Заяві на видачу Висновку (Заяві на видачу Сертифіката), перелік може містити лікарські засоби одного або декількох власників реєстраційних посвідчень.
11) засвідчені Заявником (представником Заявника) копія сертифіката лікарського засобу для міжнародної торгівлі (certificate of a pharmaceutical product), виданого компетентним органом країни виробника лікарського засобу або власника реєстраційного посвідчення (заявника), або іншим регуляторним органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною, на ринку якої розміщено лікарський засіб, із зазначенням назви документа та найменування компетентного органу, що його видав, дати видачі (для незареєстрованих лікарських засобів, які знаходяться в процесі реєстрації/внесенні змін в Україні), назви та адрес виробничих дільниць;
12) гарантійний лист згідно з додатком 8;
13) засвідчені Заявником (представником Заявника) копії реєстраційних посвідчень (marketing authorization), виданих компетентним органом країни ЄС, уповноважений орган якої видав офіційний документ щодо відповідності умов виробництва лікарських засобів вимогам GMP, або виданих компетентними органами інших країн — членів ЄС або країн, які мають угоду про взаємне визнання з ЄС або з Україною (подається для лікарських засобів, виробництво хоча б однієї стадії яких здійснюється не в країні — члені ЄС або в країні, яка не має угоди про взаємне визнання з ЄС або з Україною, включаючи контрактні виробничі дільниці).
У випадку, якщо будь-які документи із зазначеного вище переліку, які подано до Держлікслужби з метою отримання Сертифіката або Висновку, містять конфіденційну інформацію, яку виробник не може розкрити Заявнику (представнику Заявника) в Україні, виробник може надіслати такі документи безпосередньо до Держлікслужби.
3. Для виробників-резидентів підтвердження відповідності умов виробництва лікарських засобів вимогам GMP може здійснюватися Держлікслужбою за результатами планової перевірки додержання відповідних ліцензійних умов. За відсутності критичних порушень та/або численних (не менше 6 у сукупності, по конкретних лікарських засобах або процесам) суттєвих порушень відповідності вимогам GMP виробничих дільниць або окремих лікарських засобів в акті планової перевірки додержання ліцензійних умов Держлікслужба відповідно до розділу V цього Порядку видає ліцензіату Сертифікат протягом 30 робочих днів з дня отримання від нього листа щодо видачі Сертифіката із загальним переліком номенклатури продукції у паперовому вигляді та на електронному носії. Лист подається ліцензіатом до Держлікслужби протягом 15 робочих днів після проведення планової перевірки додержання ліцензійних умов.
У разі наявності критичних порушень та/або численних (не менше 6 у сукупності, по конкретних лікарських засобах або процесам) суттєвих порушень відповідності вимогам GMP виробничих дільниць або окремих лікарських засобів, виявлених під час останньої планової перевірки додержання ліцензійних умов, Держлікслужба приймає рішення про відмову у видачі Сертифіката згідно з розділом V цього Порядку. За наявності документальних підтверджень про усунення критичних порушень та/або численних (не менше 6 у сукупності, по конкретних лікарських засобах або процесам) сут євих порушень відповідності вимогам GMP виробничих дільниць або окремих лікарських засобів, виявлених під час останньої планової перевірки додержання ліцензійних умов, проводиться інспектування за місцем провадження виробництва (виробничої дільниці) на підставі поданої ліцензіатом Заяви на видачу Сертифіката.
4. Заява та документи, що додаються до неї, приймаються за описом документів згідно з додатком 9 до цього Порядку, копія якого видається Заявнику (представнику Заявника) з відміткою про дату прийняття документів Держлікслужбою та підписом відповідальної особи.
5. Відповідна заява та комплект документів для підтвердження відповідності умов виробництва лікарських засобів вимогам GMP, листи Заявників та/або представників Заявників можуть подаватися в електронному вигляді за наявності запровадженого електронного порталу Держлікслужби для подання таких документів.
6. Для проведення лабораторного аналізу якості зразків лікарських засобів, експертиз, інспектування, виконання окремих видів робіт тощо Держлікслужбою можуть залучатися підприємства, установи, організації, окремі вчені та фахівці (за їх згодою).
7. Для нерезидентів, які не мають офіційних документів щодо відповідності умов виробництва лікарських засобів вимогам GMP, виданих уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною, додатково до проведення експертизи документів здійснюється інспектування виробництва (виробничої дільниці) відповідно до розділу ІV цього Порядку.
8. При проведенні підтвердження відповідності умов виробництва лікарських засобів вимогам GMP мають бути зазначені усі проміжні виробничі дільниці (включно з контрактними дільницями), що задіяні у процесі виробництва готового лікарського засобу. Всі стадії виробництва лікарських засобів (нерозфасована продукція, первинне та вторинне пакування, сертифікація серії) повинні відповідати вимогам GMP та мати відповідні Сертифікати, видані згідно з цим Порядком, крім випадків коли в комплекті документів надані:
засвідчені підписом та печаткою (за наявності) Заявника або представника Заявника копії документів та їх переклади українською мовою, зазначені у підпунктах 2 та 4 пункту 2 цього розділу, — для проміжних виробничих дільниць, у тому числі контрактних, що задіяні у виробництві лікарського засобу та розташовані на території країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною;
засвідчені підписом та печаткою (за наявності) Заявника або представника Заявника копії документів та їх переклади українською мовою, зазначені у підпунктах 2, 4, 6, 13 пункту 2 цього розділу, — для проміжних виробничих дільниць, включно із контрактними виробничими дільницями, що задіяні у виробництві лікарського засобу та розташовані поза територією країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною.
При цьому видача Держлікслужбою окремих Висновків для проміжних виробничих дільниць (включаючи контрактні виробничі дільниці), що мають офіційні документи щодо відповідності умов виробництва лікарських засобів вимогам GMP, видані уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною, не здійснюється.
ІІІ. Строки підтвердження відповідності умов виробництва лікарських засобів вимогам GMP
1. Спеціалізована експертиза, до якої можуть залучатися фахівці або уповноважені Держлікслужбою організації, проводиться з метою складання вмотивованого висновку щодо підтвердження відповідності умов виробництва лікарських засобів вимогам GMP.
Спеціалізована експертиза за умови надання офіційного документа щодо відповідності умов виробництва лікарських засобів вимогам GMP, виданого уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною, проводиться не більше, ніж 15 робочих днів з дати реєстрації заяви на видачу Сертифікату або Висновку, крім випадків:
1) для лікарських засобів, призначених для профілактики, діагностики та лікування туберкульозу, ВІЛ/СНІД, вірусних гепатитів, онкологічних та рідкісних (орфанних) захворювань, а також для лікарських засобів, які реєструються в Україні за спрощеною процедурою реєстрації згідно Закону України «Про лікарські засоби», — не більше ніж 5 робочих днів після реєстрації Заяви на видачу Сертифіката (Заяви на видачу Висновку), у разі зазначення цієї інформації у поданій Заяві на видачу Сертифіката (Заяві на видачу Висновку);
2) для лікарських засобів, що зареєстровані за централізованою процедурою Європейським агентством з медичних продуктів (EMA), та для лікарських засобів, що пройшли процедуру прекваліфікації ВООЗ та включені до переліку ВООЗ прекваліфікованих лікарських засобів, — не більше ніж 5 робочих днів після реєстрації Заяви на видачу Сертифіката (Заяви на видачу Висновку), у разі зазначення цієї інформації у поданій Заяві на видачу Сертифіката (Заяві на видачу Висновку).
В усіх інших випадках спеціалізована експертиза проводиться не більше, ніж 20 робочих днів.
Для лікарських засобів, призначених для профілактики, діагностики та лікування туберкульозу, ВІЛ/СНІД, вірусних гепатитів, онкологічних та рідкісних (орфанних) захворювань, для лікарських засобів, що пройшли процедуру прекваліфікації ВООЗ та включені до переліку ВООЗ прекваліфікованих лікарських засобів, а також для лікарських засобів, які реєструються в Україні за спрощеною процедурою реєстрації згідно Закону України «Про лікарські засоби» у разі зазначення цієї інформації у відповідній Заяві спеціалізована експертиза здійснюється позачергово.
2. Під час проведення спеціалізованої експертизи береться до уваги інформація щодо результатів проведення інспектувань з боку Держлікслужби даної виробничої дільниці за попередні роки, крім випадків, якщо ця інформація вже була предметом перевірки з боку Держлікслужби, а також наявність документальних підтверджень усунення критичних, суттєвих та несуттєвих порушень, встановлених Держлікслужбою під час цих інспектувань та/або планової та/або позапланової перевірок додержання ліцензійних умов (для резидентів), інформація щодо вилучення з обігу в установленому порядку серій лікарського засобу, які вироблялись на даній дільниці, що пов’язані з якістю лікарського засобу.
За необхідності під час проведення експертизи Держлікслужбою може бути направлений відповідний запит до компетентного органу країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною для з’ясування питань, що виникли під час проведення експертизи, про що повідомляється Заявник (представник Заявника) протягом 10 робочих днів від дати направлення запиту. Час, потрібний для отримання відповіді на запит, не включається до строків проведення спеціалізованої експертизи.
3. У разі некомплектності документів, та/або невідповідностей, виявлених під час проведення спеціалізованої експертизи, вимогам цього Порядку доданих до Заяви на видачу Сертифіката (Заяви на видачу Висновку) документів Заявнику (представнику Заявника) надається про це письмове повідомлення. Час, необхідний для усунення Заявником (представником Заявника) виявлених порушень та/або для надання необхідних документів, не включається до строків проведення спеціалізованої експертизи.
4. Якщо Заявник (представник Заявника) протягом 30 робочих днів після отримання повідомлення не усуває виявлені порушення або не надає необхідного комплекту документів відповідно до вимог цього Порядку, Заява на видачу Сертифіката (Заява на видачу Висновку) за рішенням, яке оформляється наказом Держлікслужби, залишається без розгляду (із зазначенням причин), про що Держлікслужба повідомляє Заявника (представника Заявника). У разі необхідності продовжити строк надання необхідної інформації Заявник (представник Заявника) повинен надати відповідне обґрунтування причини і необхідного йому додаткового строку.
5. У випадку, якщо умови виробництва лікарських засобів та контролю їх якості були предметом інспектування з боку Держлікслужби за попередні роки, наступне інспектування здійснюється у разі, якщо є документальні підтвердження усунення критичних, суттєвих та несуттєвих порушень, встановлених під час попереднього(іх) інспектування(нь).
6. Під час складання плану та програми інспектування враховується необхідність перевірки усунення критичних, суттєвих та несуттєвих порушень, встановлених під час попереднього(іх) інспектування(нь) з боку Держлікслужби за попередні роки умов виробництва лікарських засобів та контролю їх якості, крім випадків, якщо ця інформація вже була предметом перевірки з боку Держлікслужби.
Копії плану та програми інспектування, які погоджуються Держлікслужбою, надсилаються за місцезнаходженням Заявника або представника Заявника у строк не пізніше ніж за 10 робочих днів до дати початку інспектування, у тому числі у разі повторного інспектування за зверненням Заявника з метою перевірки усунення порушень, виявлених під час попереднього інспектування.
У разі наявності обґрунтованих зауважень з боку Заявника щодо участі в інспектуванні окремих інспекторів ці зауваження повинні бути надіслані до Держлікслужби не пізніше ніж за 5 днів до проведення інспектування з метою їх урахування.
7. Строк проведення інспектування за місцем провадження діяльності має становити не більше 15 робочих днів.
8. Строк складання звіту за результатами інспектування виробництва становить не більше 10 робочих днів з дати закінчення інспектування.
9. Лабораторний аналіз якості зразків лікарських засобів, що були відібрані при проведенні інспектування, проводиться в строк не більше ніж 21 робочий день з дня надходження відібраних зразків до лабораторії з контролю якості та безпеки лікарських засобів або в строк, передбачений відповідними методами контролю якості лікарських засобів виробника. При цьому наявність необхідних стандартних зразків забезпечує Заявник. Час, потрібний Заявнику для забезпечення стандартними зразками, не включається до строків виконання лабораторного аналізу.
Відбір зразків лікарських засобів, що плануються до промислового випуску та знаходяться на стадії державної реєстрації, не здійснюється.
10. Розгляд Держлікслужбою на робочому засіданні звіту, складеного за результатами інспектування, з метою його оцінки та обґрунтованості викладених у ньому порушень здійснюється не пізніше 10 робочих днів від дати його складання. У разі наявності наданого до Держлікслужби письмового звернення з обґрунтованими зауваженнями з боку Заявника та/або виробника щодо проведеного інспектування, час, необхідний для розгляду результатів інспектування, може бути продовжено до закінчення розгляду цих зауважень, що становить не більше 15 робочих днів з дати їх надходження до Держлікслужби, якщо не потребується додатковий час Заявнику/виробнику для надання необхідних письмових відповідей.
11. Письмове повідомлення щодо неможливості видачі Сертифіката або Висновку становлять не більше 10 робочих днів після прийняття відповідного рішення Держлікслужбою. Час, потрібний Заявнику для усунення зауважень за результатами спеціалізованої експертизи, розгляду Держлікслужбою письмового звернення Заявника та/або виробника з обґрунтованими зауваженнями щодо проведеного інспектування (у разі його надходження після розгляду результатів інспектування), не включається до строків оформлення та видачі Сертифіката або Висновку.
12. Проект Сертифіката або Висновку з відповідним переліком лікарських засобів надсилається електронною поштою або факсом Заявнику (представнику Заявника) з метою його погодження. Заявник (представник Заявника) засвідчує підписом проект Сертифіката або Висновку з відповідним переліком лікарських засобів та надає його до Держлікслужби. Час, потрібний Заявнику для погодження проекту Сертифіката або Висновку з відповідним переліком лікарських засобів, не включається до строків оформлення та видачі Сертифіката або Висновку.
13. Оформлення внесення змін до Висновку, Сертифіката або переліку лікарських засобів до Висновку чи Сертифіката або їх переоформлення здійснюються у порядку, визначеному пунктом 11 розділу V цього Порядку, у строк не більше ніж 15 робочих днів. Час, потрібний Заявнику (представнику Заявника) для погодження проекту змін до Сертифіката, Висновку або переліку лікарських засобів, не включається до строків їх оформлення та видачі.
14. У разі наявності запровадженого електронного порталу Держлікслужби інформування з боку Держлікслужби Заявника (представника Заявника) або виробника, надання відповідних відповідей на зауваження з боку Заявника (представника Заявника) або виробника, надання плану та програми інспектування, звіту за результатами інспектування, видача або відмова у видачі Сертифікату або Висновку тощо здійснюється в електронному вигляді.
ІV. Порядок проведення інспектування на відповідність умов виробництва лікарських засобів вимогам GMP
1. Інспектування здійснюється з метою оцінки відповідності умов виробництва лікарських засобів чинним в Україні вимогам GMP.
2. За результатами проведеної спеціалізованої експертизи не пізніше ніж через 5 робочих днів після її проведення Заявнику (представнику Заявника) надсилається повідомлення щодо необхідності проведення інспектування та надання до Держлікслужби інформації про пропоновані з боку Заявника (представника Заявника) строки проведення інспектування.
Для лікарських засобів, призначених для профілактики, діагностики та лікування туберкульозу, ВІЛ/СНІД, вірусних гепатитів, онкологічних та рідкісних (орфанних) захворювань, для лікарських засобів, що пройшли процедуру прекваліфікації ВООЗ та включені до переліку ВООЗ прекваліфікованих лікарських засобів, а також для лікарських засобів, які реєструються в Україні за спрощеною процедурою реєстрації згідно Закону України «Про лікарські засоби» у разі зазначення цієї інформації у відповідній Заяві інспектування здійснюється позачергово.
У разі ненадання Заявником (представником Заявника) відповіді щодо пропозицій до проведення інспектування протягом трьох місяців з дати відправлення такого повідомлення Заявнику (представнику Заявника) Заява на видачу Сертифіката (Заява на видачу Висновку) за рішенням, яке оформлюється наказом Держлікслужби, залишається без розгляду (із зазначенням причин), про що письмово повідомляється Заявник (представник Заявника).
У разі надання Заявником (представником Заявника) без зазначення об’єктивних причин пропозицій щодо проведення інспектування у строки, що перевищують шість місяців з дати відправлення повідомлення Заявнику (представнику Заявника), Заява на видачу Сертифіката (Заява на видачу Висновку) за рішенням, яке оформлюється наказом Держлікслужби, залишається без розгляду, про що письмово повідомляється Заявник (представник Заявника).
При врахуванні Держлікслужбою наданих Заявником (представником Заявника) об’єктивних причин інспектування може бути проведено у строк не більше одного календарного року від дати подання до Держлікслужби Заяви на видачу Сертифіката (Заяви на видачу Висновку). У разі не проведення такого інспектування Заява на видачу Сертифіката (Заява на видачу Висновку) за рішенням, яке оформлюється наказом Держлікслужби, залишається без розгляду, про що письмово повідомляється Заявник (представник Заявника).
3. За необхідності під час спеціалізованої експертизи інспектори мають право ознайомитися із реєстраційними матеріалами на зареєстровані лікарські засоби та/або на лікарські засоби, що подаються на державну реєстрацію або при внесенні відповідних змін до реєстраційних матеріалів на зареєстровані в Україні лікарські засоби, про що повідомляється Заявника (представника Заявника). Спеціалізована експертиза може бути подовжена на час, який необхідний Заявнику (представнику Заявника) з метою надання таких документів, якщо про це надано письмове звернення з боку Заявника (представника Заявника). Реєстраційні матеріали подаються до Держлікслужби з супровідним листом.
У разі ненадання Заявником (представником Заявника) зазначених реєстраційних матеріалів у строки, що перевищують шість місяців з дати відправлення повідомлення Заявнику (представнику Заявника) про необхідність їх подання з метою завершення спеціалізованої експертизи та подальшого інспектування, Заява на видачу Сертифіката (Заява на видачу Висновку) за рішенням, яке оформлюється наказом Держлікслужби, залишається без розгляду, про що письмово повідомляється Заявник (представник Заявника).
4. Інспектування здійснюється у таких випадках:
у разі звернення Заявника (представника Заявника);
для нерезидентів, які не мають офіційних документів щодо відповідності умов виробництва лікарських засобів вимогам GMP, виданих уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною;
у разі відсутності копій реєстраційних посвідчень (marketing authorization), виданих компетентним органом країни ЄС, уповноважений орган якої видав офіційний документ щодо відповідності умов виробництва лікарських засобів вимогам GMP, або виданих компетентними органами інших країн — членів ЄС або країн, які мають угоду про взаємне визнання з ЄС або з Україною (подається для лікарських засобів, виробництво хоча б однієї стадії яких здійснюється не в країні — члені ЄС або в країні, яка не має угоди про взаємне визнання з ЄС або з Україною, включаючи контрактні виробничі дільниці).
у разі неможливості встановлення у звіті, складеного за результатами останньої перевірки, проведеної уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною, інформації щодо лікарських засобів, які були предметом інспектування, в тому числі інформації щодо виробничих потужностей, на яких здійснюється виробництво та яке було предметом інспектування, наявності розбіжностей у звіті та досьє виробничої дільниці (Site Master File) тощо;
у разі відмови у видачі Сертифікату або Висновку за результатами останнього інспектування, проведеного Держлікслужбою згідно цього Порядку, виробничої дільниці, що подається у Заяві на видачу Сертифікату (Заяві на видачу Висновку) з метою підтвердження відповідності умов вимогам GMP;
у разі прийняття рішень щодо вилучення з обігу в установленому порядку серій лікарського засобу, які вироблялись на даній дільниці, що пов’язані з якістю лікарського засобу;
у разі встановлення під час проведення експертизи поданих документів ознак критичних порушень вимог GMP (зокрема, можливість виробництва лікарських засобів критичного рівня ризику та/або лікарських засобів високого ступеня ризику на одних і тих самих виробничих лініях або їх виробництва в умовах, що можуть призвести до перехресної контамінації, або виробництво цих лікарських засобів не відповідає встановленим в Україні вимогам тощо), внаслідок чого виробництво може бути визнано таким, що не відповідає вимогам GMP, або ознак виробництва фальсифікованих лікарських засобів.
5. Для здійснення інспектування, експертиз, виконання окремих робіт у сфері підтвердження відповідності, вивчення окремих питань, які вимагають спеціальних знань, Держлікслужба може залучати уповноважені організації, учених та фахівців (за їх згодою).
6. Інспектування здійснюється відповідно до затверджених плану та програми інспектування, де визначено мету, об’єкти, дати проведення інспектування, призначено інспектора або групу інспекторів. Копії плану та програми інспектування надаються Заявнику або представнику Заявника або надсилаються факсом, що зазначений в Заяві на видачу Сертифіката (Заяві на видачу Висновку), або в сканованому вигляді електронною поштою за місцезнаходженням Заявника (представника Заявника) у строки, визначені розділом ІІІ цього Порядку.
При складанні плану та програми інспектування враховуються інформація щодо результатів проведення інспектувань з боку Держлікслужби, у тому числі при залученні уповноважених організацій, учених та фахівців, даної виробничої дільниці за попередні роки та наявність документальних підтверджень усунення критичних, суттєвих та несуттєвих порушень, встановлених Держлікслужбою під час цих інспектувань. Перевірка усунення порушень повинна бути включена до плану та програми інспектування, крім випадків, якщо ця інформація вже була предметом перевірки з боку Держлікслужби.
План та програма інспектування повинні охоплювати виробничі зони, зони контролю якості, у тому числі при здійсненні контролю якості за контрактом (договором) активних фармацевтичних інгредієнтів, продукції «in-bulk», стадії сертифікації серії лікарських засобів та готових лікарських засобів, складські зони, у тому числі при контрактному зберіганні (за договором) сировини, пакувальних матеріалів, продукції «in-bulk» та готових лікарських засобів тощо.
7. Під час інспектування інспектор має фіксувати факти, що підтверджують відповідність умов виробництва лікарських засобів вимогам GMP, а також виявлені в ході інспектування порушення GMP. Під час присвоєння класифікації виявленому порушенню інспектор повинен користуватися алгоритмом, викладеним у додатку 10 до цього Порядку, де наведені, як приклади, найбільш типові порушення, які виявляються під час інспектування, та їх можлива класифікація, виходячи із оцінки ризиків для пацієнта. Даний перелік не є вичерпним та за необхідності можуть додаватися додаткові порушення.
8. Якщо на виробництві, яке інспектується, випускаються лікарські засоби виключно низького рівня ризику, то виявленим порушенням, враховуючи алгоритм згідно додатку 10 до цього Порядку, зазвичай класифікація критичних порушень не присвоюється. Для таких виробництв до критичних порушень можуть бути віднесені тільки виняткові ситуації, такі як: навмисна підробка, викривлення даних, фальсифікація продукції або даних; широко поширене перехресне забруднення; зараження/інвазія або антисанітарія.
При підвищенні у звіті класифікації порушень, яка є відмінною від алгоритму згідно додатку 10 до цього Порядку, інспектором має бути представлене належне обґрунтування підвищення класифікації.
9. При виявленні в ході інспектування критичних порушень, в тому числі таких, що були зазначені в пункті 8 цього розділу, цей факт негайно доводиться інспектором до відома керівництва виробника, що інспектується. Керівництво Держлікслужби також повинно бути проінформоване про це невідкладно.
У разі виявлення критичних порушень в процесі інспектування для обговорення таких порушень проводиться нарада з представниками виробника у дні, коли такі порушення виявлені. На цій нараді виявлені порушення доводяться до відома уповноваженого представника виробництва та/або керівника виробника, що інспектується. Виробник має право надати, а інспектор зобов’язаний розглянути документи та матеріали, що підтверджують усунення порушень у ході перевірки або спростовують вищевказані порушення.
10. У разі обґрунтованого висновку інспектора та/або експерта та прийнятого відповідного рішення Держлікслужбою щодо відмови у видачі Сертифіката або Висновку Держлікслужбою можуть бути вжиті заходи відповідно до законодавства щодо зупинення обігу таких лікарських засобів до усунення порушень.
11. За результатами інспектування інспектор складає Звіт за результатами інспектування (далі — Звіт). Звіт складається українською мовою у двох примірниках за формою, наведеною в додатку 11 до цього Порядку. Додатково для нерезидентів Звіт та його переклад англійською або російською мовами можуть бути оформлені у строк не пізніше ніж 15 робочих днів з дня складання Звіту українською мовою.
Звіт повинен містити висновок щодо відповідності або невідповідності виробництва (виробничої дільниці (виробничих дільниць), та/або виробничих ліній, та/або виробництва окремих лікарських засобів тощо) вимогам GMP, у тому числі усунення або неусунення порушень відповідності вимогам GMP тощо, та рекомендації інспектора/інспекторів щодо видачі Сертифіката або Висновку або відмови у видачі Сертифіката або Висновку, які розглядаються під час прийняття рішення Держлікслужбою.
У разі ненадання Заявником та/або виробником інспектору/інспекторам всієї необхідної інформації з метою проведення інспектування, незабезпечення можливості проведення огляду виробничих, допоміжних, складських приміщень, обладнання, зон контролю якості тощо, незабезпечення можливості інтерв’ювання представників (персоналу) виробника, перешкоджання інспектору/інспекторам у проведенні інспектування, в разі присутності під час інспектування третіх осіб, що не є офіційними представниками Заявника та/або виробника, будь-яких інших дій, що вплинули на проведення інспектування в об’ємі згідно із затвердженими планом та програмою, інспектор/інспектори повинні про це зазначити у Звіті. Ця інформація повинна бути врахована під час формування у Звіті чіткого висновку щодо відповідності або невідповідності умов виробництва лікарських засобів (виробничої дільниці (виробничих дільниць), та/або виробничих ліній, та/або виробництва окремих лікарських засобів тощо) вимогам GMP.
12. Обговорення та узагальнення виявлених у процесі інспектування порушень та їх класифікація згідно з додатком 10 до цього Порядку здійснюються інспектором/інспекторами на заключній нараді під час інспектування з представниками виробника, а також оголошується загальний висновок інспектора/інспекторів щодо відповідності чи невідповідності виробництва вимогам GMP.
У разі наявності зауважень щодо проведеного інспектування та/або класифікації порушень вони можуть бути викладені Заявником (представником Заявника) та/або виробником у протоколі наради українською, російською або англійською мовами. У цьому випадку протокол наради підлягає обов’язковому розгляду та врахуванню при розгляді Держлікслужбою з метою прийняття рішення про видачу Сертифіката або Висновку або відмову в їх видачі.
13. При виявленні під час інспектування критичних порушень виробництва вимог GMP інспектору/інспекторам виробником негайно повинен бути наданий план коригувальних та запобіжних дій щодо усунення виявлених критичних порушень, який надалі додається до першого примірника звіту за результатами інспектування.
14. У разі усунення виробником порушень під час інспектування інспектору повинні бути надані копії належним чином завірених виробником документальних підтверджень їх усунення, про що повинно бути зазначено у Звіті. При цьому у Звіті викладаються опис порушення, класифікація, опис дій виробника щодо усунення цього порушення, а також зазначається про факт його усунення під час інспектування. У висновках Звіту окремо зазначаються кількість та класифікація порушень, що усунуті під час інспектування.
Надання виробником копії документальних підтверджень усунення порушень повинно враховувати час, необхідний для їх оцінки з боку інспектора, та не перешкоджати проведенню інспекції згідно із затвердженими планами та програмами інспектування.
15. Один примірник Звіту надається Заявнику (представнику Заявника). Другий примірник Звіту та матеріали інспектування зберігаються в Держлікслужбі протягом строку дії Сертифіката або Висновку, але не менше ніж три роки.
16. До другого примірника Звіту додаються:
план і програма інспектування;
протоколи наради за формою згідно з додатком 12 до цього Порядку; аудіо- та відеоматеріали (за наявності);
протокол оцінки ризиків щодо розрахунку строку наступної інспекції виробництва лікарського засобу на відповідність вимогам GMP;
копії належним чином завірених виробником документальних підтверджень усунення під час інспектування порушень (якщо про це зазначено в Звіті) тощо.
17. Якщо в процесі інспектування було відібрано зразки для лабораторного аналізу їх якості, до Звіту додаються:
акт відбору зразків продукції; направлення на випробування;
проект методів контролю якості лікарського засобу, що надається Заявником (представником Заявника);
письмовий висновок атестованої відповідно до Порядку проведення галузевої атестації лабораторій з контролю якості та безпеки лікарських засобів, затвердженого наказом Міністерства охорони здоров’я України від 14 січня 2004 року № 10, зареєстрованого у Міністерстві юстиції України 30 січня 2004 року за № 130/8729, лабораторії про якість серії лікарського засобу (після надання його зразків).
V. Порядок прийняття рішення про видачу або відмову у видачі Сертифіката або Висновку
1. У разі прийняття позитивного рішення за результатами проведеної спеціалізованої експертизи Держлікслужба надає Заявнику Висновок у строки, зазначені у розділі ІІІ цього Порядку.
Висновок Держлікслужби видається на строк дії офіційного документа щодо відповідності умов виробництва лікарських засобів вимогам GMP, виданого уповноваженим органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною.
Строк дії Висновку продовжується Держлікслужбою до шести місяців у разі надання Заявником (представником Заявника) інформації від уповноваженого органу, який видав офіційний документ щодо відповідності умов виробництва лікарських засобів вимогам GMP, про заплановане інспектування виробника на відповідність вимогам GMP або інформації щодо запланованого подовження уповноваженим органом офіційного документа щодо відповідності умов виробництва лікарських засобів вимогам GMP на основі процедури аналізу ризиків, про що Держлікслужба письмово повідомляє Заявнику (представнику Заявника) у строк не більше 20 робочих днів.
За необхідності Держлікслужбою може бути направлений відповідний запит до компетентного органу країни — члена ЄС або компетентного органу інших країн — членів ЄС або країн, які мають угоду про взаємне визнання з ЄС або з Україною, та уповноважений орган якої видав офіційний документ щодо відповідності умов виробництва лікарських засобів вимогам GMP.
2. У разі наявності обґрунтованих зауважень з боку Заявника (представника Заявника) та/або виробника щодо проведеного інспектування та/або класифікації порушень Заявник (представник Заявника) та/або виробник може направити до Держлікслужби відповідне письмове повідомлення не пізніше 5 робочих днів після проведення інспектування, яке для розгляду направляється інспекторам, які проводили інспектування, та розглядається в подальшому на засіданні робочої групи та Держлікслужбою з метою прийняття рішення щодо видачі або неможливості видачі Сертифіката або Висновку, яке приймається в строк до 15 робочих днів від дати надходження такого повідомлення до Держлікслужби.
При розгляді Держлікслужбою обґрунтованих зауважень у робочому засіданні може брати участь Заявник та/або виробник, та/або їх представник, якщо таке бажання зазначене в повідомленні. Дата та час проведення такого засідання повідомляються Держлікслужбою на офіційному сайті не пізніше, ніж за 3 робочих дні до проведення засідання.
За результатами розгляду Звіту на робочих засіданнях Держлікслужбою, враховуючи зауваження з боку Заявника, та/або виробника, та/або їх представника, розгляду додаткових, завірених Заявником, та/або виробником, та/або їх представником документальних підтверджень усунення порушень тощо Звіт може бути повернуто інспектору(ам) на доопрацювання строком не більше ніж 10 робочих днів. Час, потрібний для доопрацювання Звіту, не включається до строків оформлення та видачі Сертифіката або Висновку.
Результати розгляду зауважень повідомляються Заявнику, та/або виробнику, та/або їх представнику листом за зазначеним у Заяві на видачу Сертифіката (Заяві на видачу Висновку) місцезнаходженням не пізніше ніж через 5 робочих днів з дня прийняття рішення Держлікслужбою щодо видачі або відмови у видачі Сертифіката або Висновку. У разі незгоди з результатами зауважень Заявник (представник Заявника) та/або виробник мають право оскаржити такі дії Держлікслужби в порядку, встановленому законодавством України.
3. За результатами розгляду на робочому засіданні Держлікслужби Звіту з врахуванням висновків за результатами інспектування та рекомендацій інспектора/інспекторів, наданих документальних підтверджень усунення порушень, результатів розгляду зауважень з боку Заявника/виробника (за наявності), результатів проведених експертиз тощо Держлікслужба приймає рішення щодо видачі або відмови у видачі Сертифіката або Сертифікатів (у разі необхідності при інспектуванні різних лікарських форм тощо), або Висновку, про що письмово повідомляє Заявника або представника Заявника та видає Звіт.
Видача Сертифікату/Сертифікатів або Висновку здійснюється після надання виробником/Заявником до Держлікслужби відповідно до пункту 11 цього розділу прийнятного плану коригувальних та запобіжних дій щодо усунення виявлених порушень за результатами інспектування виробничих дільниць або окремих лікарських засобів.
Строк наступної інспекції на відповідність виробництва вимогам GMP розраховується відповідно до методології проведення аналізу згідно додатку 15 цього Порядку та зазначається в окремому протоколі, який готується інспектором/інспекторами після проведення інспектування та подається до Держлікслужби разом зі Звітом. Остаточне рішення щодо наступного строку інспектування приймається Держлікслужбою. Строк наступної інспекції письмово повідомляться Заявнику (представнику Заявника) під час надання Звіту.
Строк дії Сертифіката становить три роки від дати проведення інспектування та може бути скороченим за рекомендацією інспекторів згідно протоколу оцінки ризиків щодо розрахунку строку наступної інспекції виробництва лікарського засобу на відповідність вимогам GMP.
Не пізніше ніж за шість місяців до встановленого строку проведення інспектування згідно протоколу оцінки ризиків щодо розрахунку строку наступної інспекції виробництва лікарського засобу на відповідність вимогам GMP Держлікслужбою повідомляється про необхідність проведення інспектування Заявника (представника Заявника).
Заявник (представника Заявника) не пізніше ніж за три місяці до встановленого строку проведення інспектування повинен подати відповідно до цього Порядку Заяву та комплект документів до неї згідно пункту 2 розділу II Порядку.
У разі, якщо строк наступної інспекції визначено через рік та менше після проведення інспектування за три місяці до встановленого строку проведення інспектування Держлікслужбою повідомляється Заявника (представника Заявника) про необхідність проведення інспектування. Заявник (представника Заявника) не пізніше ніж за два місяці до встановленого строку проведення інспектування повинен подати відповідно до цього Порядку офіційний лист разом із Заявою (в додатку до листа) щодо згоди на проведення інспектування (в тому числі задіяних контрактних виробничих дільниць, лабораторій з контролю якості головних лікарських засобів, продукції in-bulk та активних фармацевтичних інгредієнтів та контрактних складів). Комплект документів до Заяви в цьому випадку не подається, водночас, Держлікслужбою можуть вимагатися реєстраційні матеріали на зареєстровані лікарські засоби в Україні, оновлений перелік лікарських засобів, у тому числі на електронному носії, тощо.
У разі неподання відповідної Заяви з метою подальшого планового інспектування або відмови у проведенні інспектування виданий Сертифікат/Сертифікати або Висновок зупиняється за рішенням Держлікслужби.
4. Про видачу Сертифіката або Висновку приймається наказ Держлікслужби. До Сертифіката або Висновку додається перелік лікарських засобів, зареєстрованих або лікарських засобів, що знаходяться в процесі реєстрації/внесенні змін, згідно з додатком 13 до цього Порядку (далі — Перелік лікарських засобів), який є невід’ємною частиною Сертифіката або Висновку.
5. Для резидентів у разі анулювання ліцензії на виробництво лікарських засобів також анулюється Сертифікат.
6. У разі негативного рішення Держлікслужба інформує Заявника про вмотивовану відмову у видачі Сертифіката або Висновку у строки, зазначені у розділі ІІІ цього Порядку.
У разі незгоди з рішенням Держлікслужби щодо відмови у видачі Сертифіката або Висновку Заявник має право оскаржити таку відмову у судовому порядку.
7. Рішення про відмову у видачі Сертифіката або Висновку оформляється наказом Держлікслужби та приймається у разі:
виявлення недостовірних відомостей у Заяві на видачу Сертифіката (Заяві на видачу Висновку) або документах, що додавалися до Заяви на видачу Сертифіката (Заяви на видачу Висновку) відповідно до цього Порядку;
виявлення в ході інспектування навмисної підробки, викривлення даних, фальсифікації продукції або даних;
встановлення критичного/критичних порушення/порушень вимог GMP за результатами інспектування та їх не усунення в ході інспектування;
якщо в ході інспектування встановлені численні (не менше 6 у сукупності, по конкретних препаратах або процесах) суттєві порушення, які свідчать про те, що виробник не контролює належним чином процеси та виробничі операції;
якщо в ході інспектування повторно встановлено, що більшість суттєвих та несуттєвих порушень, які були встановлені в ході попередніх інспектувань, не усунуті, що свідчить про те, що виробник не вживає належних запобіжних та коригувальних дій за результатами попередньої інспекції;
якщо в ході інспектування повторно встановлено, що критичні порушення, які були виявлені в ході попередніх інспектувань, не усунуті, що свідчить про те, що виробник не вживає належних запобіжних та коригувальних дій за результатами попередньої інспекції;
у випадку невиконання Заявником (представником Заявника) зобов’язань, викладених у пункті 3 розділу VIIІ цього Порядку, стосовно надання на запит інспектора всієї необхідної інформації, забезпечення можливості проведення огляду приміщень, обладнання, зон контролю якості, інтерв’ювання представників (персоналу) виробника, ознайомлення з необхідною документацією системи якості тощо, що унеможливлює надати оцінку виробництва лікарських засобів як такого, що відповідає вимогам GMP, про що зазначається у Звіті інспектора;
ненадання виробником/Заявником плану коригувальних та запобіжних дій щодо усунення виявлених порушень за результатами інспектування виробничих дільниць або окремих лікарських засобів та документальних підтверджень щодо усунення порушень у встановлені строки;
неусунення зауважень за результатами проведених експертиз у встановлені строки.
9. За наявності у Звіті критичних та/або численних (не менше 6 у сукупності, по конкретних лікарських засобах або процесам) суттєвих порушень відповідності вимогам GMP виробничих дільниць або окремих лікарських засобів виробник вживає заходів щодо їх усунення, надає до Держлікслужби план коригувальних та запобіжних дій щодо усунення виявлених порушень згідно з додатком 14 до цього Порядку у строк до 30 робочих днів від дати отримання Звіту. При цьому в плані коригувальних та запобіжних дій щодо усунення виявлених порушень повинні бути чітко зазначені заходи щодо поводження з лікарськими засобами, якщо такі вироблялись під час виявлення цих порушень.
Документальні підтвердження усунення порушень надаються до Держлікслужби відповідно до строків усунення порушень, встановлених виробником згідно з планом коригувальних та запобіжних дій щодо усунення виявлених порушень.
10. Після усунення критичних та/або численних (не менше 6 у сукупності, по конкретних лікарських засобах або процесам) суттєвих порушень відповідності вимогам GMP умов виробництва лікарських засобів або окремих лікарських засобів та надання до Держлікслужби документальних підтверджень про усунення Заявник/представник Заявника/виробник може надати до Держлікслужби письмове звернення щодо проведення інспектування з метою перевірки.
У разі підтвердження за результатами інспектування повного усунення цих порушень та у разі відсутності підстав щодо відмови у видачі Сертифіката або Висновку відповідно до пункту 7 розділу V цього Порядку Держлікслужба приймає рішення про видачу Сертифіката згідно з додатком 3 до цього Порядку або Висновку згідно з додатком 4 до цього Порядку.
У разі підтвердження за результатами цього інспектування не усунення порушень та/або за наявності підстав щодо відмови у видачі Сертифіката або Висновку відповідно до пункту 7 розділу V цього Порядку Держлікслужба приймає рішення про відмову у видачі Заявнику Сертифіката або Висновку.
Інспектування з метою перевірки усунення порушень може здійснюватися декілька разів, але у строк не більше одного календарного року від дати подання до Держлікслужби Заяви на видачу Сертифіката (Заяви на видачу Висновку) та при своєчасному наданні до Держлікслужби відповідних планів коригувальних та запобіжних дій щодо усунення виявлених порушень та документальних підтверджень про усунення порушень.
11. За наявності у Звіті суттєвих (менше 6 у сукупності, по конкретних лікарських засобах або процесам) та несуттєвих порушень вимог GMP виробник повинен вжити заходів щодо приведення виробництва у відповідність до вимог GMP, надати на експертизу до Держлікслужби план коригувальних та запобіжних дій щодо усунення виявлених порушень згідно з додатком 14 до цього Порядку у строк до 90 робочих днів від дати отримання Звіту. Експертиза наданих документів здійснюється протягом 15 робочих днів, результати якої повідомляються Заявнику/представнику Заявника/виробнику.
Строки усунення порушень встановлюються виробником, враховуючи рекомендації інспекторів згідно зі Звітом та результатами проведеної експертизи та планом коригувальних і запобіжних дій щодо усунення виявлених порушень, зазначаються в плані коригувальних і запобіжних дій та не можуть перевищувати один рік від дати проведення інспектування.
Документальні підтвердження усунення порушень надаються до Держлікслужби відповідно до строків усунення порушень, встановлених в плані коригувальних та запобіжних дій щодо усунення виявлених порушень.
У разі ненадання виробником/Заявником плану коригувальних та запобіжних дій щодо усунення виявлених порушень у встановлені строки та/або відсутність його позитивної експертизи з боку Держлікслужби, Держлікслужба приймає рішення про зупинення дії Сертифікату/Сертифікатів або Висновку відповідно до пункту 7 розділу V цього Порядку. План коригувальних та запобіжних дій щодо усунення виявлених порушень може подаватися декілька разів, однак у строк, що не перевищує 90 робочих днів від дати отримання Звіту.
У разі ненадання виробником/Заявником документальних підтверджень щодо усунення порушень у встановлені строки Держлікслужба приймає рішення про зупинення дії Сертифіката або Висновку відповідно до пункту 4 розділу VІ цього Порядку.
12. У разі виникнення необхідності у Заявника (представника Заявника) внесення змін та/або переоформлення Висновку чи Сертифіката та/або Переліку лікарських засобів, який додається до Висновку чи Сертифіката (з метою реєстрації лікарських засобів або внесення змін до реєстраційної документації, у тому числі зміни назви лікарського засобу, дозування, найменування виробника тощо, з метою виправлення технічних помилок), Заявник (представник Заявника) подає до Держлікслужби письмове звернення щодо необхідності внесення змін чи переоформлення Висновку чи Сертифіката.
З письмовим зверненням до Держлікслужби надаються: загальний оновлений перелік номенклатури продукції у паперовому вигляді та на електронному носії інформації, засвідчений Заявником (представником Заявника), копія сертифіката лікарського засобу для міжнародної торгівлі (certificate of a pharmaceutical product), виданого компетентним органом країни виробника лікарського засобу або власника реєстраційного посвідчення (заявника), або іншим регуляторним органом країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною, на ринку якої розміщено лікарський засіб, із зазначенням назви документа та найменування компетентного органу, що його видав, дати видачі (для незареєстрованих лікарських засобів, які знаходяться в процесі реєстрації/внесенні змін в Україні), назви та адрес виробничих дільниць (для незареєстрованих лікарських засобів, які плануються до реєстрації в Україні); засвідчені Заявником (представником Заявника) копії реєстраційних посвідчень (marketing authorization), виданих компетентним органом країни ЄС, уповноважений орган якої видав офіційний документ щодо відповідності умов виробництва лікарських засобів вимогам GMP, або виданих компетентними органами інших країн — членів ЄС або країн, які мають угоду про взаємне визнання з ЄС або з Україною (подається для лікарських засобів, виробництво хоча б однієї стадії яких здійснюється не в країні — члені ЄС або в країні, яка не має угоди про взаємне визнання з ЄС або з Україною, включаючи контрактні виробничі дільниці); копії інших документів, що підтверджують достовірність змін, які потребують внесення.
У разі внесення додаткової виробничої дільниці, виробничої операції повинні бути надані відповідні документи згідно пункту 2 розділу ІІ цього Порядку.
При змінах, пов’язаних із зазначенням номерів реєстраційних посвідчень на лікарські засоби, на загальному оновленому переліку номенклатури продукції достатньо підпису та печатки (за наявності) Заявника (представника Заявника).
При проведенні експертизи у разі розширення Переліку лікарських засобів, який додається до Сертифіката або Висновку, беруться до уваги результати інспектування виробничої дільниці, на якій виробляються або плануються до виробництва лікарські засоби, що подаються на розширення, надання до Держлікслужби плану коригувальних та запобіжних дій щодо усунення виявлених порушень та його виконання з боку виробника тощо.
Для лікарських засобів, що пройшли процедуру прекваліфікації ВООЗ, інспектування у разі розширення Переліку лікарських засобів не здійснюється.
У разі відсутності підстав для відмови у переоформленні або внесенні змін до Сертифіката, Висновку або Переліку лікарських засобів, в тому числі враховуючи пункт 7 цього розділу та пункт 3 розділу VI цього Порядку, Держлікслужба протягом 15 робочих днів з дня отримання відповідних документів та їх спеціалізованої експертизи у разі позитивного висновку оформлює проект оновленого Переліку лікарських засобів, у разі переоформлення Сертифіката або Висновку — оформлює проект оновленого Сертифіката або Висновку з відповідним Переліком лікарських засобів. Проекти надсилаються електронною поштою або факсом Заявнику (представнику Заявника) з метою їх погодження. Заявник (представник Заявника) засвідчує підписом проекти документів та надає їх до Держлікслужби. Переоформлений Сертифікат або Висновок видається Заявнику (представнику Заявника).
Відповідні звернення, комплекти документів, листи Заявників та/або представників Заявників можуть подаватися в електронному вигляді за наявності запровадженого електронного порталу Держлікслужби для подання таких документів.
VI. Контроль за дотриманням вимог належної виробничої практики протягом строку дії Сертифіката або Висновку
1. Заявник є відповідальним за дотримання вимог GMP у виробництві лікарських засобів протягом строку дії Сертифіката або Висновку.
2. Виробництво лікарських засобів, яке згідно з цим Порядком отримало підтвердження відповідності вимогам GMP, підлягає позаплановому інспектуванню на дотримання вимог GMP протягом строку дії Сертифіката або Висновку за наявності відповідних підстав, зазначених у пункті 3 цього розділу.
3. Підставами для позапланового інспектування виробництва (або окремих виробничих дільниць) (спрямована інспекція) з метою перевірки дотримання виробником вимог GMP можуть бути:
звернення Заявника/представника Заявника/виробника;
наявність документально підтверджених фактів або ознак виробництва лікарських засобів, які можуть бути небезпечними для здоров’я і життя людей або можуть призвести до тяжких наслідків для здоров’я людей внаслідок можливих порушень технології виробництва та/або методів контролю якості;
розширення номенклатури лікарських засобів, що виробляються на сертифікованому виробництві (у тому числі якщо виробництво цих лікарських засобів створює ризик контамінації або перехресної контамінації);
у разі внесення змін до переліку лікарських засобів після реєстрації в Україні (внесення номерів реєстраційних посвідчень), за умови, що інспектування проводилося Держлікслужбою до реєстрації цих лікарських засобів в Україні та/або реєстраційні матеріали не були доступні інспекторам;
ненадання до Держлікслужби документальних підтверджень усунення порушень відповідно до строків, встановлених виробником згідно з планом коригувальних та запобіжних дій щодо усунення виявлених порушень, та/або їх невиконання в встановлені строки;
наявність приписів про повну заборону обігу та/або рішень органів державного нагляду (контролю) щодо вилучення з обігу лікарських засобів трьох серій (для інфузійних та ін’єкційних лікарських засобів — однієї серії), які вироблялись на даній дільниці, якщо дефект якості лікарського засобу можливо пов’язаний із технологією виробництва та/або методами його контролю;
наявність фактів виробництва неякісних або фальсифікованих лікарських засобів (у тому числі у разі підозри щодо можливого виробництва фальсифікованої продукції);
встановлення факту виробництва лікарських засобів, які можуть бути шкідливими для здоров’я та життя людини, при їх виробництві на одних і тих самих виробничих лініях (наприклад, лікарські засоби з використанням субстанції та/або продукції «іn-bulk», які вважаються високосенсибілізуючими, сильнодіючими або високотоксичними, лікарські засоби, які містять живі клітини, гормони, сульфаніламіди, бета-лактамні антибіотики, такі як пеніциліни, цефалоспорини, пенеми, карбацефеми, монобактами, інші високосенсибілізуючі матеріали, патогенні організми, цитотоксини) або які мають певну небезпеку (наприклад, радіофармацевтичні);
введення в технологічну схему виробництва нової технологічної стадії, технологічної операції або нової одиниці критичного технологічного обладнання;
технічне переоснащення виробництва із заміною одиниць обладнання, суттєва модернізація або реконструкція виробничої дільниці, у тому числі систем забезпечення виробництва технологічними середовищами;
зміни технологічних параметрів критичних технологічних стадій, якщо заявником не надано матеріалів валідаційних досліджень;
перенесення виробничої дільниці на інші площі або в інші будівлі;
невиконання гарантійних зобов’язань щодо повідомлення стосовно змін щодо перенесення виробничої дільниці на інші площі або в інші будівлі, у тому числі для проміжних виробничих дільниць, включно із контрактними виробничими дільницями, що задіяні у виробництві лікарського засобу, тощо.
4. У разі виявлення невідповідності виробництва лікарських засобів вимогам GMP, наявність фактів виробництва неякісних або фальсифікованих лікарських засобів (у тому числі у разі підозри щодо можливого виробництва фальсифікованої продукції), наявність документально підтверджених фактів або ознак виробництва лікарських засобів, які можуть бути небезпечними для здоров’я і життя людей або можуть призвести до тяжких наслідків для здоров’я людей внаслідок можливих порушень технології виробництва та/або методів контролю якості, ненадання Заявником/представником Заявника/виробником плану коригувальних та запобіжних дій щодо усунення виявлених порушень та документальних підтверджень щодо усунення порушень вимог GMP у строки, визначені у розділі V цього Порядку, та/або його невиконання, невиконання гарантійних зобов’язань, встановлення факту виробництва лікарських засобів, які можуть бути шкідливими для здоров’я та життя людини, при виробництві на одних і тих самих виробничих лініях (гормони, цитостатики тощо) та згідно інших підстав пункту 3 цього розділу Держлікслужба приймає рішення про зупинення дії або анулювання Сертифіката або Висновку, про що повідомляє Заявнику (представнику Заявника) протягом 10 робочих днів після прийняття відповідного рішення. Після усунення порушень вимог GMP у встановлені строки Держлікслужба приймає рішення щодо поновлення дії Сертифіката або Висновку (у разі зупинення їх дії).
5. При здійсненні позапланового інспектування з підстав, визначених розділом VI цього Порядку, інспектуванню підлягають лише ті питання, необхідність перевірки яких стала підставою для проведення позапланового інспектування, з обов’язковим зазначенням цих питань у документах на проведення інспектування. Не підлягає перевірці період, який уже був предметом під час перевірки, крім випадків, встановлених за рішенням суду, яке набрало законної сили, або з метою перевірки усунення порушень, що були виявлені під час останнього інспектування. У разі відмови Заявника (представника Заявника) від проведення інспектування за рішенням Держлікслужби, що оформлюється наказом, зупиняється дія Сертифікату або Висновку.
VII. Права та обов’язки інспектора при проведенні процедури підтвердження відповідності вимогам GMP
1. Інспектор при здійсненні інспектування виробництва має право:
ознайомлюватися з усіма необхідними для проведення інспектування документами, які стосуються вимог GMP і матеріалів реєстраційного досьє, та одержувати від Заявника, та/або виробника, та/або їх представника необхідні відомості з питань, що належать до його компетенції;
безперешкодно відповідно до Закону України «Про основні засади державного нагляду (контролю) у сфері господарської діяльності» проводити огляд виробничих, складських, допоміжних приміщень (зон), приміщень (зон) контролю якості та інших приміщень (зон) Заявника з метою їх інспектування для з’ясування питань, зазначених в плані та програмі інспектування;
відповідно до Закону України «Про основні засади державного нагляду (контролю) у сфері господарської діяльності» фіксувати процес здійснення інспектування чи кожну окрему дію засобами аудіо- та відеотехніки, не перешкоджаючи здійсненню такого заходу та не порушуючи комерційну таємницю Заявника, про що Заявник та/або виробник має бути попереджений під час вступної наради;
вимагати припинення дій, які перешкоджають здійсненню процедури підтвердження відповідності умов виробництва лікарських засобів вимогам GMP;
призначати експертизу, одержувати пояснення, довідки, документи, матеріали, відомості з питань, що виникають під час процедури підтвердження відповідності вимогам GMP;
відбирати зразки лікарських засобів для лабораторної перевірки їх якості згідно з чинним законодавством України в разі встановлення порушень технології виробництва, вимог щодо якості, умов та/або правил зберігання лікарських засобів, що можуть призвести до виробництва неякісних лікарських засобів та/або оптової, роздрібної торгівлі неякісними лікарськими засобами;
отримувати належним чином завірені копії необхідних документів (витягів з документів), пов’язаних з інспектуванням виробництва на відповідність вимогам GMP;
одержувати від Заявника письмові пояснення з питань, що виникають під час інспектування.
2. Інспектор повинен дотримуватись вимог щодо:
дотримання конфіденційності щодо інформації про Заявника, та/або виробника, та/або їх представника, у тому числі отриманої від нього під час інспектування;
принципів класифікації виявлених під час інспектування порушень;
забезпечення об’єктивності, повноти та достовірності інформації результатів інспектування, в тому числі інформації, що міститься у Звіті та є підставою для прийняття рішення про видачу Сертифіката або Висновку;
відсутності конфлікту інтересів при проведенні робіт із підтвердження відповідності умов виробництва лікарських засобів вимогам GMP, у тому числі під час інспектування;
нерозголошення конфіденційної інформації та інформації, що є комерційною таємницею Заявника та/або виробника, що стає доступною інспектору у процесі підтвердження відповідності умов виробництва лікарських засобів вимогам GMP;
виконання законодавства України, у тому числі положень цього Порядку, та рекомендацій PIC/S.
3. Інспектор зобов’язаний:
дотримуватися ділової етики у взаємовідносинах із Заявником, та/або виробником, та/або їх представником;
не втручатися і не перешкоджати здійсненню господарської діяльності під час здійснення процедури підтвердження відповідності умов виробництва лікарських засобів вимогам GMP, якщо це не вимагається обставинами, які безпосередньо свідчать про виробництво лікарських засобів з критичними порушеннями;
надати Звіт в Держлікслужбу в строки, передбачені розділом IІІ цього Порядку.
VIII. Права та обов’язки Заявника та/або виробника при проведенні процедури підтвердження відповідності умов виробництва лікарських засобів вимогам GMP
1. Заявник та/або виробник є відповідальними за достовірність наданої інформації.
2. Заявник та/або виробник мають право:
вимагати від інспекторів додержання вимог чинного законодавства України;
не допустити інспектора/інспекторів до інспектування без пред’явлення ним/ними документа, що засвідчує особу;
перевіряти наявність в інспектора документа, що засвідчує особу, і одержувати копії документів на проведення інспектування;
бути присутніми під час здійснення інспектування;
вимагати нерозголошення конфіденційної інформації та інформації, що є комерційною таємницею Заявника та/або виробника;
фіксувати процес здійснення інспектування чи кожну окрему дію засобами аудіо- та відеотехніки, не перешкоджаючи здійсненню такого заходу, про що інспектор має бути попереджений під час вступної наради;
надавати обґрунтовані зауваження щодо проведеного інспектування та за бажанням бути присутнім при розгляді результатів інспектування;
одержувати Звіт, а також інші передбачені цим Порядком документи у строки, встановлені цим Порядком;
звернутися до Держлікслужби з проханням залишити без розгляду Заяву про видачу Сертифіката (Заяву про видачу Висновку) або повідомлення щодо необхідності внесення змін чи переоформлення Переліку лікарських засобів;
у всіх випадках незгоди з результатами проведених експертиз, процесом інспектування та результатами інспектування, включаючи, але не обмежуючись, розгляд зауважень та заперечень Заявника та/або виробника, прийняті рішення щодо видачі Сертифіката або Висновку або відмови у їх видачі, зупинення або анулювання дії Сертифіката або Висновку тощо, оскаржувати їх в установленому законодавством порядку.
3. Заявник та/або виробник зобов’язаний:
повідомляти Держлікслужбу про зміни, які стосуються виробничої дільниці (у тому числі про зміну ліцензії на виробництво лікарських засобів, зміну найменування Заявника, його місцезнаходження, перенесення виробничої дільниці на інші площі тощо);
надавати для ознайомлення на запит Держлікслужби та/або залучених уповноважених організацій, учених та фахівців перед проведення інспектування та під час інспектування реєстраційні матеріали на зареєстровані лікарські засоби та/або на лікарські засоби, що подаються на державну реєстрацію або при внесенні відповідних змін до реєстраційних матеріалів на зареєстровані в Україні лікарські засоби;
надавати на запит інспектора всю необхідну інформацію, забезпечувати можливість проведення огляду виробничих, допоміжних, складських приміщень, обладнання, зон контролю якості, інтерв’ювання представників (персоналу) виробника, ознайомлення з необхідною реєстраційної документацією та документацією системи якості тощо, ознайомлення та огляд яких необхідні для з’ясування викладених у плані та програмі інспектування питань;
виконувати вимоги Держлікслужби щодо усунення виявлених порушень вимог GMP та чинного законодавства України;
виконувати передбачені цим Порядком вимоги Держлікслужби щодо усунення виявлених під час первинної або спеціалізованої експертиз або під час інспектування порушень вимог чинного законодавства України;
надавати копії документів, зразки продукції, пояснення, довідки, відомості, матеріали з питань, що виникають під час процедури підтвердження відповідності вимогам GMP.
Додаток 1
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 5 розділу І)
Державна служба України з лікарських засобів та контролю за наркотиками
ЗАЯВА
на видачу сертифіката відповідності умов виробництва лікарських засобів вимогам належної виробничої практики
Дата надходження: «__» ________ 20__ року
Зареєстровано за № ___
| ЗАЯВНИК (найменування ліцензіата або власника реєстраційного посвідчення (торговельної ліцензії) на лікарський засіб або найменування/прізвище, ім’я, по батькові представника заявника) _______________________________________________________________
Місцезнаходження (місце проживання) _____________________________________________________________________________________________ П. І. Б., посада керівника заявника __________________________________________________________________________________________________ Контактні телефони, факс ___________________________________________________________________________________________________________ |
| ВИРОБНИК (найменування суб’єкта господарювання та найменування виробничої(их) дільниці(ць), заявленої(их) для процедури підтвердження відповідності вимогам GMP (за наявності)) ___________________________________________________________________
Місце провадження діяльності виробника _____________________________________________________________________________________ DUNS номер адреси виробника та GPS координати _________________________________________________________________________ П. І. Б., посада керівника підприємства, тел./факс _________________________________________________________________________________ П. І. Б., посада керівника підрозділу з виробництва, тел./факс ____________________________________________________________________ П. І. Б., посада керівника служби якості (Уповноваженої особи), тел./факс _______________________________________________________ П. І. Б., посада керівника відділу контролю якості, тел./факс ______________________________________________________________________ Інші контактні адреси (у тому числі e-mail) __________________________________________________________________________________________ Місце провадження діяльності, DUNS номери та GPS координати лабораторій, що здійснюють контроль якості за контрактом (договором), складських зон за контрактом (договором) _________________________________________________ Згода на проведення інспектування керівника суб’єкта господарювання, де здійснюється контроль якості за контрактом, зберігання лікарських засобів тощо: П. І. Б., посада керівника, підпис ____________________________________________________________________________________________________ |
Достовірність наданої інформації гарантую:
____________________________________________________________________________________________ (П. І. Б., посада і підпис керівника (представника) заявника)
Дата складання «__» _________ 20__ року
М. П. (за наявності)
Додаток 2
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 5 розділу І)
Державна служба України з лікарських засобів та контролю за наркотиками
ЗАЯВА
на видачу висновку щодо підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики
Дата надходження: «__» _____ 20__ року
Зареєстровано за № ___
| ЗАЯВНИК (найменування ліцензіата або власника реєстраційного посвідчення (торговельної ліцензії) на лікарський засіб або найменування/прізвище, ім’я, по батькові представника заявника) ________________________________________________________________
Місцезнаходження (місце проживання) _____________________________________________________________________________________________ П. І. Б., посада керівника заявника __________________________________________________________________________________________________ Контактні телефони, факс ____________________________________________________________________________________________________________ |
| ВИРОБНИК (найменування суб’єкта господарювання та найменування виробничої(их) дільниці(ць), заявленої(их) для процедури підтвердження відповідності вимогам GMP (за наявності))_____________________________________________________________________
Місце провадження діяльності виробника ______________________________________________________________________________________ П. І. Б., посада керівника підприємства, тел./факс __________________________________________________________________________________ П. І. Б., посада керівника підрозділу з виробництва, тел./факс _____________________________________________________________________ П. І. Б., посада керівника служби якості (уповноваженої особи), тел./факс _________________________________________________________ П. І. Б., посада керівника відділу контролю якості, тел./факс _______________________________________________________________________ Інші контактні адреси (у тому числі e-mail) ___________________________________________________________________________________________ |
Примітки (в тому числі зазначаються підстави для позачергової експертизи та/або інспектування):
___________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Достовірність наданої інформації гарантую:
____________________________________________________________________________________________ (П. І. Б., посада і підпис керівника (представника) заявника)
Дата складання «__» _________ 20__ року
М. П. (за наявності)
Додаток 3
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 6 розділу І)
Державний Герб України
УКРАЇНА
UKRAINE
СЕРТИФІКАТ ВІДПОВІДНОСТІ УМОВ ВИРОБНИЦТВА ЛІКАРСЬКИХ ЗАСОБІВ ВИМОГАМ НАЛЕЖНОЇ ВИРОБНИЧОЇ ПРАКТИКИ
CERTIFICATE OF GMP COMPLIANCE
Сертифікат №://Certificate No:
Строк дії до://Valid till: дд мм рррр
| Частина 1 | Part 1 |
|---|---|
| Державна служба України з лікарських засобів (Держлікслужба України) засвідчує:
Найменування виробника, місцезнаходження: Найменування виробничої(их) дільниці(ць): Місце провадження діяльності: Ліцензія на виробництво лікарських засобів від « __» ______ № __ Місце виробництва систематично проходить інспектування зі встановленою періодичністю на відповідність вимогам GMP згідно зі встановленим порядком. За результатами інспектування цього виробника, останнє з яких було проведене … …/… …/… … (дата), встановлено, що він відповідає вимогам належної виробничої практики, зазначеним в _________________________________________ (нормативний акт), що відповідають вимогам належної практики при виробництві і контролі якості Системи співробітництва фармацевтичних інспекцій (PIC/S), директивам ЄC та рекомендаціям Всесвітньої організації охорони здоров’я відносно продукції, що призначена для торгівлі та дистрибуції в країні походження або для експорту. Цей сертифікат відображає стан виробничої дільниці на момент інспектування, зазначеного вище, і не може використовуватися для підтвердження відповідності, якщо з моменту проведення цього інспектування пройшло більше ніж 3 роки. Цей сертифікат включає частини 1, 2 та додаток. Чинність цього сертифіката може бути підтверджена органом, що його видав. |
State Administration of Ukraine on Medicinal Products (SAUMP) confirms the following:
Manufacturer’ s name, registered place of business: Name(s) of manufacturing site(s): Manufacturing site address: Manufacturing authorization for medicinal products _________ __________ No. ________ Facilities of above mentioned manufacturer are subject to GMP inspections at suitable intervals in accordance with the National certification procedure. From the knowledge gained during inspection of this manufacturer, the latest of which was conducted on… …/… …/… … (date), it is considered that it complies with the Good Manufacturing Practice requirements1 referred to in the ______________________________________ (name of regulation document) which is harmonized with the requirements of Good practices in the manufacture and quality control of the Pharmaceutical Inspection Convention/Co-operation Scheme (PIC/S), EU Directives, and World Health Organization recommendations in respect of products to be sold or distributed within the county of origin or to be exported. This certificate reflects the status of the manufacturing site at the time of the inspection noted above and should not be relied upon to reflect the compliance status if more than three years have elapsed since the date of that inspection. This certificate includes the Parts 1, 2 and Annex. The authenticity of this certificate may be verified with the issuing authority. |
| Частина 2 | Part 2 |
| Лікарські засоби для людини | Human Medicinal Products |
| 1. ВИРОБНИЧІ ОПЕРАЦІЇ — ЛІКАРСЬКІ ЗАСОБИ*
1.1. Стерильні продукти 1.1.1. Асептично виготовлені (виробничі операції для наступних лікарських форм) 1.1.1.1. Рідини в упаковках великого об’єму 1.1.1.2. Ліофілізати 1.1.1.3. М’які 1.1.1.4. Рідини в упаковках малого об’єму 1.1.1.5. Тверді та імплантанти 1.1.1.6. Інші асептично виготовлені продукти (зазначити) 1.1.2. Що піддаються кінцевій стерилізації (виробничі операції для наступних лікарських форм) 1.1.2.1. Рідини в упаковках великого об’єму 1.1.2.2. М’які 1.1.2.3. Рідини в упаковках малого об’єму 1.1.2.4. Тверді та імплантанти 1.1.2.5. Інші продукти, що піддаються кінцевій стерилізації (зазначити) 1.1.3. Сертифікація серій 1.2. Нестерильні продукти 1.2.1. Нестерильні продукти (виробничі операції для наступних лікарських форм) 1.2.1.1. Капсули, тверді 1.2.1.2. Капсули, м’які 1.2.1.3. Жувальні гуми 1.2.1.4. Імпрегновані матриці 1.2.1.5. Рідини для зовнішнього застосування 1.2.1.6. Рідини для внутрішнього застосування 1.2.1.7. Медичні гази 1.2.1.8. Інші тверді лікарські форми 1.2.1.9. Препарати під тиском 1.2.1.10. Генератори радіонуклідів 1.2.1.11. М’яка 1.2.1.12. Супозиторії 1.2.1.13. Таблетки 1.2.1.14. Трансдермальні пластирі 1.2.1.15. Стоматологічні матеріали 1.2.1.16. Інші нестерильні лікарські засоби (зазначити) 1.2.2. Сертифікація серій 1.3. Біологічні лікарські засоби 1.3.1. Біологічні лікарські засоби 1.3.1.1. Препарати крові 1.3.1.2. Імунобіологічні лікарські засоби 1.3.1.3. Лікарські засоби клітинної терапії 1.3.1.4. Лікарські засоби генної терапії 1.3.1.5. Біотехнологічні лікарські засоби 1.3.1.6. Препарати, екстраговані з тканин людини або тварин 1.3.1.7. Лікарські засоби тканинної інженерії 1.3.1.8. Інші біологічні лікарські засоби (зазначити) 1.3.2. Сертифікація серій (перелік) 1.3.2.1. Препарати крові 1.3.2.2. Імунобіологічні лікарські засоби 1.3.2.3. Лікарські засоби клітинної терапії 1.3.2.4. Лікарські засоби генної терапії 1.3.2.5. Біотехнологічні лікарські засоби 1.3.2.6. Препарати, екстраговані з тканини людини або тварини 1.3.2.7. Лікарські засоби тканинної інженерії 1.3.2.8. Інші біологічні лікарські засоби (зазначити) 1.4. Інші продукти або виробнича діяльність 1.4.1. Виробництво: 1.4.2. Продукти з рослинної сировини 1.4.3. Гомеопатичні препарати 1.4.4. Інші (зазначити) 1.4.2. Стерилізація активних речовин/допоміжних речовин/готової продукції1. 4.2.1. Фільтрація 1.4.2.2. Сухожарова стерилізація 1.4.2.3. Стерилізація паром 1.4.2.4. Хімічна 1.4.2.5. Гамма-випромінювання 1.4.2.6. Електронно-променева 1.4.3. Інші (зазначити) 1.5. Пакування 1.5.1. Первинне пакування 1.5.1.1. Капсули, тверді 1.5.1.2. Капсули, м’які 1.5.1.3. Жувальні гуми 1.5.1.4. Імпрегновані матриці 1.5.1.5. Рідини для зовнішнього застосування 1.5.1.6. Рідини для внутрішнього застосування 1.5.1.7. Медичні гази 1.5.1.8. Інші тверді лікарські форми 1.5.1.9. Препарати під тиском 1.5.1.10. Генератори радіонуклідів 1.5.1.11. М’які 1.5.1.12. Супозиторії 1.5.1.13. Tаблетки 1.5.1.14. Трансдермальні пластирі 1.5.1.15. Стоматологічні матеріали 1.5.1.16. Інші нестерильні лікарські засоби (зазначити) 1.5.2. Вторинне пакування 1.6. Проведення випробувань в рамках контролю якості 1.6.1. Мікробіологічні: стерильність 1.6.2. Мікробіологічні: мікробіологічна чистота 1.6.3. Фізичні/хімічні 1.6.4. Біологічні 1. ВИРОБНИЧІ ОПЕРАЦІЇ — АКТИВНІ ФАРМАЦЕВТИЧНІ ІНГРЕДІЄНТИ* Активний(і) фармацевтичний(і) інгредієнт(и): 2.1. Виробництво активної речовини шляхом хімічного синтезу 2.1.1. Виробництво активних проміжних речовин 2.1.2. Виробництво неочищеного активного фармацевтичного інгредієнта 2.1.3. Солеутворення/очищення: (зазначити) (наприклад, кристалізація) 2.1.4. Інші (зазначити) 2.2. Отримання активного фармацевтичного інгредієнта з природних джерел 2.2.1. Отримання речовини з рослин 2.2.2. Отримання речовини з тварин 2.2.3. Отримання речовини з людського джерела 2.2.4. Отримання речовини з мінерального джерела 2.2.5. Модифікація отриманої речовини (зазначити джерело) 2.2.6. Очищення отриманої речовини (зазначити джерело) 2.2.7. Інше (зазначити) 2.3. Виробництво активного фармацевтичного інгредієнта з використанням біологічних процесів 2.3.1. Ферментація 2.3.2. Культура клітин (зазначити тип клітин) (наприклад, ссавців/бактеріальні) 2.3.3. Виділення/Очищення 2.3.4. Модифікація 2.3.5. Інше (зазначити) 2.4. Виробництво стерильного активного фармацевтичного інгредієнта (розділи 3.1, 3.2, 3.3, заповнюються за необхідності) 2.4.1. Асептично виготовлені 2.4.2. Препарати, що піддаються кінцевій стерилізації 2.5. Ступені загальної обробки 2.5.1. Ступені фізичної обробки (зазначити) (наприклад, сушіння, подрібнення/мікронізація, просіювання) 2.5.2. Первинне пакування (закупорювання/герметизація активного фармацевтичного інгредієнта пакувальним матеріалом, який знаходиться в прямому контакті з речовиною) 2.5.3. Вторинне пакування (розміщення герметичної первинної упаковки всередині зовнішнього пакувального матеріалу або контейнера. Це також включає в себе будь-яке маркування матеріалу для ідентифікації або простежуваності (нумерація серії) активного фармацевтичного інгредієнта) 2.5.4. Інше (зазначити) (для операцій, не описаних вище) 2.6 Проведення випробувань в рамках контролю якості 2.6.1. Фізичні/хімічні випробування 2.6.2. Мікробіологічні випробування (виключаючи випробування стерильності) 2.6.3. Мікробіологічні випробування (включаючи випробування стерильності) 2.6.4. Біологічні випробування 3. ІНША ДІЯЛЬНІСТЬ — АКТИВНІ ФАРМАЦЕВТИЧНІ ІНГРЕДІЄНТИ (зазначити) *Будь-які обмеження або пояснення, що мають відношення до цього сертифіката:……………………………………….. |
1. MANUFACTURING OPERATIONS — MEDICINAL PRODUCTS* 1.1. Sterile products 1.1.1. Aseptically prepared (processing operations for the following dosage forms) 1.1.1.1. Large volume liquids 1.1.1.2. Lyophilisates 1.1.1.3. Semi-solids 1.1.1.4. Small volume liquids 1.1.1.5. Solids and implants 1.1.1.6. Other aseptically prepared products <free text> 1.1.2. Terminally sterilised (processing operations for the following dosage forms) 1.1.2.1. Large volume liquids 1.1.2.2. Semi-solids 1.1.2.3. Small volume liquids 1.1.2.4. Solids and implants 1.1.2.5. Other terminally sterilised prepared products <free text> 1.1.3. Batch certification 1.2. Non-sterile products 1.2.1. Non-sterile products (processing operations for the following dosage forms) 1.2.1.1. Capsules, hard shell 1.2.1.2. Capsules, soft shell 1.2.1.3. Chewing gums 1.2.1.4. Impregnated matrices 1.2.1.5. Liquids for external use 1.2.1.6. Liquids for internal use 1.2.1.7. Medicinal gases 1.2.1.8. Other solid dosage forms 1.2.1.9. Pressurised preparations 1.2.1.10. Radionuclide generators 1.2.1.11. Semi-solids 1.2.1.12. Suppositories 1.2.1.13. Tablets1.2.1.14. Transdermal patches 1.2.1.15. Intraruminal devices 1.2.1.16. Other non-sterile medicinal product <free text> 1.2.2. Batch certification 1.3. Biological medicinal products 1.3.1. Biological medicinal products 1.3.1.1. Blood products 1.3.1.2. Immunological products 1.3.1.3. Cell therapy products 1.3.1.4. Gene therapy products 1.3.1.5. Biotechnology products 1.3.1.6. Human or animal extracted products 1.3.1.7. Tissue engineered products 1.3.1.8. Other biological medicinal products <free text> 1.3.2. Batch certification (list of product types) 1.3.2.1. Blood products 1.3.2.2. Immunological products 1.3.2.3. Cell therapy products 1.3.2.4. Gene therapy products 1.3.2.5. Biotechnology products 1.3.2.6. Human or animal extracted products 1.3.2.7. Tissue engineered products 1.3.2.8. Other biological medicinal products <free text> 1.4. Other products or processing activity 1.4.1. Manufacture of: 1.4.1.1. Herbal products 1.4.1.2. Homoeopathic products 1.4.1.3. Other <free text> 1.4.2. Sterilisation of active substances/excipients/finished product: 1.4.2.1. Filtration 1.4.2.2. Dry heat 1.4.2.3. Moist heat 1.4.2.4. Chemical 1.4.2.5. Gamma irradiation 1.4.2.6. Electron beam 1.4.3. Others <free text> 1.5. Packaging 1.5.1. Primary packing 1.5.1.1. Capsules, hard shell 1.5.1.2. Capsules, soft shell 1.5.1.3. Chewing gums 1.5.1.4. Impregnated matrices 1.5.1.5. Liquids for external use 1.5.1.6. Liquids for internal use 1.5.1.7. Medicinal gases 1.5.1.8. Other solid dosage forms 1.5.1.9. Pressurised preparations 1.5.1.10. Radionuclide generators 1.5.1.11. Semi-solids 1.5.1.12. Suppositories 1.5.1.13. Tablets 1.5.1.14. Transdermal patches 1.5.1.15. Intraruminal devices 1.5.1.16. Other non-sterile medicinal products <free text> 1.5.2. Secondary packing 1.6. Quality control testing 1.6.1. Microbiological: sterility 1.6.2. Microbiological: non-sterility 1.6.3. Chemical/Physical 1.6.4. Biological 2. MANUFACTURING OPERATIONS – ACTIVE SUBSTANCES Active Substance(s): 2.1. Manufacture of Active Substance by Chemical Synthesis 2.1.1. Manufacture of active substance intermediates 2.1.2. Manufacture of crude active substance 2.1.3. Salt formation/Purification steps : <free text> (e.g. crystallisation) 2.1.4. Other <free text> 2.2. Extraction of Active Substance from Natural Sources 2.2.1. Extraction of substance from plant source 2.2.2. Extraction of substance from animal source 2.2.3. Extraction of substance from human source 2.2.4. Extraction of substance from mineral source 2.2.5. Modification of extracted substance <specify source> 2.2.6. Purification of extracted substance <specify source> 2.2.7. Other <free text> 2.3. Manufacture of Active Substance using Biological Processes 2.3.1. Fermentation 2.3.2. Cell Culture <specify cell type> (e.g. mammalian/bacterial) 2.3.3. Isolation/Purification 2.3.4. Modification 2.3.5. Other <free text> 2.4. Manufacture of sterile active substance (sections 3.1, 3.2, 3.3. to be completed as applicable) 2.4.1. Aseptically prepared 2.4.2. Terminally sterilized 2.5. General Finishing Steps 2.5.1. Physical processing steps <specify> (e.g. drying, milling/micronisation, sieving) 2.5.2. Primary Packaging (enclosing/sealing the active substance within a packaging material which is in direct contact with the substance) 2.5.3. Secondary Packaging (placing the sealed primary package within an outer packaging material or container. This also includes any labelling of the material which could be used for identification or traceability (lot numbering) of the active substance) 2.5.4. Other <free text> (for operations not described above) 2.6. Quality Control Testing 2.6.1. Physical/Chemical testing 2.6.2. Microbiological testing (excluding sterility testing) 2.6.3. Microbiological testing (including sterility testing) 2.6.4. Biological Testing 3. OTHER ACTIVITIES — ACTIVE SUBSTANCES <free text> Any restrictions or clarifying remarks related to the scope of this certificate:………………………………………. |
| підпис відповідальної особи, М.П.
……./……./……. [дата] Державна служба України з лікарських засобів Місцезнаходження: Тел.: Факс: Електронна адреса: www.diklz.gov.ua ___________________ *Залишити потрібне |
signature of the Executive officer (see left)
……./……./……. [date] State Administration of Ukraine on Medicinal Products Address: Phone: Fax: e-mail: www.diklz.gov.ua This English translation is for reference only and is not part of the official certificate Номер сторінки/Загальна кількість сторінок |
Додаток 4
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 6 розділу І)
Бланк Державної служби України з лікарських засобів та контролю за наркотиками
ВИСНОВОК
щодо підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики
На підставі розгляду заяви від «___» ____ 20__ року № ___________________________________________________________________________________________________________________(найменування заявника та/або виробника), документів, що додаються до неї, та, враховуючи позитивні результати проведеної спеціалізованої експертизи (експертний висновок від «__» _____ 20__ року № __), сертифікат відповідності виробництва лікарських засобів вимогам GMP № __ від «__» _____ 20__ року, виданий
_______________________________________________________________________________________________(найменування уповноваженого органу країни — члена ЄС або країни, яка має угоду про взаємне визнання з ЄС або з Україною)
виробничій діяльності ___________________________________________________________________________(найменування виробника та/або виробничої дільниці)
адреса місця провадження діяльності: ____________________________________________________________,
строком дії до «__» ____ 20__ року, вважається таким, що підтверджує відповідність виробництва лікарських засобів чинним в Україні вимогам GMP.
Дія цього висновку розповсюджується на такі форми лікарських засобів, як:
_______________________________________________________________________________________________.
Перелік лікарських засобів, що виробляються на дільниці
_________________________________________________________________________________________________(найменування, місце провадження (адреса) виробника (виробничої дільниці)), наведено в додатку до цього висновку і є його невід’ємною частиною.
________________ ______________________ ____________________________(посада) (підпис) (прізвище, ім’я по батькові)
«__» ____________ 20__ року
М.П.
Додаток 5
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(підпункт 8 пункту 2 розділу IІ)
ДОВІДКА
про якість продукції, що виробляється
на__________________________________________________________________________________________(найменування виробничої діяльності, що вказана в заяві)
______________________________________________________________________________________________(найменування заявника)
з 20____ по 20____ р. (навести дані за останні три роки, рахуючи від дати подання заяви)
| № з/п | Відомості про претензії і відкликання продукції | Усього | Перелік найменувань і номерів серій лікарських засобів |
|---|---|---|---|
| 1 | Кількість обґрунтованих претензій до якості продукції: | ||
| 1.1 | за результатами державного контролю | ||
| 1.2 | за зверненнями споживачів | ||
| 2 | Кількість відкликань продукції з мережі реалізації в Україні: | ||
| 2.1 | за приписами державних органів контролю | ||
| 2.2 | за рішенням виробника | ||
| 3 | Кількість відкликаної продукції з мережі реалізації країни, де розташоване виробництво (для нерезидентів): | ||
| 3.1 | за приписами державних органів країни, де розташоване виробництво | ||
| 3.2 | за рішенням виробника | ||
| 4 | Кількість відкликаної продукції з мережі реалізації в інших країнах: | ||
| 4.1 | за приписами відповідних державних органів країн, де здійснювалася реалізація продукції | ||
| 4.2 | за рішенням виробника |
Дата складання «___» ___________ 20____ року
Керівник підприємства ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
М.П. (за наявності)
Керівник служби якості (уповноважена особа) ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
Додаток 6
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(підпункт 9 пункту 2 розділу IІ)
ДОВІДКА
про результати перевірок виробничої дільниці, проведених органами державного контролю
____________________________________________________________________________________(найменування виробничої дільниці та найменування заявника)
з 20___ по 20___ р. (указати за останні три роки до дати подання заяви)
| № з/п | Найменування уповноваженого органу | Вид перевірки (інспектування) (планова, позапланова) | Період, дата(и) перевірки | Звіт/акт від(дата)№ | Результат перевірки (навести посилання на документ та надати короткі висновки, наведені у звіті/акті) |
|---|---|---|---|---|---|
| 1 | Орган державного контролю лікарських засобів в Україні | ||||
| 2 | Державний або уповноважений орган у сфері контролю лікарських засобів країни, де розташоване виробництво (для нерезидентів) | ||||
| 3 | Уповноважені органи у сфері контролю лікарських засобів, країни яких є членами ЄС або мають угоди про взаємне визнання з ЄС або з Україною | ||||
| 4 | Уповноважені органи у сфері контролю лікарських засобів інших країн |
Дата складання «__» ____________ 20__ року
Керівник виробника ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
М.П. (за наявності)
Керівник служби якості (уповноважена особа) ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
Додаток 7
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(підпункт 10 пункту 2 розділу IІ)
(Формула 1)
ЗАГАЛЬНИЙ ПЕРЕЛІК НОМЕНКЛАТУРИ ПРОДУКЦІЇ1
________________________________________________________________________________(найменування виробничої дільниці)
________________________________________________________________________________(місце провадження діяльності)
________________________________________________________________________________(країна виробника)
| № з/п | Торговельна назва2 | Міжнародна непатентована назва (МНН)3 | Реєстраційне посвідчення в Україні | Реєстраційне посвідчення (торгова ліцензія) в країні, де здійснюється виробництво від «__» ____ № __ | Інформація про виробників4 | |||||||||
| торговельна назва (укр.) | торговельна назва (англ.) | лікарська форма (укр.) | лікарська форма (англ.) | номер | рік видачі | місяць видачі | день видачі | виробник нерозфасованого продукту | первинна упаковка | вторинна упаковка | дозвіл на випуск серій5 | |||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
Дата складання «__» _______________ 20__ року
Керівник виробника ________ __________ ___________________________(посада) (підпис) (прізвище, ім’я, по батькові)
М. П. (за наявності)
Керівник служби якості/Уповноважена особа ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
Керівник виробництва ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
(Форма 2)
ЗАГАЛЬНИЙ ПЕРЕЛІК НОМЕНКЛАТУРИ ПРОДУКЦІЇ1
________________________________________________________________________________(найменування виробничої дільниці)
________________________________________________________________________________(місце провадження діяльності)
________________________________________________________________________________(країна виробника)
| № з/п | Торговельна назва2 | Міжнар одна непатентована назва (МНН)3 | Код АТ С | Реєстраційне посвідчення в Україні | Реєстраційне посвідчення (торгова ліцензія) в країні, де здійснюється виробництво від «__» _____ № __ | Інформація про виробників4 | Інформація про контрактні лабораторії, складські зони6 | |||||||||
| торговельна назва (укр.) | торговельна назва (англ.) | лікарська форма (укр.) | лікарська форма (англ.) | номер | рік видачі | місяць видачі | день видачі | виробник нерозфасованого продукту | первинна упаковка | вторинна упаковка | дозвіл на випуск серій5 | |||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 |
Дата складання «__» _______________ 20__ року
Керівник виробника ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
М. П. (за наявності)
Керівник служби якості/Уповноважена особа ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
Керівник виробництва ________ __________ _________________________(посада) (підпис) (прізвище, ім’я, по батькові)
Додаток 8
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(підпункт 12 пункту 2 розділу IІ)
(Бланк заявника)
Державна служба України з лікарських засобів та контролю за наркотиками
ГАРАНТІЙНИЙ ЛИСТ
про суттєві зміни, які стосуються виробничої дільниці,
що пройшла підтвердження відповідності умов виробництва лікарських засобів вимогам GMP
Від ___________________________________________________________________________________________(заявник), в особі ___________________________________________________________________________________________, що діє на підставі ____________________________________________________________________________________________ гарантує, що Держлікслужбу буде повідомлено про зміни щодо виробничої дільниці, що пройшла підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики (GMP), включаючи зміни у проміжних (контрактних) виробничих дільницях, які задіяні у виробництві лікарського засобу (у тому числі про зміну ліцензії на виробництво лікарських засобів, зміну найменування виробника, його місцезнаходження (юридичної адреси) та/або адреси виробничих потужностей тощо) ________________________________________________________________________________________________.
ВИРОБНИК ________________________________________________________________________________________________(найменування суб’єкта господарювання, місце провадження діяльності)
Контактні дані керівника Заявника/керівника представника Заявника: ________________________________________________________________________________________________(телефон, факс, електронна адреса)
До гарантійного листа додається:
копія документа, що підтверджує повноваження особи, яка підписала гарантійний лист.
Керівник Заявника/представника Заявника ___________ _____________________________(підпис) (ініціали та прізвище)
М.П. (за наявності)
Додаток 9
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 4 розділу IІ)
Державна служба України з лікарських засобів та контролю за наркотиками
ОПИС № ________
документів, що додаються до заяви на видачу сертифіката відповідності умов виробництва лікарських засобів вимогам належної виробничої практики (до заяви на видачу висновку щодо підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики)
від _________________________________________________________________________________________(найменування заявника)
Дата і номер реєстрації заяви «__» __________ 20__ року № __
| № з/п | Найменування документа | Кількість аркушів у документі | Відмітка про наявність документів (наявні, відсутні) | Примітки |
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 |
Прийняв _________ документів ____________________
__________________ __________________________________ _________________(цифрами і словами) (підпис відповідальної особи) (П.І.Б.)
«__»______________ 20__ року
Копію опису отримав _____________________________________ ________________________(підпис представника заявника) (П.І.Б.)
«__»______________ 20__ року
Додаток 10
До Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 8 розділу ІV)
Алгоритм з класифікації порушень
При класифікації порушення як «Критичне» інспектори повинні визначити чи існують чіткі докази, враховуючи ризик шкоди, як зазначено у визначенні (приклад наведений у блок-схемі, рисунок 1).
Коли «Критичне» порушення не є чітко очевидним, порушення може бути оцінене як «Критичне», «Суттєве» або «Несуттєве». Необхідно визначити класифікацію, за якою можна дотримуватися зазначених нижче вказівок.
Провести детальну оцінку порушення для визначення початкової класифікації згідно з блок-схемою, рисунки 2–5.
Провести оцінку факторів, які або збільшують, або зменшують ризик, незалежно від початкової класифікації, як описано в поясненнях щодо факторів, що можуть вплинути на підвищення або зменшення ризику.
Прийняти рішення щодо того, чи може початкова класифікація ризику бути такою, як описано в блок-схемі, рисунок 1:
- підвищити за рахунок ефектів, які збільшують ризик, тобто впливу, що збільшує ризик,
- залишити без змін, або
- знизити внаслідок ефектів, які зменшують ризик, тобто впливають на зниження ризику.
Формат написання та групування порушень також може бути чинником, що впливає на класифікацію порушення.
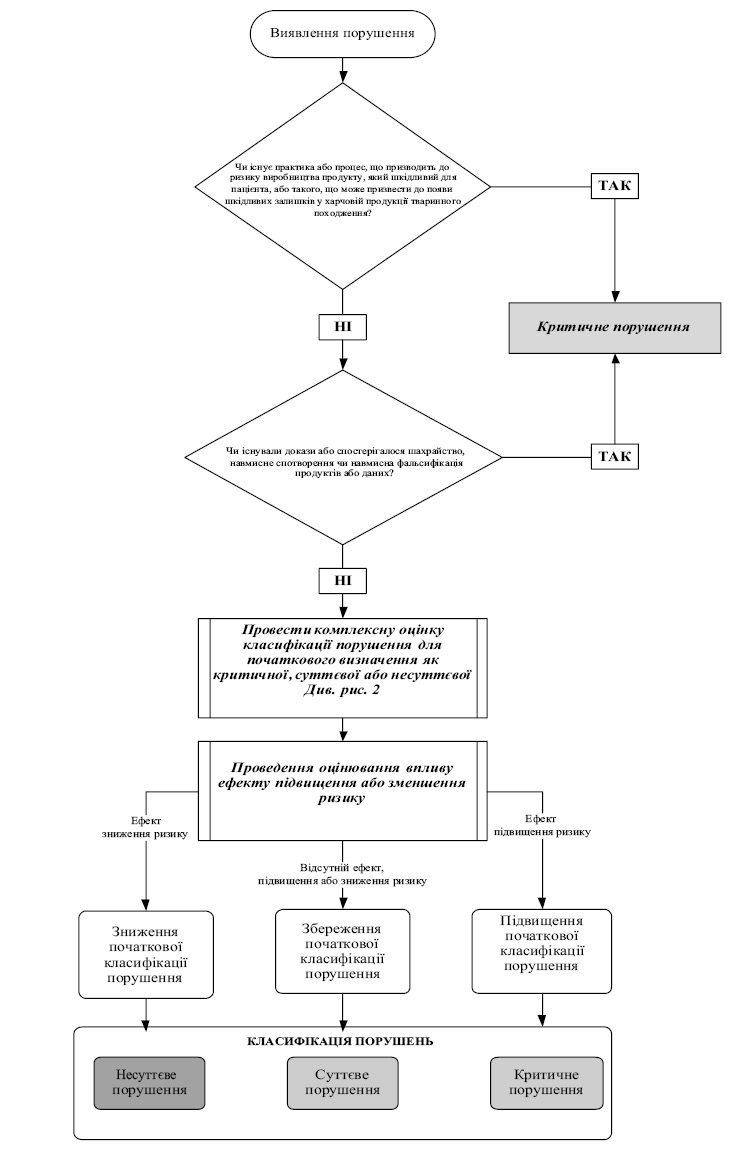
Блок-схема, рисунок 1 — Процес класифікації — Огляд
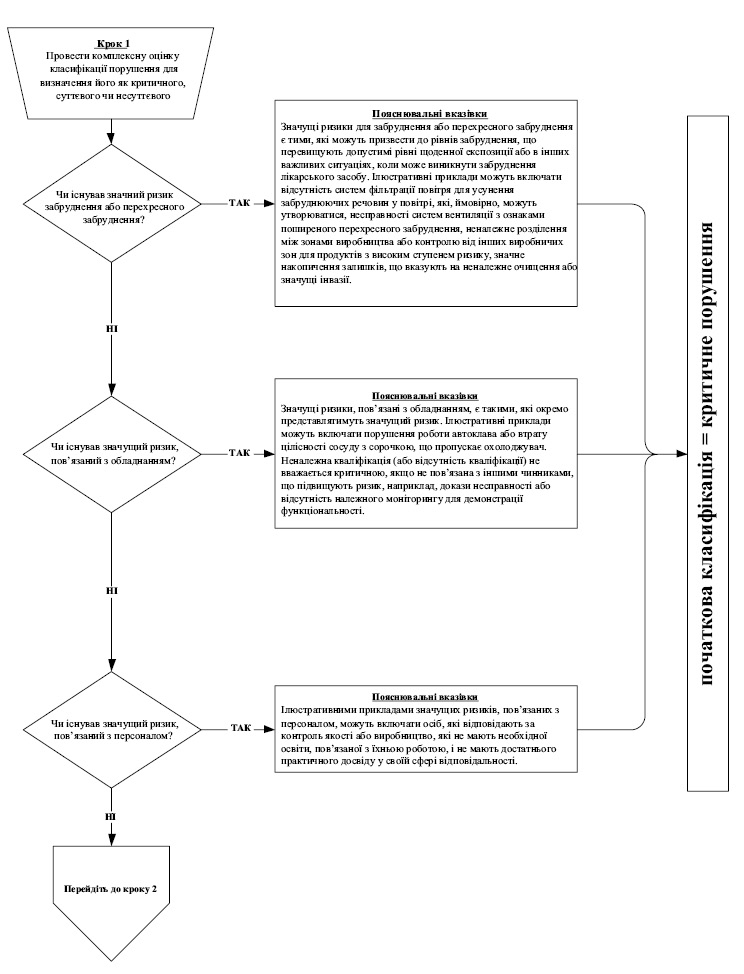
Блок-схема, рисунок 2 — Процес класифікації — Огляд (продовження)
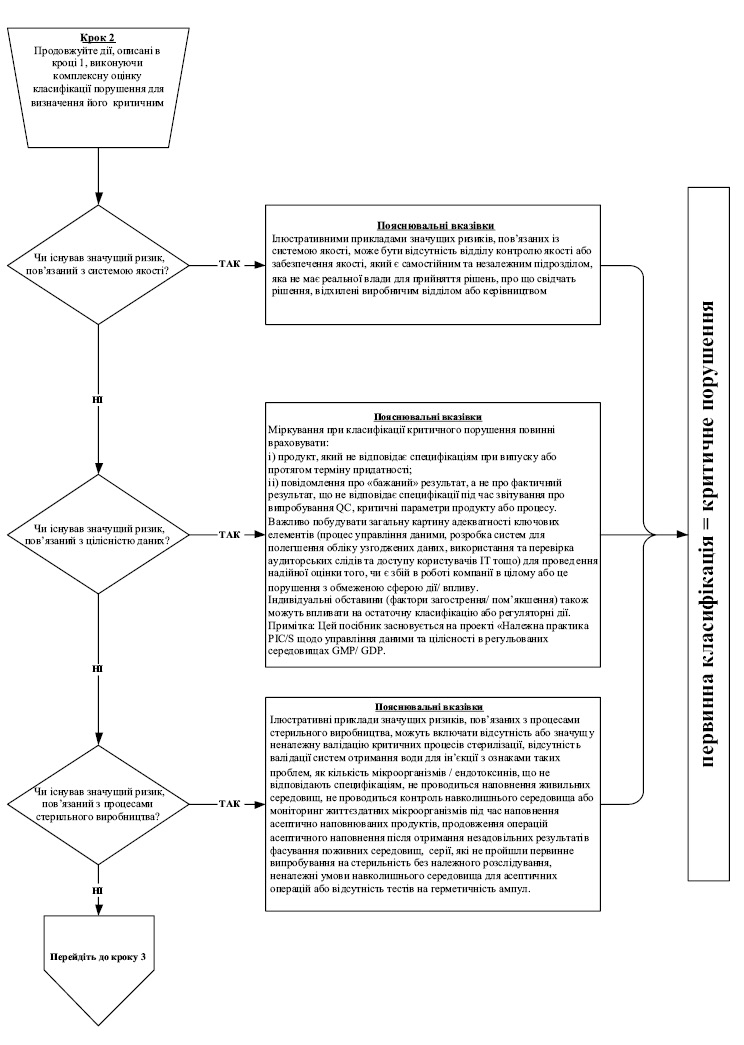
Блок-схема, рисунок 3 — Процес класифікації — Огляд (продовження)
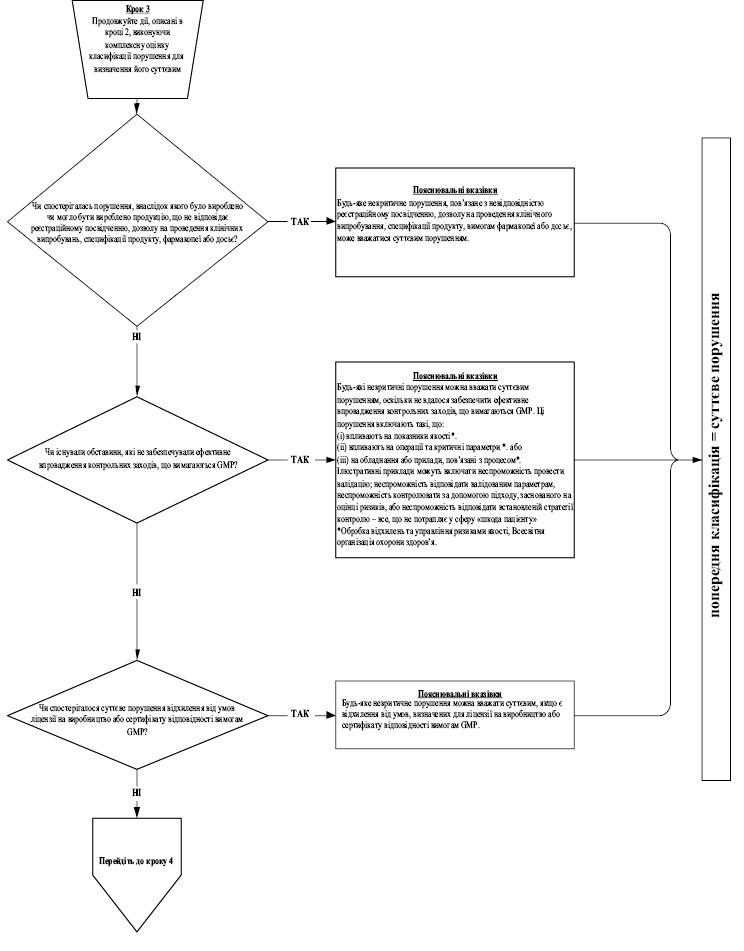
Блок-схема, рисунок 4 — Процес класифікації — Огляд (продовження)
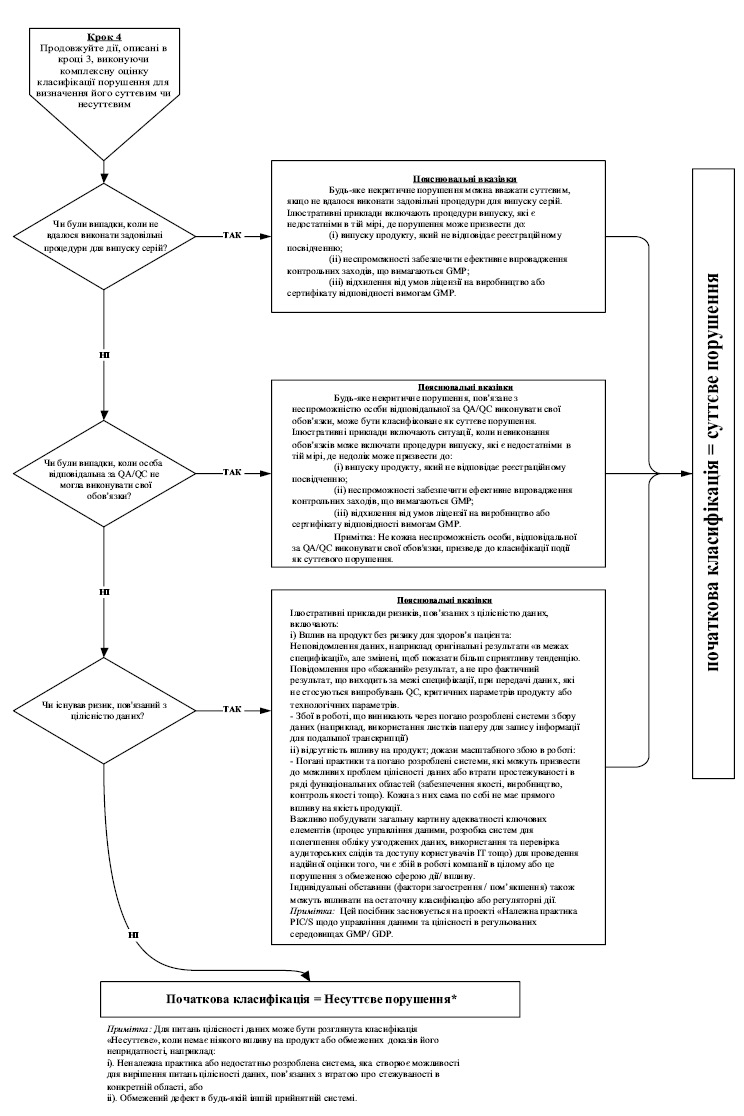
Блок-схема, рисунок 5 — Процес класифікації — Огляд (продовження)
ПОЯСНЕННЯ ЩОДО ФАКТОРІВ, ЩО МОЖУТЬ ВПЛИНУТИ НА ПІДВИЩЕННЯ АБО ЗМЕНШЕННЯ РИЗИКУ
1. Фактори, що збільшують ризик підвищення початкової класифікації
«Суттєві» та «Несуттєві» порушення можуть бути підвищені на один рівень до «Критичного» або «Суттєвого» порушення, відповідно, коли можуть існувати умови, що відповідають сутності визначення підвищеної класифікації ризиків. Вважається, що це досягається, коли існують певні фактори, що підвищують ризик.
Фактори збільшення ризику включають:
повторне або постійне порушення (пункт 3 пояснень);
групу або комбінацію порушень (пункт 4 пояснень);
ризик, пов’язаний з продуктом (пункт 5 пояснень);
нездатність керівництва виробника визначити та вжити обґрунтованих заходів для зменшення ризику для пацієнта до прийнятного рівня для продукції, що перебуває в дистрибуції, і для майбутнього виробництва внаслідок невідповідної практики або процесу.
2. Фактори, що зменшують ризик — зниження початкової класифікації
«Критичне» та «Суттєве» порушення можуть бути знижені на один рівень до «Суттєвого» або «Несуттєвого» порушення, відповідно, коли можуть існувати умови для зниження класифікації ризиків. Вважається, що це досягається, коли існують певні фактори, що знижують ризик.
При розгляді факторів, що знижують ризик, важливо забезпечити, щоб ці фактори були послідовними та ефективними.
Фактори, що знижують ризик, включають:
мінімізацію ризику, пов’язаного з продуктом (пункт 5 пояснень);
мінімізацію ризику шкоди для пацієнта;
інші фактори зниження ризику (пункт 6 пояснень);
дії, вжиті виробником, наприклад, план попереджувальних і коригувальних дій для зниження ризику дефіциту.
Вплив продукту, який вже постачається на ринок, слід враховувати при зниженні категорії критичного порушення.
3. Повторні або повторювані порушення — підвищення початкової класифікації
Повторні або часто повторювані порушення — це порушення, які також були виявлені при попередній перевірці, коли відповідні попереджувальні або коригувальні дії не були вжиті.
У певних випадках повторювані порушення можуть розглядатися як такі, що підвищують ризик, що дозволяє підвищити початкову класифікацію ризику, зокрема, якщо очевидно, що існують навмисні або незадовільні зусилля для усунення порушення. Ефект, що підвищує ризик, слід враховувати, коли:
існує серйозний недолік в системі якості, за допомогою якої не можна задовільно визначити потенційні першопричини невідповідності або не можна адекватно розглянути ці причини без наявності інших факторів, що зменшують ризик, або
існують й інші фактори для розгляду, які підпадають під визначення підвищеної класифікації ризиків, наприклад, необґрунтовано тривале впровадження коригувальних дій.
Примітка: Очікується, що підвищення ризику повторюваної невідповідності потребуватиме розуміння потенційних факторів, які, можливо, призвели до повторення.
4. Група або комбінація порушення — оновлення початкової класифікації
Різні питання/проблеми, виявлені під час перевірки, можуть бути згруповані або об’єднані в одне порушення, якщо кожне питання/проблема супроводжує або стосується зазначеного основного порушення.
Ефект підвищення ризику, може бути застосований для підвищення початкової класифікації ризику на один рівень, коли визначення підвищеної класифікації ризику було застосоване.
Приклади декількох «Несуттєвих» порушень, жодне з яких саме по собі не може бути «Суттєвим», але які разом можуть представляти «Суттєве» порушення, слід пояснити і повідомити про це.
5. Ризик, пов’язаний з продуктом — підвищення або зниження початкової класифікації
На деяких виробничих дільницях є продукти та процеси, які пов’язані зі значно більшими ризиками, ніж інші.
Визначення класифікації ризиків, пов’язаних з продуктом:
продукти з високим ступенем ризику, які мають високу чутливість до забруднення в процесі виробництва, включаючи термін придатності, наприклад мікробного або хімічного;
продукти з низьким ступенем ризику, які мають меншу ймовірність забруднення в процесі виробництва, включаючи термін зберігання.
Фактор, що збільшує ризик, і той що знижує ризик, можуть застосовуватися після розгляду ризиків, пов’язаних з продуктом, таким чином:
для деяких продуктів з високим ступенем ризику певні порушення, що класифікуються, як «Суттєве» або «Несуттєве», можуть бути відповідно підвищені до «Критичного» або «Суттєвого» порушення. Це може бути застосовано, коли обставини порушення, що розглядаються, відповідають інтерпретації визначення «Критичного» порушення;
для деяких продуктів з низьким ступенем ризику певні порушення, що класифікуються як «Критичні» або «Суттєві», можуть бути відповідно знижені до «Суттєвого» порушення або «Несуттєвого» порушення. Для продуктів з низьким ступенем ризику «Критичне» порушення може бути знижено до «Суттєвого», якщо воно не буде підпадати під визначення «Критичного» порушення.
6. Інші фактори, що зменшують ризик
Коли інші фактори, що зменшують ризик, очевидні для мінімізації ризику, пов’язаного з порушенням, то оцінка ризику може бути знижена.
Інші фактори, що знижують ризик, зазвичай можна розглядати лише тоді, коли існує вторинна система, яка може мінімізувати ризики, пов’язані з порушеннями. Наприклад, кваліфікована система пакування з системою візуальної інспекції упаковки, яка забезпечує 100% контроль кожного упакованого продукту, може розглядатися як фактор, що знижує ризик для порушення, пов’язаного головним чином з друкованими матеріалами, що зберігаються невпорядковано, що може призвести до переплутування.
Якщо існує ряд факторів, що збільшують ризик і зменшують ризик, слід розглянути одночасно всі фактори ризику, а потім визначити загальну оцінку ризику для підвищення або зниження початкового ризику.
ПРИКЛАДИ КЛАСИФІКАЦІЇ ПОРУШЕНЬ
Наданий список є допоміжним інструментом і не є вичерпним, або обов’язковим.
1. Приклади критичних порушень:
відсутність валідації стерилізації (стосується всіх стерильних продуктів);
відсутність належних заходів контролю, що призводять до фактичного або значного ризику перехресної контамінації, що перевищує максимально допустимий рівень впливу у наступних продуктах;
докази зараження паразитами/шкідниками (стосується всіх виробників); фальсифікація або хибне подання результатів аналізів чи записів (стосується всіх виробників);
неможливість забезпечити якість та/або ідентичність вихідної сировини (стосується всіх виробників);
відсутність основних документів щодо виготовлення серії (стосується всіх виробників);
відсутність, фальсифікація або хибне подання записів щодо виробництва та пакування (стосується всіх виробників);
не проведена валідація системи водопостачання для стерильних продуктів (стосується виробників стерильних продуктів);
не проведена валідація системи вентиляції та кондиціювання повітря для стерильних продуктів (стосується виробників стерильних продуктів);
непридатні приміщення з високим або ймовірним ризиком контамінації (стосується всіх виробників);
відсутні підтвердження, що процедури санкціонованого відкликання були дотримані (стосується всіх виробників).
2. Приклади суттєвих невідповідностей:
не проведена валідація для критичних процесів (застосовується до всіх лікарських засобів, але може бути перекваліфіковане в «критичне» для продуктів з низькою дозою/сильнодіючих; зокрема для процесів стерилізації для стерильних продуктів);
відсутність або явно неналежна фільтрація повітря (застосовується до всіх виробників лікарських засобів — може бути перекваліфіковане в «критичне», коли забруднюючі речовини можуть становити проблему безпеки та є «критичними» для стерильних лікарських засобів);
відсутні або неефективні заходи контролю, що забезпечують належну впевненість у тому, що перехресна контамінація буде контролюватися у відповідних межах допустимого впливу на здоров’я для наступних продуктів (буде «критичним», якщо перехресна контамінація перевищує чи може перевищити допустимий рівень впливу);
пошкодження (отвори, тріщини, відшаровування фарби) на стінах/стелі у виробничих приміщеннях, де продукт знаходиться в нестерильних зонах;
конструкція виробничих зон, що не дозволяє ефективно проводити очищення;
невідповідне виробниче приміщення, що може призвести до переплутування;
відсутні місця відбору проб вихідної сировини для виробників лікарських засобів (якщо вжито належних запобіжних заходів, можна класифікувати як «Несуттєве»);
для виробництва рідини/крему не використовується технічна арматура у санітарному виконанні;
обладнання, що зберігається, не захищено від забруднення;
особи, відповідальні за контроль якості/виробництво, не відповідають кваліфікації за освітою, компетентністю та досвідом;
неналежна початкова та поточна підготовка персоналу та/або відсутність записів з навчання;
процедури очищення не документовані та/або відсутні записи про очищення;
не проведена валідація процедури очищення виробничого обладнання;
скорочено контроль якості вхідної сировини без відповідних підтверджуючих даних від постачальників;
неповне випробування/тестування сировини;
не проведена валідація методів випробування;
не проведена валідація складних виробничих процесів для некритичних продуктів;
незатверджені/незареєстровані зміни до технологічного регламенту або еквівалентних документів;
відхилення від інструкцій не схвалені;
відсутня або неналежна програма самоінспекцій;
відсутність належного випуску для процедури постачання;
продукт перероблений без належного схвалення;
немає системи/процедури розгляду скарг або повернення продуктів;
неналежне випробування/тестування пакувальних матеріалів;
немає поточної програми випробування стабільності та/або недоступні дані про стабільність для всіх продуктів;
недостатнє освітлення у виробничих зонах або зонах контролю;
не ідентифіковані контейнери, з яких були відібрані проби;
не здійснюється моніторинг і відсутня аварійна/сигнальна система сигналізації щодо значень температур для критичних зон зберігання з контролем температур;
неналежна система управління змінами;
неналежна система управління відхиленнями;
не виконуються розслідування стосовно аварійних сигналів та виходів температури за межі відхилень від вимог щодо зберігання та транспортування.
Додаток 11
до Порядку проведення
підтвердження відповідності умов
виробництва лікарських засобів
вимогам належної виробничої
практики
(пункт 11 розділу ІV)
ЗВІТ
ЗА РЕЗУЛЬТАТАМИ ІНСПЕКТУВАННЯ
І. Загальні положення
| Виробник: | найменування виробника, місцезнаходження (юридична адреса); найменування виробничої(их) дільниці(ць), місце провадження діяльності, DUNS номер та GPS координати |
| Лабораторії, що здійснюють контроль якості за контрактом (договором), складські зони за контрактом (договором) | найменування, місце провадження діяльності, DUNS номери та GPS координати |
Види діяльності, які інспектувались: (зазначити потрібне знаком «Х»)
| Виробництво активних фармацевтичних інгредієнтів (субстанцій) | |
| Виробництво готових лікарських засобів (ГЛЗ) | |
| Виробництво досліджуваних лікарських засобів | |
| Виробництво проміжних продуктів, нерозфасованих продуктів (продукції «in bulk») | |
| Упаковка (тільки) | |
| Лабораторний контроль якості лікарських засобів | |
| Контроль кожної серії та дозвіл на реалізацію серій лікарських засобів | |
| Виробництво та аналіз за контрактом (зовнішня (аутсорсингова) діяльність) | |
| Зберігання, реалізація та транспортування (дистрибуція) | |
| Інше |
Дата проведення інспектування:_________________________________________________________(за місцями розташування виробничих дільниць, контрактних лабораторій та складських зон)
Інспектори: _______________________________________________________________________________(П.І.Б., посади інспекторів)
На виконання наказу від ________________ №____________________________________________________________________________________________(інформація щодо конфіденційності)
Нормативна база: Чинна версія GMP ЄС. Настанова «Лікарські засоби. Належна виробнича практика», затверджена МОЗ України, яка гармонізована з чинною версією GMP ЄС.
Вступ
(навести коротку інформацію про підприємство, яке було проінспектовано, включаючи стислі відомості щодо найменування і адреси виробництва (виробничих дільниць), лабораторії, у тому числі що здійснюють контроль якості за контрактом (договором), складські зони, наявність системи забезпечення якості відповідно до чинників GMP (за розділами досьє виробничої дільниці), у тому числі щодо дозвільних документів, змін, що відбулися, тощо) ________________________________________________________________________________________________________________________________________________________________________________________________
Відомості про ліцензовану діяльність, сертифікати GMP ________________________________________________________________________________________________________________________________________________________________________________________________
Зміни, що відбулися на виробництві з моменту попереднього інспектування (за наявності) ________________________________________________________________________________________________________________________________________________________________________________________________
Інформація про хід інспектування
Мета інспектування (номер, дата і короткий зміст наказу про інспектування) ________________________________________________________________________________________________________________________________________________________________________________________________
Масштаб і об’єкти
(навести перелік об’єктів (процеси, служби та системи), які були проінспектовані згідно із програмою та планом інспектування, а також тих, що не були предметом інспекції; навести назви лікарських форм, виробництво яких було проінспектовано (наприклад: стерильні лікарські засоби (перелік форм), нестерильні лікарські засоби (перелік форм); навести інформацію щодо відхилень від програми (перестановки пунктів програми між днями та/або інтервалами часу тощо), якщо такі мали місце під час інспекції, та про причини, що їх зумовили) ________________________________________________________________________________________________________________________________________________________________________________________________
Працівники підприємства, які брали участь в інспектуванні (навести перелік із зазначенням посад) ________________________________________________________________________________________________________________________________________________________________________________________________
II. Результати інспектування
Загальний опис виробництва та спостереження під час інспектування
| Фармацевтична система якості | |
| Персонал | |
| Приміщення та обладнання | |
| Документація | |
| Технологічний процес | |
| Контроль якості | |
| Зовнішня (аутсорсингова) діяльність | |
| Рекламації та відкликання продукції | |
| Самоінспекції | |
| Зберігання, реалізація татранспортування продукції (дистрибуція) | |
| Досьє виробничої дільниці (Site Master File, SMF) | Відповідність SMF вимогам, встановленим до його змісту та оформлення, загальна оцінка відповідності фактичного стану виробничих дільниць інформації, наведеній у відповідних частинах SMF, та вимогам GMP |
| Інші специфічні питання (за наявності) | |
| Відбір зразків під час інспекції (якщо такий проводився)
Результати виконання заходів щодо усунення порушень, виявлених під час попередньої інспекції (за наявності) |
Інформація про відбір зразків, виконаний під час інспекції |
Детальний перелік установлених фактів невідповідності вимогам GMP та їх класифікація*:
| № з/п | Пункт настанови | Детальний опис виявленого порушення | Класифікація порушення (критичне, суттєве, несуттєве) |
|---|---|---|---|
| Кількість копій звіту та їх розповсюдження | |
| Додатки до звіту | План і програма інспекції тощо |
| Висновки |
За результатами інспекції можна констатувати наявність невідповідностей вимогам GMP, які викладені і класифіковані в цьому звіті, всього: з них: критичних — _______ суттєвих — _______ несуттєвих — _______ Якщо в процесі інспектування порушення усунуті, про це повинно бути зазначено. Наявний стан виробництва та наявність виявлених під час інспектування невідповідностей дають/не дають можливість надати оцінку підприємству як такому, що відповідає/не відповідає вимогам GMP При повторному інспектуванні зазначаються класифікація та кількість неусунутих порушень. |
| Рекомендації інспекторів** |
Інспектори: ____________ ______________ ___________________(посада) (підпис, дата) (П.І.Б)
____________ ______________ ___________________(посада) (підпис, дата) (П.І.Б)
Додаток 12
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 17 розділу ІV)
Протокол наради/Meeting minutes
вступна нарада/opening meeting
Дата/Date ______________
заключна нарада/final meeting
| Загальна інформація щодо даного інспектування/General information on given inspection | |
| Найменування суб’єкта господар ської діяльності/Name of the business entity | |
| Місце провадження діяльності/Address of the business activity | |
| Мета перевірки/Object of the inspection | |
| Інспектори/Inspectors | |
| Ця частина протоколу зустрічі заповнюється на заключній зустрічі виключно посадовими особами, що здійснюють перевірку суб’єкта господарювання/Only public servants conducting the inspection of business entity fill this part of the meeting minutes during the final meeting | Так/Yes | Ні/No |
|---|---|---|
| Класифікацію порушень (невідповідностей), виявлених під час інспектування, оголошено/Is the classification of deficiencies (nonconformities) identified during the inspection announced? | | |
| Надано відповідні документальні підтвердження усунення порушень (невідповідностей) протягом проведення інспектування/Is the relevant documentary evidence for deficiencies (nonconformities) correction during the inspection provided?(надаються у разі усунення порушень (невідповідностей) протягом проведення інспектування)/(provided in the case of deficiencies (nonconformities) correction during the inspection) | | |
| Надано план коригувальних та запобіжних дій щодо усунення виявлених під час інспектування критичних порушень (невідповідностей)/Is the corrective and preventive action plan (САРА) on correction of critical deficiencies (nonconformities) revealed during the inspection provided? (вимагається за наявності критичних порушень (невідповідностей))/(required in the case of critical deficiencies (nonconformities)) | | |
| Наявні зауваження щодо проведеного інспектування та/або класифікації порушень (невідповідностей) з боку Заявника та/або виробника/Are there comments on conducted inspection and/or classification of deficiencies (nonconformities) by the Applicant and/or manufacturer? | | |
| Перелік значущих спостережень, які були оцінені в порядку значимості/List of significant observations ranked in order of significance. | |
| П.І.Б. інспекторів/Inspectors Full Name | Підпис/Signature |
| Опис зауважень щодо проведеного інспектування та/або класифікації порушень (невідповідностей) з боку Заявника та/або виробника/Comments on conducted inspection and/or classification of deficiencies (nonconformities) by the Applicant and/or manufacturer | ||
| Посада представника Заявника та/або виробника/Position of the Applicant’s and/or manufacturer’s representative | П.І.Б./Full Name | Підпис/Signature |
| Особи, що беруть участь у заході/Participants of the event: | ||
| П.І.Б. учасника зустрічі Full name of the meeting participant | Посада/Position | Підпис/Signature |
| Примітки/Notes |
Додаток 13
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 4 розділу V)
ПЕРЕЛІК ЛІКАРСЬКИХ ЗАСОБІВ, ЗАРЕЄСТРОВАНИХ АБО ЗАПЛАНОВАНИХ ДО РЕЄСТРАЦІЇ В УКРАЇНІ1
LIST O F PRO DUCTS, AUTHORISED OR PLANNED TO BE AUTHORISED IN UKRAINE
Додаток до Висновку/до Сертифіката від
Annex to Conclusion/to Certificate from
виробник/manufacturer
до Висновку/до Сертифіката
to Conclusion/to Certificate
строком дії до/valid till
| № з/п | Назва лікарського засобу | Реєстраційне посвідчення (торгова ліцензія) № __ (в Україні) | ||
| назва та лікарська форма лікарського засобу (українською мовою) | назва та лікарська форма лікарського засобу (англійською мовою) | міжнародна непатентована назва діючої(их) речовини(ин), у тому числі їх перелік — для багатокомпонентних (комбінованих) (англійською мовою) | ||
____________ ______________ ___________________(посада) (підпис, дата) (П.І.Б)
«__» _____________20__ року
Додаток 14
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 13 розділу ІV)
ПЛАН
коригувальних та запобіжних дій щодо усунення виявлених порушень
| № з/п | Порушення | Класифікація порушення (критичне, суттєве, несуттєве) | Коригувальні/запобіжні дії | Строк виконання | Відмітка про виконання | Примітки |
Дата складання «__» ___________ 20__ року
Керівник виробника ____________ ______________ ___________________(посада) (підпис, дата) (прізвище, ім’я, по батькові)
М. П. (за наявності)
Керівник служби якості (уповноважена особа) ____________ ______________ ___________________(посада) (підпис, дата) (прізвище, ім’я, по батькові)
Додаток 15
до Порядку проведення підтвердження відповідності
умов виробництва лікарських засобів вимогам належної виробничої практики
(пункт 3 розділу V)
Методологія проведення оцінки ризиків щодо розрахунку строку наступної інспекції виробництва лікарського засобу на відповідність вимогам GMP
Розрахунок строку проведення наступної інспекції є наслідком дати проведення останньої інспекції і процесу оцінювання інспекційним органом ризику згідно з цією процедурою.
1. Розрахунок внутрішнього ризику.
Внутрішній ризик, пов’язаний з виробничою дільницею, складається з оцінювання складності виробничої дільниці, її процесів і продукції та критичності лікарських засобів, що виробляється виробничою дільницею.
Визначення складності процесів та критичності лікарських засобів:
Складність виробничої дільниці, її процесів та продукції
Існують три можливі оцінки: А, В і С.
Загальними показниками складності виробничої дільниці є:
• Розмір виробничої дільниці — великі виробничі дільниці оцінюються як більш складні ніж менші за розмірами дільниці
• Кількість різних виробничих процесів, що застосовуються на виробничій дільниці — більші кількості, як правило, обумовлюють більшу складність
• Рівень спеціалізації обладнання та приміщень (наприклад, установки повітря підготовки), наявний на виробничій дільниці — виробничі дільниці з низьким рівнем спеціалізації вважаються складнішими за інші дільниці
• Кількість персоналу на виробничій дільниці — більші кількості, як правило, обумовлюють більшу складність
• Кількість комерційних ринків/країн, до яких виробнича дільниця здійснює постачання — більші кількості, як правило, обумовлюють більшу складність
• Кількість постачальників для виробничої дільниці — більша кількість як правило, обумовлює більшу складність
• Якщо виробнича дільниця є контрактним виробником чи контрактною лабораторією,
вона вважається відносно складною
Загальними показниками складності процесів є:
• Стерильні та асептичні виробничі процеси завжди вважаються процесами високої складності.
• Операції з параметричного випуску — як правило, вважаються процесами високої складності.
• Кількість критичних етапів, що повинні бути під контролем в межах процесу — як правило, процеси з великою кількістю критичних етапів можуть вважатись складнішими процесами.
• Тип продукції, що виробляється — деякі типи продукції, наприклад, лікарські форми низької концентрації/сильнодіючі лікарські форми і лікарські форми з уповільненим вивільненням можуть бути складнішими з огляду виробництва ніж інші типи продукції (як наприклад, таблетки зі швидким вивільненням), і складність їх виробничого процесу повинна оцінюватись вище в такому випадку.
• Кількість типових операцій у нестерильному виробничому процесі — більші кількості, як правило, обумовлюють більшу складність.
• Операції з перепакування — перепакування вже запакованої серії може вважатись процесом від середньої до високої складності.
• Ступінь обробки чи переробки, що відбувається на виробничій дільниці: такі операції можуть додати складності процесу
• Біологічні процеси
• Ступінь субпідряду, що використовується виробничою дільницею, — значне використання контрактних виробників або контрактних лабораторій як правило, обумовлюють більшу складність.
Загальними показниками складності продукції є:
• Кількість компонентів, що складають будь-яку одиницю пакування продукту — більші кількості компонентів в одиниці пакування, як правило, обумовлюють більшу складність продукту. Наприклад, одиниця пакування ін’єкційного продукту може мати 4 компонента в своєму складі (флакон з ліофілізатом, флакон з розчинником, голкаперехідник і інструкція з застосування, в той час, як одиниця пакування таблетованого продукту може містити блістер і інструкцію з застосування).
• Продукти, що вимагають спеціальних умов зберігання та дистрибуції: (наприклад, управління продуктами холодового ланцюга і продуктами, що швидко псуються, як радіофармацевтичні препарати, може бути складним).
Критичність лікарських засобів
лікарський засіб критичного рівня ризику
лікарський засіб високого рівня ризику
лікарські засоби низького рівня ризику
2. Оцінювання ризику, пов’язаного з відповідністю вимогам (оцінювання виявлених порушень)
| Виявлені порушення протягом інспектування | Оцінка ризику, пов’язаного з відповідністю вимогам |
|---|---|
| 1 чи більше критичних порушень та/або шість та більше суттєвих порушень по різним процесам та лікарським засобам | високий |
| Від 1 до 5 суттєвих порушень | середній |
| Відсутність суттєвих та критичних порушень | низький |
3. Визначення категорії ризику виробничої дільниці
| Внутрішній ризик | |||
| Оцінка ризику, пов’язаного з відповідністю вимогам | високий | середній | низький |
| низький | A | А | В |
| середній | А | В | С |
| високий | В | С | С |
Рекомендації з оцінювання:
Оцінку А присвоюють виробничим дільницям з низьким загальним рівнем складності
Оцінку В присвоюють дільницям з середнім загальним рівнем складності
Оцінку С присвоюють дільницям з високим загальним рівнем складності
Примітка: при присвоєнні загальної оцінки складності слід обрати оцінку (А, В чи С), що найбільше відображає різні окремі оцінки складності, що були присвоєні складності дільниці, процесів та продукції. Це подібно до вирахування середнього значення з усіх окремих оцінок складності, що бути присвоєні.
У випадках, коли немає достатньо інформації чи знань про складність, що пов’язана з виробничою дільницею, її процесами та продукцією, слід присвоїти середню оцінку В.
4. Рекомендована періодичность інспекцій на виробничій дільниці
| Категорія ризику | Рекомендована періодичність інспектування |
|---|---|
| А | Скорочена періодичність, від 2 до 3 років |
| В | Середня періодичність, від 1 до 2 років |
| С | Підвищена періодичність, <1 року |
- Виробничі дільниці з категорію ризику «А» мають як мінімум одну оцінку низького ризику стосовно складності виробничої дільниці, її процесів та продукції або ризику порушення вимог. Під час планування наступних інспекцій такі виробничі дільниці можуть інспектуватись зі скороченою періодичністю, наприклад, із періодичністю менше ніж раз на два роки (наприклад, одна інспекція кожні 2,5 роки);
- Виробничі дільниці з категорією ризику «С» мають як мінімум одну оцінку високого ризику стосовно складності виробничої дільниці, її процесів та продукції або ризику порушення вимог. Під час планування наступних інспекцій такі виробничі дільниці можуть інспектуватись із підвищеною періодичністю, наприклад, як мінімум щорічно чи навіть частіше;
- Виробничі дільниці з категорією ризику «В» знаходяться між «А» та «С», і протягом планування наступних інспекцій такі виробничі дільниці можуть інспектуватись із середньою періодичністю, наприклад, у проміжок між 12 та 24 місяцями.
- Фактична періодичність інспектування, що призначена виробничій дільниці в межах будь-якої категорії ризику (A, B чи C), повинна відображати кількість та тип невідповідностей, що були виявлені протягом останньої інспекції. Наприклад, якщо двом виробничим дільницям присвоєно категорію ризику B, однак, якщо на одній виробничій дільниці результат останньої інспекції гірший за іншу виробничу дільницю (наприклад, п’ять суттєвих порушень у порівнянні з однією суттєвим порушенням) точна періодичність інспектування, призначена першій виробничій дільниці, повинна, як правило, бути у більш обмежувальному кінці часового діапазону (тобто періодичність інспекції ближче до одного року ніж до двох років);
- Крім того, періодичність інспектування, призначена виробничим дільницям з однаковими категоріями ризику може враховувати окремі оцінки внутрішнього ризику та ризику порушення вимог. Наприклад, коли виробнича дільниця має високий внутрішній ризик і високий ризик порушення вимог, що призведе до отримання категорії загального ризику С, призначена періодичність інспектування (наприклад, 9 місяців) може бути вищою за періодичність виробничої дільниці з високим внутрішнім ризиком, але середнім ризиком порушення вимог, що також призводить до отримання категорії загального ризику С.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим