МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ
НАКАЗ
від 22.01.2020 р. № 142
Про затвердження методичних рекомендацій із застосування Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02 жовтня 2013 року № 753, Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02 жовтня 2013 року № 754 та Технічного регламенту щодо активних медичних виробів, які імплантують, затвердженого постановою Кабінету Міністрів України від 02 жовтня 2013 року № 755
Відповідно до абзацу третього частини другої статті 8 Закону України «Про технічні регламенти та оцінку відповідності», пункту 1 постанови Кабінету Міністрів України від 16 грудня 2015 року № 1057 «Про визначення сфер діяльності, в яких центральні органи виконавчої влади здійснюють функції технічного регулювання», абзацу п`ятдесятого підпункту 10 пункту 4 та пункту 8 Положення про Міністерство охорони здоров’я України, затвердженого постановою Кабінету Міністрів України від 25 березня 2015 року № 267,
НАКАЗУЮ:
Затвердити такі, що додаються:
Методичні рекомендації «Класифікація медичних виробів»;
Методичні рекомендації «Медичні вироби. Допоміжні засоби. Виробник.»;
Методичні рекомендації «Однорідні партії»;
Методичні рекомендації «Субпідряд — зв’язок з системою управління якістю»;
Методичні рекомендації «Дата «використати до»;
Методичні рекомендації «Сфера застосування Технічного регламенту щодо активних медичних виробів, які імплантують, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 755»;
Методичні рекомендації «Уповноважений представник».
Контроль за виконанням цього наказу залишаю за собою.
Перший заступник МіністраА. Семиволос
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Класифікація медичних виробів»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.4/1, редакція 9, червень 2010 та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753.
Цей документ містить рекомендації для застосування правил класифікації медичних виробів, відповідно до додатку 2 до Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753 (далі — ТР щодо МВ).
Можливе відступлення від правил класифікації в рамках технічного прогресу або інформації, отриманої після виходу виробу на ринок.
| Зміст | Сторінки |
| 1. Цілі і принципи класифікації медичних виробів
2. Практичне значення класифікації 1) Загальні вимоги 2) Оцінка відповідності 3) Клінічне оцінювання та дослідження 4) Інструкція із застосування 5) Різне 3. Здійснення класифікації 1) Основні визначення 2) Застосування правил класифікації 3) Як користуватися правилами 4) Практичний приклад 5) Вирішення проблем інтерпретації 4. Роз’яснення спеціальних правил класифікації медичних виробів 1) Графічний перелік — схема вказівок з класифікації медичних виробів для первинної ідентифікації ймовірного класу виробу 2) Загальні роз’яснення правил, практичних питань, приклади |
3 3 3 3 4 4 4 4 5 7 8 8 9 9 9 1 7 |
1. Цілі і принципи класифікації медичних виробів
Піддавати усі загалом медичні вироби найсуворішим процедурам перевірки недоцільно економічно і невиправдано на практиці. Ступінчаста система контролю є більш доцільною. У такій системі рівень контролю відповідає рівню потенційної небезпеки, властивої типу виробу. Класифікація медичних виробів необхідна для визначення застосування відповідної процедури оцінки відповідності до медичних виробів.
З метою гарантування ефективного функціонування оцінки відповідності згідно з ТР щодо МВ виробники повинні бути в змозі визначити класифікацію свого виробу якомога раніше, на стадії проектування.
Класифікація медичних виробів заснована на вразливості людського тіла з урахуванням потенційних ризиків, пов’язаних з виробами. Цей підхід дозволяє використовувати набір критеріїв, які можна об’єднати в різний спосіб для визначення, наприклад, тривалості контакту з тілом, ступінь інвазивності і співвідношення локального і системного ефекту. Ці критерії можна застосовувати до широкого кола різних медичних виробів і технологій. Критерії класифікації медичних виробів викладені в додатку 2 до ТР щодо МВ.
Слід визнати, що, хоча існуючі правила будуть належним чином класифікувати переважну більшість виробів, все ж може бути важко класифікувати певну їх кількість. Такі випадки, зокрема, включають в себе вироби, що межують між двома різними класами. Крім того, існують вироби, які неможливо класифікувати згідно з діючими правилами через їх незвичайну природу або ситуації, коли класифікація призведе до неправильного рівня оцінки відповідності згідно з небезпекою виробу (див. також розділ 3.5).
2. Практичне значення класифікації
1) Загальні вимоги
Незалежно від класу виробу вони мають:
- відповідати основним вимогам, включаючи вимоги щодо надання інформації, яка буде поставлятися виробником (додаток 1 до ТР щодо МВ);
- підпадати під вимоги щодо надання звітності відповідно до системи контролю за медичними виробами;
- мати маркування знаком відповідності (за винятком виробів, виготовлених на замовлення і виробів, призначених для клінічних досліджень).
2) Оцінка відповідності
Класифікація медичних виробів впливатиме на хід оцінки відповідності, яку повинен здійснити виробник для нанесення маркування знаком відповідності на медичний виріб.
Технічна документація щодо виробів класу IIa і IIb, повинна бути розглянута органом з оцінки відповідності на основі програми представницької вибірки в контексті додатків 3, 6 і 7 до ТР щодо МВ.
| Процедури оцінки відповідності | Класи | |||||
|---|---|---|---|---|---|---|
| Додатки | I | I Стерильні | I вимір | IIa | IIb | III |
| 3 (+ перевірка проекту) | √ | |||||
| 3 (- перевірка проекту) | √ | √ | √ | √ | ||
| 4 | √ | √ | ||||
| 5 | √ | √ | √ | √ | √ | |
| 6 | √ | √ | √ | √ | √ | |
| 7 | √ | √ | √ | √ | ||
| 8 | √ | √ | √ | √ | ||
Клінічне оцінювання та дослідження
В рамках Основних вимог, клінічне оцінювання відповідно до додатку 10 проводиться щодо всіх медичних виробів.
У додатку 10 до ТР щодо МВ йдеться, що в якості загального правила підтвердження відповідності вимогам щодо характеристик і функціональних властивостей, при звичайних умовах використання виробу, оцінка небажаних побічних ефектів повинна базуватися на клінічних даних.
Крім цього, відповідно до додатку 10 до ТР щодо МВ клінічні дослідження виробів, що імплантують і виробів класу III повинні проводитися, крім випадків, коли належним чином обґрунтоване використання існуючих клінічних даних.
Клінічні дослідження виробів класу III, виробів, що імплантують і довготермінових інвазивних виробів, що відносяться до класу IIa і IIb, можуть бути розпочаті через 60 днів після повідомлення виробником компетентного органу, крім випадків, коли компетентний орган повідомив про протилежне рішення.
Інструкція із застосування
Інструкція із застосування не є обов’язковою для виробів класу I і IIa, якщо ці вироби можуть безпечно використовуватися без будь-яких інструкцій.
Різне
Виробник медичних виробів класу I і його відповідний уповноважений представник, призначений ним, повинен повідомити свою зареєстровану адресу і опис виробів компетентному органу.
3) Здійснення класифікації
Виробник повинен спочатку визначити, чи є продукт медичним виробом, або допоміжним засобом до такого медичного виробу.
Основні визначення
Правила класифікації базуються на різних умовах, таких як тривалість контакту з пацієнтом, рівень інвазивності і частина тіла, яка перебуває під впливом виробу.
Цільове призначення
Цільове призначення визначається виробником.
Тривалість
За тривалістю застосування медичні вироби поділяються на:
тимчасові — медичні вироби, призначені для безперервного застосування протягом не більш ніж 60 хвилин;
короткотермінові — медичні вироби, призначені для безперервного застосування протягом не більш ніж 30 днів;
довготермінові — медичні вироби, призначені для безперервного застосування протягом більш ніж 30 днів.
У певних випадках тривалість ефекту виробу слід розглядати як тривалість використання. Наприклад, нанесення крему для шкіри займає всього кілька секунд, але крем може залишатися на шкірі протягом багатьох годин. Тому тривалість використання не повинна розглядатися як час, необхідний для застосування виробу, а скоріше як час, протягом якого виріб досягає проставленої мети.
Поняття неперервного використання
При розрахунку тривалості безперервне використання встановлюють як час безперервного фактичного використання виробу за призначенням. Однак, коли використання виробу припиняється з метою заміни його подібним або ідентичним виробом, це слід розглядати як продовження постійного використання виробу.
Наприклад, скальпель може бути використаний на одному і тому ж пацієнті протягом операції, яка може тривати кілька годин. Безперервне використання за призначенням, тобто розрізання тканин, як правило, триває не більше декількох секунд. Тому скальпель є виробом тимчасового користування. Однак, коли використання виробу припиняється з метою заміни його подібним або ідентичним виробом (наприклад, заміна уретрального катетера), це слід розглядати як продовження постійного використання виробу.
Якщо не можна довести, що компоненти виробу були ліквідовані в період між використанням, то це вважається негайною заміною.
Інвазивність
Слід відзначити наступне:
Хірургічно створена стома, що використовується для уростомії, колостомії та ілеостомії або постійної трахеостомії, вважається отвором тіла. Тому вироби, що вводяться в такі стоми, не є хірургічно інвазивними. Хірургічно створений отвір для забезпечення доступу до системи кровообігу, навпаки, не слід вважати «отвором тіла». Вироби, введені в такий отвір, вважаються хірургічно інвазивними.
Виріб, який поставляє енергію до тіла, не слід розглядати як інвазивний, якщо тільки енергія проникає в тіло, а не сам виріб. Енергія сама по собі не є виробом, тому не може бути класифікована. Тільки виріб, що генерує енергію, повинен бути класифікований. Однак, якщо виріб вводить речовину, незалежно від того чи є ця речовина лікарським засобом або медичним виробом, така речовина повинна оцінюватися окремо (наприклад, речовини, що вводяться за допомогою струменевого інжектора).
Будь-який виріб, який повністю або частково проникає всередину тіла через природні отвори або через поверхні тіла, є інвазивним виробом. Хірургічно інвазивний виріб завжди передбачає проникнення через штучно створений отвір, такий як хірургічний розріз, або це може бути отвір, створений за допомогою голки. Тому хірургічні рукавички і голки, що використовуються зі шприцами, є хірургічно інвазивними.
Поняття хірургічно інвазивний слід розуміти також як такий, що охоплює рідини, які знаходяться в контакті з органами, тканинами або іншими частинами тіла, якщо доступ для таких рідин здійснюється через хірургічно створені отвори.
Вироби, що імплантуються
Одним з ключових елементів у визначенні виробу для імплантації є поняття «процедури». Таким чином, такі вироби повинні залишатися в організмі пацієнта після процедури. Під «процедурою» слід розуміти в цьому контексті хірургічну процедуру, в ході якої імплантат поміщається в тіло, і негайний післяопераційний догляд, пов’язаний з процедурою. Поняття «процедури» не поширюється на завершення терапевтичного лікування, наприклад, видалення імплантату має розглядатися як інша процедура. Таким чином, пластина, яка використовується для зменшення перелому кістки, являє собою імплантат, навіть якщо його потім виймають. У цьому випадку введення пластини і її вилучення — дві різні хірургічні операції.
Деякі вироби, що імплантуються частково, вважаються імплантатами. Наприклад, якщо здійснюються операції для введення порту для внутрішньовенних вливань, то такий порт буде залишатися в тілі протягом як мінімум 30 днів і, отже, буде імплантатом. Однак, безтунельний центральний венозний катетер, який використовується для тимчасового доступу до судин і призначений для вилучення після 7–10 днів — не є довготерміновим виробом для імплантації. Також і шов для закриття шкірних ран, який витягується протягом 30 днів, не вважається імплантатом.
Активні медичні вироби
Дія щодо перетворення енергії стосується перетворення енергії в виробі і/або перетворення на межі між виробом і тканинами або в тканинах.
Наприклад, електрод не є активним виробом за цією системою класифікації до тих пір, доки кількість енергії на вході і виході однакова. Наприклад, опір в проводі, який викликає незначні зміни між входом і виходом, не може вважатися складовою значної зміни. Проте, електроди, що використовуються в електрохірургії для розрізу тканини або припікання, є активними виробами, тому що їх робота залежить від енергії, що виробляється генератором, і їх дія досягається шляхом перетворення енергії на межі між пристроєм і тканиною або в тканинах. Електроди, призначені для ЕКГ або ЕЕГ, зазвичай не вважаються активними виробами тому, що вони, як правило, не працюють за рахунок перетворення енергії.
Застосування правил класифікації
Стосовно подальшої інтерпретації правил класифікації слід враховувати наступне.
Клас виробу визначає цільове призначення, а не конкретні технічні характеристики виробу, якщо вони не мають прямого відношення до призначення. Наприклад, введення допоміжної речовини, тканин тваринного походження і т.д.
Саме цільове, а не випадкове, призначення виробу визначає клас виробу. Наприклад, шовний органайзер, призначений для підтримки в порядку шовних ниток, що використовуються в операції на відкритому серці, не слід розглядати як інвазивний виріб, якщо в звичайному використанні він може перебувати поза пацієнтом. Подібним чином, якщо лікар використовує виріб в порядку, не передбаченому виробником, це не змінює клас виробу в рамках оцінки відповідності. Проте, якщо звичайне клінічне застосування виробу змінюється з плином розвитку клінічної практики, яка змінює цільове призначення і класифікацію виробу, то виробник повинен провести нову оцінку відповідності для цільового призначення.
Цільове призначення, присвоєне виробником виробу, а не клас, присвоєний подібним виробам, визначає клас виробу. Наприклад, два шва, що мають однаковий склад, можуть мати різне призначення.
В якості альтернативи класифікації системи в цілому, визначення класу певного виробу може здійснюватися по відношенню до найпростішої конфігурації, з урахуванням його власних функціональних особливостей. Виріб, який є частиною системи, наприклад, трубка набору для штучного кровообігу може бути класифікований як окремий виріб, а не як частина системи. Однак, в такому випадку виріб повинен мати власне маркування знаком відповідності.
Подібні комбіновані вироби з частинами, що мають різне функціональне призначення, можна аналізувати окремо по відношенню до кожної з цих частин. Наприклад, дренажний виріб буде мати інвазивну трубку і неінвазивний виріб для збору. Ці компоненти можуть бути класифіковані окремо, за умови, що вони також мають окреме маркування знаком відповідності.
Якщо виріб можна класифікувати за кількома правилами, то застосовується максимально можливий клас.
Якщо виріб не призначений для використання виключно або головним чином на певній частині тіла, він повинен розглядатися, виходячи з самого критичного спеціального використання. Класифікація повинна бути визначена на основі вимог, що містяться в інформації, що надається разом із виробом. Виробник повинен навести в якості мінімальної вимоги позитивні і негативні показання до застосування.
Для того, щоб виріб мав «цільове призначення», вказане в певному правилі класифікації, виробник повинен чітко вказати, що виріб призначений для такого спеціального використання та інформацію, що супроводжує виріб. В іншому випадку, буде вважатися, що він має цільове призначення, яке застосовується і прийняте в широкій медичній практиці.
Як користуватися правилами
Виробник повинен враховувати всі правила для встановлення належної класифікації свого виробу. Цілком можливо, наприклад, що одне із загальних правил, які не є специфічним для активних виробів, все одно відноситься до цього виробу. Всі характеристики виробів повинні прийматися до уваги. Характеристика або комбінація характеристик відповідно до цільового призначення виробу, що має найвищий клас, визначає клас виробу в цілому.
При відступі від правил класифікації, викладених у додатку 2 до ТР щодо МВ, виробники грудних імплантатів та замінників тазостегнового, колінного та плечового суглобів, а також медичних виробів, виготовлених з використанням тканин тваринного походження, застосовують більш жорстку класифікацію.
Практичний приклад
| Простий виріб для дренажу ран складається з трьох компонентів, які слід взяти до уваги: канюля, трубка і виріб для збору. Якщо виріб продається без канюлі, то класифікація канюлі не повинна братися до уваги. Крім того, передбачається, що зібрані рідини не призначені ні для повторного вливання в організм, ні для перероблення для можливого повторного вливання, і що виріб не призначений для підключення до всмоктувальної системи. | Правило/пункт ТР щодо МВ | Клас |
| Хірургічна інвазивна канюля для розміщення в рані в плевральній порожнині для дренажу порожнини. | 15 | IIa |
| Неінвазивна трубка для видалення рідин організму до збирача. | 9 | I |
| Неінвазивний збирач для надходження рідин організму. | 9 | I |
Однозначним висновком тут є те, що виробник матиме вибір в застосуванні класу ІІа до всього виробу або проведенні окремих процедур оцінки відповідності для канюлі з одного боку і трубки та збирача — з іншого.
Вирішення проблем інтерпретації
У разі, якщо виробник не впевнений, як класифікувати свої вироби, йому слід спочатку звернутися до органу з оцінки відповідності.
У разі, якщо залишилися якісь сумніви або непогодження з органом з оцінки відповідності, слід звернутися до відповідного компетентного органу в сфері технічного регулювання.
4) Роз’яснення спеціальних правил класифікації медичних виробів
Цей розділ починається з графічного переліку правил, як передмова до підрозділів спеціальних правил класифікації медичних виробів. Кожний підрозділ починається з загального пояснення правила з подальшим поданням правила у табличному вигляді та прикладів виробів, до яких вони застосовуються. Слід підкреслити, що якщо конкретний тип виробу наведено як приклад, це не означає, що такі вироби у всіх випадках відносяться до класу, наведеному у прикладі. Цілком можливо, що деякі виробники визначають до такого виробу зовсім інше призначення, ніж те, яке було використане в контексті виробу.
Графічний перелік — схема вказівок з класифікації медичних виробів для первинної ідентифікації ймовірного класу виробу
Примітка: Завжди перевіряйте точну класифікацію, уважно прочитавши всі правила, та використовуйте додаткову допомогу у вигляді цього керівного документа, як це передбачено в формі загальних пояснень правил та прикладів виробів (див. пункт 4 підпункт 2).
| ТЕМИ |
| Неінвазивні вироби — Правила/пункти 9, 10, 11, 12 ТР щодо МВ
Інвазивні вироби — Правила/пункти 13, 14, 15, 16 ТР щодо МВ Активні вироби — Правила/пункти 17, 18, 19, 20 ТР щодо МВ Спеціальні правила — Правила/пункти 21, 22, 23, 24, 25, 26 ТР щодо МВ |
Загальні роз’яснення правил, практичних питань, приклади
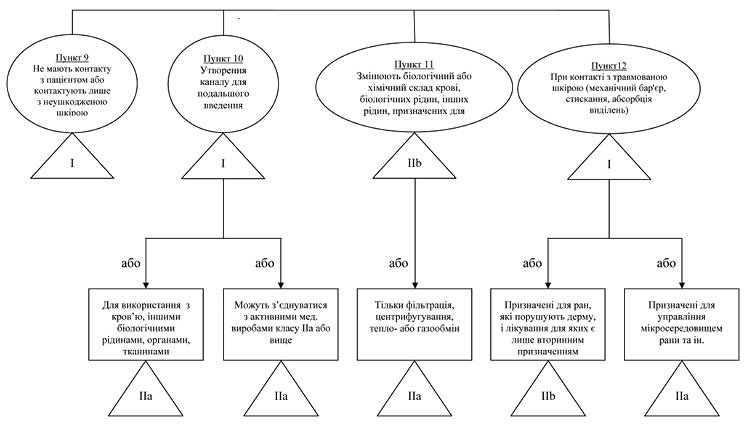
Правило/пункт 9 — Вироби, що не мають контакту з пацієнтом або контактують лише з неушкодженою шкірою.
Загальне пояснення правила
Це правило, що застосовується до всіх виробів, які не охоплюються більш вузьким правилом.
Це правило застосовується в цілому до виробів, що вступають в контакт лише з неушкодженою шкірою або не мають контакту з пацієнтом.
| Правило/пункт 9 | Приклади |
|---|---|
| Всі неінвазивні вироби відносяться до Класу I, якщо не можна застосувати одне з викладених нижче правил. | – Вироби для збору рідин організму призначені для використання без зворотного потоку рідин (наприклад, для збору відходів організму, такі як пляшки для збору сечі, стомні мішки, урологічні прокладки або пакети, що використовуються з виробами для дренажу ран). Вони можуть бути підключені до пацієнта катетерами або трубками
– Вироби, що використовуються для знерухомлення частин тіла та/або застосування сили до них чи стискання (наприклад, нестерильні пов’язки для допомоги лікування розтягнень, гіпс, шийні коміри, вироби для витягування, компресійний трикотаж) – Вироби, призначені в основному для зовнішньої підтримки пацієнта (наприклад, лікарняні ліжка, підйомники для пацієнтів, милиці, інвалідні візки, носилки, стоматологічні крісла для пацієнтів) – Коригувальні окуляри та оправи – Стетоскопи для діагностики – Пластирі для ока – Тканина для закриття ран – Гелі-провідники – Неінвазині електроди (електроди для ЕКГ або ЕЕГ) – Екрани для покращення зображення – Постійні магніти для видалення очного бруду |
Практичні проблеми класифікації
Деякі неінвазивні вироби непрямо контактують з тілом і можуть впливати на внутрішній фізіологічний процес шляхом зберігання, передачі або лікування крові, інших рідин організму або рідин, що повернуті або введені в організм, або шляхом генерування енергії, що надходить до організму. Вони повинні бути виключені зі сфери застосування цього Правила і застосовуватися відповідно до іншого правила через небезпеку, властиву таким непрямим контактам.
Правило/пункт 10 — Переливання або зберігання для подальшого введення
Загальне пояснення правила
Ці види виробів необхідно розглядати окремо від безконтактних виробів правила/пункту 9, адже вони можуть бути непрямо інвазивними. Вони направляють або зберігають речовини, які в подальшому будуть введені в організм. Як правило, такі вироби використовуються при переливанні, вливанні, штучному кровообігу і введенні анестезуючих газів та кисню.
У деяких випадках вироби, що підпадають під це правило, — це дуже прості вироби для введення.
| Правило/пункт 10 | Приклади |
|---|---|
| Усі неінвазивні медичні вироби, призначені для переливання або зберігання крові, рідин і тканин тіла, а також рідин або газів з метою подальшої інфузії, застосування або введення в організм, відносяться до класу IIа:
якщо вони можуть бути під’єднані до активних медичних виробів класу IIа або вищого класу; |
– Вироби, призначені для використання в якості каналів в активних системах введення ліків, наприклад, трубки, що використовуються з інфузійними насосами.
– Вироби, що використовуються для направлення, наприклад, антистатичні трубки для анестезії, анестезійних дихальних контурів, індикатору тиску, виробів обмеження тиску. – Шприци для інфузійних насосів. |
| якщо вони призначені для зберігання або переливання крові чи інших рідин тіла або для зберігання органів, частин органів або тканин організму. | – Вироби, призначені для направлення крові (наприклад, при переливанні, штучному кровообігу).
– Вироби, призначені для тимчасового зберігання або транспортування органів для трансплантації (тобто контейнери, сумки та аналогічні вироби). – Вироби, призначені для довгострокового зберігання біологічних речовин та тканин, таких як роговиця, сперма, ембріони людини і т.д. (наприклад, контейнери, сумки та аналогічні вироби). – Холодильники, спеціально призначені для зберігання крові, тканин тощо. |
| У всіх інших випадках зазначені вироби відносяться до класу I. | – Вироби, що забезпечують просту функцію направлення за допомогою гравітації, що забезпечує транспортування рідини, наприклад, введення набору для інфузії.
– Вироби, призначені для тимчасового утримання або зберігання, наприклад, чашки і ложки, спеціально призначені для введення ліків.- Шприци без голок. |
Практичні проблеми класифікації
Мішки з кров’ю як виняток підпадають під окреме правило (див. Правило/пункт 26).
Якщо виріб, наприклад, трубки, можна використовувати для цілей, що призведуть до підключення його до активного виробу, такий виріб автоматично перейде до Класу IIa, якщо виробник не вказує, що його не слід підключати до активного виробу Класу IIa та вище.
Пояснення спеціальних понять
Примітка: «Може бути підключений до активного виробу».
Таке підключення існує між неактивним виробом та активним виробом, коли неактивний виріб утворює зв’язок при передачі речовини між пацієнтом та активним виробом, та один із виробів контролює безпеку і роботу іншого. Наприклад, це відноситься до трубки в системі штучного кровообігу, що знаходиться нижче насоса для крові та контуру кровотоку, але не має прямого контакту з насосом.
Правило/пункт 11 — Неінвазивні вироби, що змінюють біологічний або хімічний склад крові, рідин організму або інших рідин, призначених для введення в організм.
Загальне пояснення правила
Ці типи виробів слід розглядати окремо від безконтактних виробів Правила/пункт 9 тому, що вони є непрямо інвазивними. Це правило поширюється, головним чином, на більш складні елементи наборів для штучного кровообігу, систем діалізу та систем автотрансфузії, а також на вироби для штучної обробки рідин організму, що мають бути введені до організму, коли пацієнт не знаходиться в замкнутій системі з виробом.
| Правило/пункт 11 | Приклади |
|---|---|
| Усі неінвазивні медичні вироби, призначені для зміни біологічного або хімічного складу крові, інших рідин тіла або інших рідин, призначених для переливання в тіло, відносяться до класу IIб. | – Вироби, призначені для видалення небажаних речовин з крові за допомогою обміну розчинів, такі як виріб для гемодіалізу.
– Вироби, призначені для поділу клітин за допомогою фізичних засобів, наприклад, градієнтне середовище для поділу сперми. – Концентрати для гемодіалізу. |
| У разі коли лікування проходить шляхом фільтрації, центрифугування, газо- або теплообміну, зазначені вироби відносяться до класу IIа. | – Часткова фільтрація крові в системі штучного кровообігу. Використовуються для видалення частинок і емболій з крові.
– Центрифугування крові для підготовки його до переливання або автотрансфузії. – Видалення двоокису вуглецю з крові та/або додавання кисню. – Нагрівання або охолодження крові в системі штучного кровообігу. |
Практичні проблеми класифікації
Ці вироби зазвичай використовуються в поєднанні з активним медичним виробом відповідно до Правила/пункт 17 або Правила/пункт 19.
Фільтрацію та центрифугування в контексті цього правила слід розуміти як виключно механічні методи.
Правило/пункт 12 — Неінвазивні вироби, що контактують з ушкодженою шкірою
Загальне пояснення правила
Це правило в першу чергу поширюється на пов’язки для ран незалежно від глибини рани. Дія традиційних типів продукції, таких як ті, що використовуються в якості механічного бар’єру, добре зрозуміла та ці вироби не призводять до великої небезпеки. Можна споглядати швидкий технологічний розвиток в цій сфері, з появою нових видів пов’язок для ран, стосовно яких робляться нетрадиційні твердження, наприклад, управління мікросередовищем рани, покращення природного процесу загоєння.
Більш амбітні твердження стосуються механізму загоєння, що з’явився як вторинний намір, такий як вплив на основні механізми гранулювання або формування епітелію. Деякі вироби, що використовуються на порушеній дермі, можуть навіть мати за мету підтримку або порятунок життя, наприклад, коли присутня повна руйнація великої площі шкіри або систематичний ефект.
Вироби, що містять лікарські засоби, які діють як допоміжні речовини до виробів, підпадають під Клас III.
| Правило/пункт 12 | Приклади |
|---|---|
| Усі неінвазивні медичні вироби, що вступають у контакт з пошкодженою шкірою:
– відносяться до класу I — якщо призначені для використання як механічний бар’єр для компресії або для абсорбції ексудатів; |
– Ранові пов’язки, такі як: поглинаючі прокладки, вата, ранові смужки, пластирі (лейкопластирі) та марлеві пов’язки, що діють як бар’єр, підтримують рану або поглинають ексудати з рани |
| – відносяться до класу IIб — якщо призначені для використання переважно під час лікування ран, проникають під шкіру та лікування для яких є лише вторинним призначенням; | – Головним чином призначені для використання на серйозних ранах зі значним і широким порушенням дерми, і де процес загоєння може бути тільки вторинним наміром, такі як:
– пов’язки для хронічних ран з виразками; – пов’язки для важких опіків з обширно порушеною дермою; – пов’язки для серйозних пролежнів; – пов’язки, що поєднують засоби збільшення тканини та забезпечення тимчасової заміни шкіри; |
| – відносяться до класу IIа — у всіх інших випадках (зокрема медичні вироби, призначені переважно для управління мікросередовищем рани). | – Мають специфічні властивості, призначені для сприяння процесу загоєння, контролюючи рівень вологості в рані під час процесу загоєння і в цілому для регулювання вологості та температури середовища, рівнів кисню та інших газів і рівню pH, або шляхом впливу на процес іншими фізичними засобами.
– Ці вироби можуть мати специфічні властивості загоєння, які при цьому не призначені для великих ран, що потребують загоєння. – Лейкопластирі для місцевого застосування. – Полімерні пов’язки. – Гідрогелеві пов’язки. – Просочені марлеві пов’язки без лікарських засобів. |
Практичні проблеми класифікації
Продукти, охоплені цим правилом, чутливі до класифікації, наприклад, полімерна пов’язка буде відноситись до Класу IIa, якщо її призначення — управління мікросередовищем рани, або до Класу I, якщо її призначення обмежене збереженням інвазивної канюлі в рані. Отже, не можна стверджувати, апріорі, конкретний клас пов’язки, не знаючи її призначення, визначеного виробником. Більшість пов’язок класу IIa чи IIb, також виконують функції Класу I, наприклад, в якості механічного бар’єру. Такі вироби класифікуються відповідно до призначення за вищим класом.
Для виробів, що поєднують лікарський засіб або похідні людської крові, потрібно керуватися Правилом/пунктом 21, або для тих, що поєднують тканини тваринного походження або нежиттєздатні похідні — керуватися Правилом/пунктом 25.
Пояснення:
– Пошкоджена дерма: рана розкриває принаймні частково підшкірну тканину.
– Вторинний намір: рана загоюється спочатку шляхом заповнення грануляційною тканиною, а потім епітелій відростає поверх грануляційної тканини, і рана звужується. На відміну від первинного наміру, який передбачає стягування рани, наприклад, шляхом накладання швів, щоб рана загоїлася.
– Шкіру слід вважати «ушкодженою» внаслідок патологічних змін (наприклад, діабетичні виразки) чи зовнішніх факторів (наприклад, опіки).
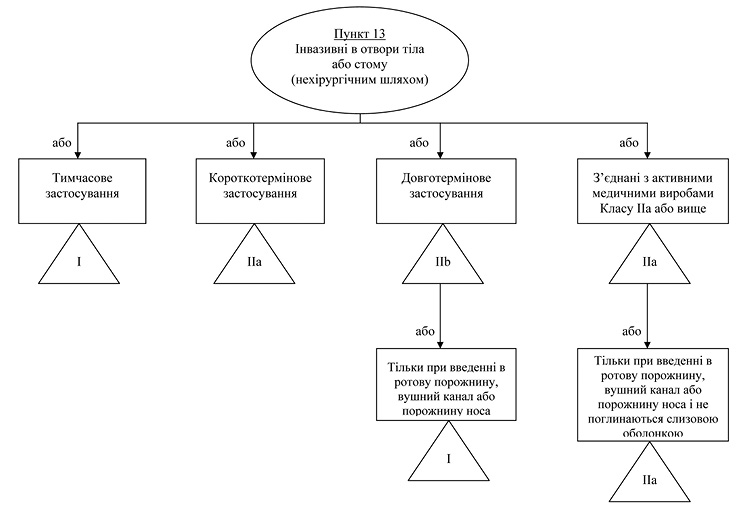
Правило/пункт 13 — Вироби, що вводяться в отвори тіла
Загальне пояснення правила
Інвазивність відносно отворів тіла (вухо, рот, ніс, очі, анус, уретра, піхва) слід розглядати окремо від проникнення через розріз на поверхні тіла (хірургічна інвазивність). Для довготермінового застосування слід виділити інвазивність відносно менш вразливих передніх частин вуха, рота і носа та інших анатомічних ділянок, що доступні через природні отвори тіла.
Хірургічно створену стому, що, наприклад, дозволяє видалити сечу або кал, також слід розглядати як отвори тіла.
Вироби, що підпадають під це правило, зазвичай є діагностичними та терапевтичними виробами, які використовуються вузькими спеціальностями (отоларингологія, офтальмологія, стоматологія, проктологія, урологія та гінекологія).
| Правило/пункт 13 | Приклади |
|---|---|
| Усі інвазивні медичні вироби, призначені для введення в отвори тіла, крім хірургічних інвазивних медичних виробів, що не призначені для підключення до активних медичних виробів або що призначені для підключення до активних медичних виробів класу I: | |
| – відносяться до класу I — якщо призначені для тимчасового застосування; | – Дзеркала, що використовуються в стоматології для стоматологічної діагностики та хірургії.
– Матеріали для зубних зліпків. – Трубки для шлунку. – Відтиски. – Вироби для клізми. – Оглядові рукавички. – Сечовий катетер, призначений для тимчасового застосування. – Дилатаційний катетер для передміхурової залози. |
| – відносяться до класу IIа — якщо призначені для короткотермінового застосування. | – Короткотермінові коригувальні контактні лінзи.
– Трахейні трубки. – Стенти. – Вагінальні песарії. – Постійні сечові катетери, призначені для короткотермінового застосування. |
| У разі застосування у ротовій порожнині, носоглотці, у слуховому проході до барабанної перетинки або в носовій порожнині такі вироби відносяться до класу I; | – Перев’язувальні матеріали для носових кровотеч
– Матеріали для виготовлення зубних протезів. |
| – відносяться до класу IIб — якщо призначені для довготривалого застосування. | – Уретральні стенти.
– Довготермінові коригувальні контактні лінзи. – Трахейні канюлі. – Сечові катетери, призначені для довготривалого застосування. |
| У разі коли такі вироби використовуються у ротовій порожнині, носоглотці, у слуховому проході до барабанної перетинки або в носовій порожнині і не призначені для поглинання слизовою оболонкою, вони відносяться до класу IIа. | – Ортодонтичні проводи.
– Фіксовані зубні протези. – Герметики для фіссур. |
| Усі інвазивні медичні вироби, призначені для введення в отвори тіла (крім хірургічних інвазивних медичних виробів) з підключенням до активних медичних виробів класу IIа або більш високого класу, відносяться до класу IIа. | – Трахеостомічні або трахейні трубки, підключені до апарату штучної вентиляції легенів.
– Аналізатори крові, поміщені під повіки. – Носові іригатори. – Носоглоткові дихальні шляхи. – Волоконна оптика в ендоскопах, з’єднана з хірургічними лазерами. – Дренажні катетери або трубки для дренування шлунка. – Стоматологічні аспіраторні наконечники. |
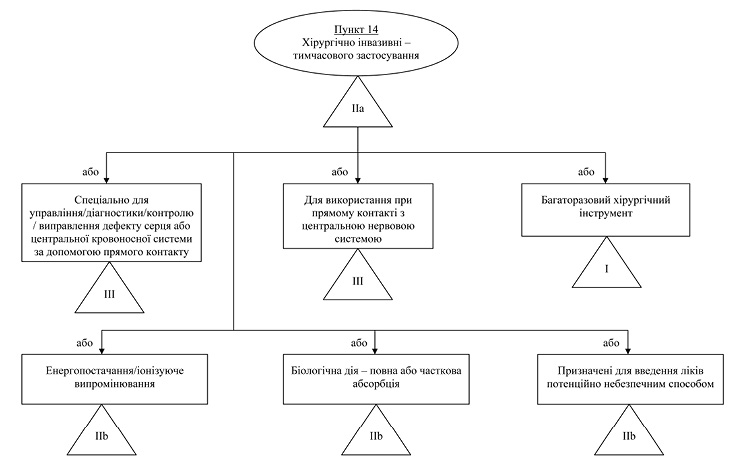
Правило/пункт 14 — Хірургічно інвазивні вироби, призначені для тимчасового використання (<60 хвилин)
Загальне пояснення правила
Це правило, в першу чергу, поширюється на три основні групи виробів: вироби для створення провідника через шкіру (голки, канюлі тощо), хірургічні інструменти (скальпелі, пили тощо) та різні типи катетерів, дренажів тощо.
| Правило/пункт 14 | Приклади |
|---|---|
| Усі хірургічні інвазивні медичні вироби, призначені для тимчасового застосування, відносяться до класу IIа, крім виробів, що: | – Голки для накладання швів.
– Голки шприців. – Ланцети. – Одноразові скальпелі і одноразові леза скальпелів. – Допоміжні вироби в офтальмологічній хірургії. – Степлери. – Хірургічні тампони. – Свердла, підключені до активних виробів. – Хірургічні рукавички. – Тестер штучних клапанів серця. – Тампони для зразків ексудатів. – Одноразові аортальні трансплантати (див. примітку 2). |
| – призначені спеціально для контролю, діагностики, моніторингу або корекції пороків2 серця або центральної системи кровообігу1 шляхом прямого контакту з цими частинами тіла. Такі вироби відносяться до класу III3 | – Серцево-судинні катетери (наприклад, катетери балонної ангіопластики, катетери/системи для введення стента), в тому числі пов’язані дротові напрямувачі катетера, інкубатори та одноразові4 серцево-судинні хірургічні інструменти, наприклад, електрофізіологічні катетери, електроди для електрофізіологічної діагностики та абляції.
– Катетери, що містять або з’єднують герметичне джерело радіоізотопів, коли радіоактивний ізотоп не призначений для вивільнення в організм при використанні в центральній системі кровообігу. |
| – багаторазові хірургічні інструменти, в цьому випадку вони відносяться до Класу I3 | – Скальпелі та ручки скальпелів.
– Розвертки. – Свердла. – Пили, що не призначені для підключення до активного виробу. – Ретракторні щипці, екскаватори та зубила. – Ретрактори для грудини для тимчасового використання. |
| – призначені спеціально для застосування в безпосередньому контакті з центральною нервовою системою. Такі вироби відносяться до класу III; | – Нейроендоскопи.
– Мозкові шпателі. – Канюлі для прямої стимуляції. – Ретрактори спинного мозку. – Спинальні голки. |
| – призначені для передачі енергії у вигляді іонізуючого випромінювання. Такі вироби відносяться до класу IIб; | – Катетери, що містять або з’єднують герметичне джерело радіоізотопів коли радіоактивний ізотоп не призначений для вивільнення в організм при використанні в системі кровообігу, за винятком центральної системи. |
| – призначені для біологічного впливу5 або для повного чи часткового поглинання6. Такі вироби відносяться до класу IIб; | |
| – призначені для введення лікувальних засобів за допомогою системи доставки, якщо це виконується у спосіб, що є потенційно небезпечним7 з урахуванням методу застосування. Такі вироби відносяться до класу IIб. | – Вироби для повторного самостійного застосування, коли рівні та характер медичного виробу є критичними, наприклад, шприц-ручки з інсуліном. |
Пояснення спеціальних понять
Примітка 1: Хірургічні інструменти, з’єднані з активним медичним виробом, не слід вважати «багаторазовим хірургічним інструментом».
Примітка 2: Вироби для виправлення дефекту не є виробами, що використовуються як допоміжні в кардіохірургічних операціях, наприклад, затискачі, перфоратор аорти. Перший абзац цього правила не поширюється на перфоратори аорти та аналогічні ріжучі інструменти, що функціонують подібно скальпелю.
Примітка 3: Хірургічні інструменти, спеціально не призначені для цілей, описаних в першому абзаці, і незалежно від місця застосування, відносяться до Класу IIa, якщо вони призначені для одноразового використання, та до Класу I, якщо вони є багаторазовими.
Примітка 4: Призначений означає, що цільове призначення виробу або допоміжного виробу — контролювати, діагностувати, спостерігати або коригувати дефект серця або центральної кровоносної системи.
Примітка 5: Біологічна дія: Всі матеріали і вироби можуть впливати на тканини після застосування в хірургічно інвазивній операції. Вважається, що матеріал має біологічну дію, якщо він активно і навмисно викликає, змінює чи запобігає відповіді від тканин, оточених специфічними реакціями на молекулярному рівні. Такий виріб можна віднести до біологічно активних.
Примітка 6: Поглинаються повністю або частково: термін абсорбції (поглинання) відноситься до розпаду матеріалу всередині організму та метаболічного усунення продуктів розпаду з організму.
Примітка 7: Поняття «потенційно небезпечний спосіб» пов’язане з характеристиками виробу, а не уміннями користувача.
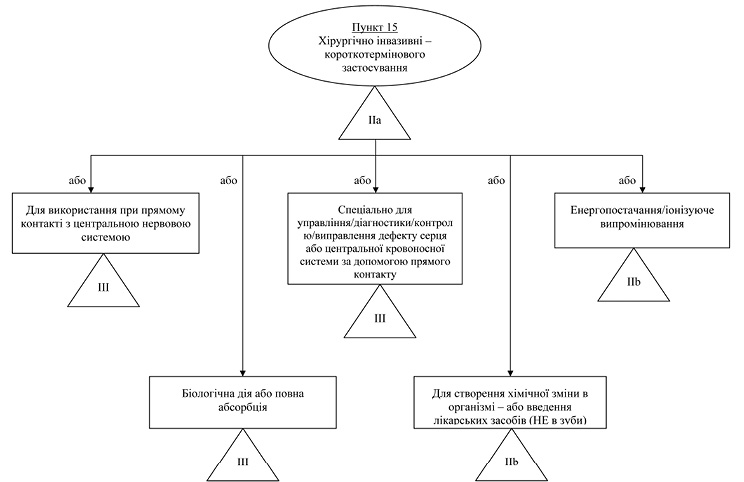
Правило/пункт 15 — Хірургічно інвазивні вироби для короткотермінового використання (>60 хвилин, <30 днів)
Загальне пояснення правила
В основному, ці вироби використовуються під час операції або в період післяопераційного догляду (наприклад, затискачі, дренажі), використання інфузійних виробів (канюлі, голки) та катетерів різних типів.
| Правило/пункт 15 | Приклади |
|---|---|
| Усі хірургічні інвазивні медичні вироби, призначені для короткотермінового застосування, відносяться до класу IIа, крім виробів, що призначені: | – Затискачі.
– Інфузійні канюлі. – Вироби для закриття шкіри. – Тимчасові стоматологічні матеріали. – Стабілізатори2 тканини, що використовуються в кардіохірургії. |
| – спеціально для контролю, діагностики, моніторингу або корекції пороків2 серця або центральної системи кровообігу шляхом прямого контакту з цими частинами тіла. Такі вироби відносяться до класу III; | – Серцево-судинні катетери.
– Серцеві зонди. – Дроти тимчасового кардіостимулятора. – Шунти сонної артерії. – Аблаційний катетер. |
| – спеціально для використання в прямому контакті з центральною нервовою системою. Такі вироби відносяться до класу III; | – Неврологічні катетери.
– Кортикальні електроди. |
| – для передачі енергії у вигляді іонізуючого випромінювання. Такі вироби відносяться до класу IIб; | – Вироби для брахітерапії. |
| – для здійснення біологічного впливу або для повного або часткового поглинання. Такі вироби відносяться до класу III; | – Шви, що розсмоктуються.
– Біологічні клеї. |
| – для проведення хімічних змін у тілі (крім випадків, коли медичні вироби розташовані в зубах) або для введення лікарських засобів1. Такі вироби відносяться до класу IIб. | – Клеї. |
Практичні проблеми класифікації
Примітка 1: Введення лікарських засобів також включає показники зберігання та/або впливу на обсяг та темп введення ліків. Імплантовані капсули для повільного введення ліків є лікарськими засобами, а не медичними виробами.
Примітка 2: Вироби для виправлення дефекту не є виробами, що використовуються як допоміжні в кардіохірургії, наприклад, стабілізатори тканини.
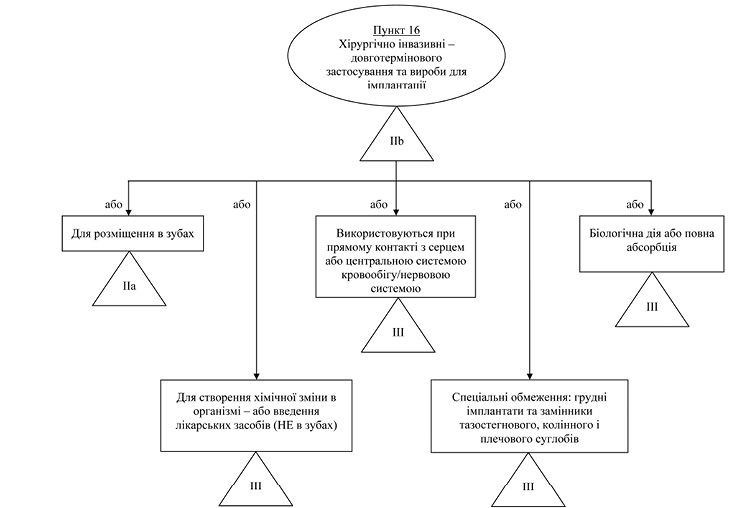
Правило/пункт 16 — Вироби для імплантації та хірургічно інвазивні вироби довготермінового використання (>30 днів)
Загальне пояснення правила
В основному, це ортопедичні, стоматологічні, офтальмологічні, серцево-судинні імплантати, а також імплантати м’яких тканин, що використовуються, наприклад, в пластичній хірургії.
| Правило/пункт 16 | Приклади |
|---|---|
| Усі медичні вироби, які імплантуються, а також хірургічні інвазивні медичні вироби довготривалого застосування відносяться до класу IIб, крім виробів, що призначені для: | – Ортопедичні замінники суглобів, крім імплантованих складових частин систем загальної заміни суглоба, які призначені для забезпечення функції, подібної до функції природного кульшового суглоба, природного колінного суглоба або природного плечового суглоба.
– Зв’язки. – Шунти. – Стенти і клапани (наприклад, легеневі). – Внутрішньоочні лінзи. – Імплантати для нарощення тканини. – Периферичні судинні катетери. – Периферійні судинні протези та стенти. – Імплантати для статевого органу. – Шви, що не розсмоктуються, кістковий цемент та щелепно-лицеві імплантати, в’язкопружні хірургічні вироби, призначені спеціально для зовнішніх офтальмологічних хірургічних операцій 1. |
| – розташування в зубах3. Такі вироби відносяться до класу IIа; | – Мости і коронки.
– Стоматологічні пломбувальні матеріали та штифти. – Стоматологічні сплави, кераміка та полімери. |
| – використання в безпосередньому контакті з серцем, центральною системою кровообігу або центральною нервовою системою. Такі вироби відносяться до класу III; | – Протези клапанів серця.
– Кліпси для аневризмів. – Судинні протези та стенти. – Центрально-судинні катетери. – Стенти для спинного хребта. – Електроди для ЦНС. – Серцево-судинні шви. – Постійні та тимчасові фільтри порожнистої вени. – Вироби для оклюзії – Внутрішньоаортальний балон-насос. – Допоміжні вироби роботи зовнішнього лівого шлуночка. |
| – здійснення біологічного впливу або повного чи часткового поглинання. Такі вироби відносяться до класу III; | – Шви, що розсмоктуються |
| – проведення хімічних змін4 у тілі (крім випадків, коли медичні вироби розташовані в зубах) або введення лікарських засобів1. Такі вироби відносяться до класу III. | – Акумуляторні системи введення неактивних лікарських засобів. |
| випадки відступів від цих правил, вироби слід відносити до класу III | – Грудні імплантати |
| випадки відступів від правил, вироби слід відносити до класу III | – Системи повної заміни стегнового, колінного та плечового суглобів і компоненти системи1. |
Практичні проблеми класифікації
Примітка 1: Ці продукти є імплантатами, тому що за нормальних умов значна кількість речовини залишається на місті операції після процедури. Якщо такі вироби містять тваринні тканини або їх похідні, на них поширюється Правило/пункт 25.
Примітка 2: Для закриття артеріотомії в периферичній судинній системі.
Примітка 3: Імплантати без біоактивних поверхонь, призначені для захисту зубів або протезів верхньо- чи нижньощелепної кістки, відносяться до Класу IIb та підпорядковуються загальному правилу. Гідроксиапатит вважається таким, що має біологічну дію, тільки якщо це стверджується та демонструється виробником.
Примітка 4: Пункт про хімічні зміни за цим правилом не відноситься до таких продуктів як кістковий цемент, коли хімічна зміна відбувається під час розміщення і не є довготерміновою.
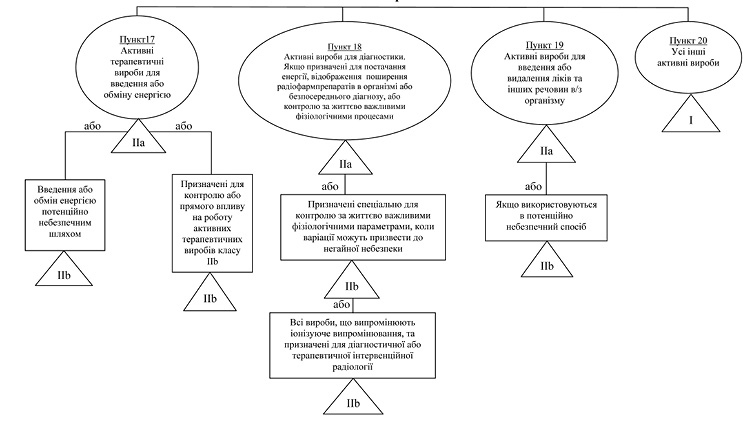
Правило/пункт 17 — Активні терапевтичні вироби для введення або обміну енергії
Загальне пояснення правила
Вироби, класифіковані за цим правилом, в основному, електрообладнання, що використовується в хірургії, таке як лазери та хірургічні генератори. Крім того, існують вироби для спеціалізованого лікування, такі як променева терапія. До іншої категорії відносяться вироби для стимуляції, проте, не всі вони можуть вважатися такими, що мають небезпечний рівень енергії, який надходить до тканини.
| Правило/пункт 17 | Приклади |
|---|---|
| Усі активні медичні вироби для терапії, призначені для передачі або обміну енергією, відносяться до класу IIа, | Електрична та/або магнітна та електромагнітна енергія:
– М’язові стимулятори. – Зовнішні стимулятори росту кісток – Очні електромагніти. – Електрична акупунктура. Термальна енергія: – Обладнання для кріохірургії. – Теплообмінники, за винятком типів, описаних нижче. Механічна енергія: – Підключені дерматоми. – Підключені свердла. – Стоматологічні наконечники. Світло: – Фототерапія для догляду за шкірою і для догляду за новонародженими. Звук: – Слухові апарати. Ультразвук: – Обладнання для фізіотерапії. |
| крім виробів, характеристики яких дають змогу подавати енергію до людського тіла або здійснювати енергообмін в потенційно небезпечний спосіб1, з урахуванням властивостей, інтенсивності та місця застосування енергії. У такому разі вироби відносяться до класу IIб. | Кінетична енергія:
– Апарати для штучної вентиляції легенів – Інкубатори для новонароджених – Ковдри для зігрівання – Підігрівачі крові – Теплообмінники з електричним приводом (наприклад, такі, що використовуються для пацієнтів, що не мають можливості відчувати) Електроенергія: – Високочастотні хірургічні генератори і електрокоагуляційне обладнання, в тому числі їхні електроди. – Зовнішні кардіостимулятори та дефібрилятори. – Обладнання для електросудомної терапії. Когерентне світло: – Хірургічні лазери. Ультразвук: – Літотріптери, хірургічні ультразвукові вироби. Іонізуюче випромінювання: – Радіоактивні джерела для терапії подальшого введення. – Терапевтичні циклотрони та лінійні прискорювачі. – Терапевтичні рентгенівські джерела. |
| Усі активні медичні вироби, призначені для контролю або моніторингу характеристик активних медичних виробів для терапії класу IIб або призначені для здійснення безпосереднього впливу на характеристики таких медичних виробів, відносяться до класу IIб. | – Зовнішні системи зворотного зв’язку для активних терапевтичних виробів.
– Системи контролю для подальшого введення. |
Пояснення спеціальних понять
Примітка 1: Поняття «потенційно небезпечний» залежить від типу технології та призначення виробу для пацієнта. Наприклад, всі вироби, призначені для іонізуючого випромінювання, всі апарати для штучної вентиляції легенів та літотріптери, слід відносити до Класу IIb.
Правило/пункт 18 — Активні вироби для діагностики
Загальне пояснення правила
В першу чергу поширюється на цілий ряд обладнання широкого використання в різних областях, наприклад, ультразвукова діагностика, забір фізіологічних сигналів та лікувально-діагностична радіологія.
| Правило/пункт 18 | Приклади |
|---|---|
| Активні медичні вироби, призначені для діагностики, відносяться до класу IIа:якщо вони призначені для передачі енергії, яка буде поглинатися людським тілом, крім медичних виробів, що використовуються для освітлення тіла споживача у видимій частині спектра; | – Магнітно-резонансне обладнання.
– Апарати для одонтодіагностики. – Стимулятори викликання реакції. – Діагностичний ультразвук. |
| – якщо вони призначені для отримання зображення розподілу радіофармацевтичних препаратів in vivo; | – Гамма камери.
– Позитронно-емісійна томографія та емісійно-комп’ютерна томографія. |
| – якщо вони призначені для прямого діагностування або моніторингу життєво важливих фізіологічних процесів1. | – Електрокардіографи.
– Електроенцефалографи. – Кардіоскопи з чи без показників пульсу. – Електронні термометри. – Електронні стетоскопи. – Електронне обладнання для вимірювання тиску. |
| – крім тих, що спеціально призначені для моніторингу життєво важливих фізіологічних параметрів, де властивість енергії є такою, що може створювати безпосередню небезпеку для споживача, зокрема призводити до змін серцевої діяльності, дихання, діяльності центральної нервової системи. Такі вироби відносяться до класу IIб. | – Вироби для інтенсивного моніторингу та сигналізації (наприклад, кров’яний тиск, температура, насичення крові киснем).
– Біологічні сенсори. – Аналізатори газів крові, що використовуються в операціях на відкритому серці. – Кардіоскопи. – Дихальний монітор, в тому числі дихальний монітор для домашнього використання. |
| Активні медичні вироби, призначені для випромінювання іонізуючої радіації3, а також для діагностичної та терапевтичної інтервенційної радіології, включаючи медичні вироби, призначені для контролю або моніторингу4 таких медичних виробів, або такі, що безпосередньо впливають на їх роботу, відносяться до класу IIб. | – Діагностичні рентгенівські джерела. |
Пояснення спеціальних понять
Примітка 1: Життєві фізіологічні процеси і параметри включають, наприклад, дихання, частоту серцевих скорочень, церебральні функції, склад газів крові, кров’яного тиску і температуру тіла. Медичні вироби, призначені для тривалого спостереження за життєво важливими фізіологічними процесами при анестезії, інтенсивній терапії або невідкладній медичній допомозі, відносяться до Класу IIb, в той час як медичні вироби, призначені для отримання показань життєвих фізіологічних сигналів при звичайних та самостійних оглядах, відносяться до Класу IIa. Тепловізійний виріб, призначений для моніторингу кровотоку, не вважається пристроєм для вимірювання температури.
Примітка 2: Вироби, спеціально призначені для моніторингу активних медичних виробів, які імплантують, підпадають під Технічний регламент щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
Примітка 3: Терапевтична інтервенційна радіологія відноситься до діагностики під час хірургічних операцій.
Примітка 4: Це відноситься до активних виробів для контролю, моніторингу або впливу на іонізуюче випромінювання, а не до подальшої обробки, запису чи розгляду отриманого зображення.
Правило/пункт 19 — Активні вироби, призначені для введення та/або видалення лікарських засобів, рідин організму чи інших речовин в або з організму
Загальне пояснення правила
Це правило, в першу чергу, поширюється на системи введення ліків та обладнання для анестезії.
| Правило/пункт 19 | Приклади |
|---|---|
| Усі активні медичні вироби, призначені для введення лікарських засобів, рідин тіла або інших речовин в тіло та/або видалення їх з тіла, відносяться до класу IIа. | – Вакуумне обладнання.
– Насос для годування. – Безголкові шприци для вакцинації. – Небулайзери для використання на притомних та спонтанно дихаючих пацієнтах, коли неможливість введення відповідної дози не є потенційно небезпечною. |
| У разі коли зазначене введення або видалення здійснюється у спосіб, що є потенційно небезпечним з урахуванням характеру застосованих речовин, відповідної частини тіла та способу застосування, такі медичні вироби відносяться до класу IIб. | – Інфузійні насоси.
– Вентилятори. – Апарати для анестезії. – Випарники для наркозу. – Обладнання для діалізу. – Насос для крові для апарату «серце-легені». – Гіпербаричні камери. – Регулятори тиску для медичних газів. – Медичні змішувачі газів. – Обмінники вологи в дихальних контурах при використанні на непритомних або спонтанно не дихаючих пацієнтах. – Небулайзери, коли неможливість введення відповідної дози може бути небезпечною. |
Правило/пункт 20 — Всі інші активні вироби
Загальне пояснення правила
Це правило є додатковим та поширюється на всі активні вироби, не охоплені попереднім правилом.
| Правило/пункт 20 | Приклади |
|---|---|
| Решта активних медичних виробів відносяться до класу I. | – Активні діагностичні вироби, призначені для освітлення тіла пацієнта у видимій області спектра, такі як ліхтарики для огляду, або для оптичного огляду тіла, такі як хірургічні мікроскопи.
– Вироби, призначені в основному для зовнішньої підтримки пацієнта (наприклад, лікарняні ліжка, підйомники пацієнта, інвалідні крісла, стоматологічні крісла). – Активні діагностичні вироби, призначені для термографії. – Стоматологічні світловідбивачі. |
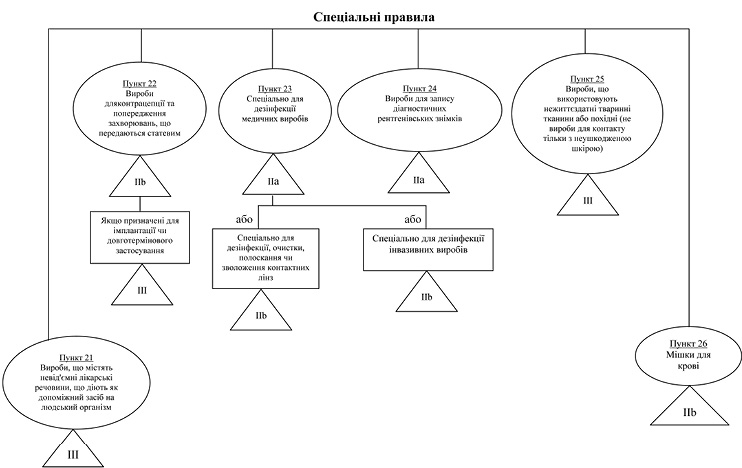
Спеціальні правила
Правило/пункт 21 — Вироби, що містять невід’ємні лікарські речовини або похідні людської крові
Загальне пояснення правила
Це правило поширюється на комбіновані вироби, що містять лікарську речовину для сприяння функціонуванню цього виробу. Проте, це правило не поширюється на ті вироби, що містять речовини, що можуть вважатися лікарськими речовинами, але включені у виріб виключно для підтримки певних характеристик виробу та не мають впливати на організм людини. Основна функція виробу не залежить від фармакологічної, метаболічної або імунологічної дії лікарського засобу.
| Правило/пункт 21 | Приклади |
|---|---|
| Усі медичні вироби, які містять як невід’ємну частину речовину, яка в разі її окремого використання може розглядатися як лікарський засіб згідно з визначенням, наведеним у Законі України «Про лікарські засоби», і дія якої на організм є допоміжною порівняно з дією медичного виробу, відносяться до класу III. | – Кістковий цемент з антибіотиками.
– Презервативи зі сперміцидами. – Катетери з гепариновим покриттям. – Ендодонтичні матеріали з антибіотиками. – Офтальмологічні іригаційні розчини, призначені для іригації, що містять компоненти для підтримки метаболізму ендотеліальних клітин рогівки. – Перев’язувальні матеріали, що містять протимікробні засоби, коли мета такого засобу — допоміжна дія стосовно рани. – Протизаплідні внутрішньоматкові спіралі, що містять мідь або срібло. – Стенти з лікарськими засобами, наприклад, коронарні, легеневі. |
| Усі медичні вироби, які містять як невід’ємну частину похідні крові людини, відносяться до класу III. | – Хірургічні герметики, що містять сироватковий альбумін людини. |
Примітка 1: «Невід’ємна частина» означає, що виріб та лікарська речовина фізично або хімічно поєднані під час введення (використання, імплантації, застосування тощо) пацієнту.
Правило/пункт 22 — Вироби для контрацепції та попередження захворювань, що передаються статевим шляхом
Загальне пояснення правила
Вироби призначені для використання в окремих випадках людської вразливості, на яку не поширюється загальні критерії часу, інвазивності та функцій органів.
Хоча це правило поширюється на два дуже різні способи застосування виробів, деякі вироби можуть виконувати обидві функції, наприклад, презервативи. На вироби, призначені для запобігання передачі ВІЛ статевим шляхом, також поширюється це правило.
| Правило/пункт 22 | Приклади |
|---|---|
| Усі медичні вироби, які використовуються для контрацепції та профілактики передачі венеричних захворювань, відносяться до класу IIб. | – Презервативи.
– Контрацептивні діафрагми. |
| Медичні вироби, які імплантуються, або інвазивні медичні вироби довготермінового застосування, які використовуються із зазначеною метою, відносяться до класу III. | – Протизаплідні внутрішньоматкові спіралі1. |
Примітка 1: Внутрішньоматкові контрацептиви, основною метою яких є виділення прогестинів, не є медичними виробами
Правило/пункт 23 — Спеціально для дезінфекції, очистки та промивання виробів
Загальне пояснення правила
Це правило, в першу чергу, поширюється на різні розчини для контактних лінз. Воно також охоплює речовини та інше обладнання, що використовується в основному в медичному середовищі для дезінфекції медичних виробів.
| Правило/пункт 23 | Приклади |
|---|---|
| Усі медичні вироби, спеціально призначені для дезінфекції, очищення, промивання або в разі потреби — зволоження контактних лінз, відносяться до класу IIб. | – Розчини для контактних лінз.
– Розчини для дезінфекції. |
| Усі медичні вироби, призначені спеціально для дезінфекції медичних виробів, відносяться до класу IIа. | – Дезінфекційні засоби, спеціально призначені для неінвазивних медичних виробів та обладнання, такі як стерилізатори, призначені для стерилізації медичних виробів в медичному середовищі та мийки-дезінфектори.
– Мийки-дезінфектори призначені для дезінфекції неінвазивних медичних виробів. |
| Медичні вироби, що призначені спеціально для дезінфекції інвазивних медичних виробів, відносяться до класу IIб. | – Дезінфекційні засоби для зубних протезів.
– Мийки-дезінфектори для ендоскопів. – Дезінфекційні засоби для трубок обладнання для гемодіалізу. – Дезінфекційні засоби для очних протезів, хірургічного обладнання та інвазивного стоматологічного обладнання. |
| Зазначені вимоги не поширюються на медичні вироби, призначені для фізичного очищення медичних виробів (крім контактних лінз)1. |
Практичні проблеми класифікації
Примітка 1: Це правило не поширюється на механічні засоби очищення виробів, такі як кисті та ультразвук. Такі вироби підпадають під ТР щодо МВ тільки, якщо вони спеціально призначені для використання з медичними виробами.
Правило/пункт 24 — Вироби для запису діагностичних рентгенівських знімків
| Правило/пункт 24 | Приклади |
|---|---|
| Медичні вироби, спеціально призначені для запису діагностичних рентгенівських знімків, відносяться до класу IIа. | – Рентгенівські знімки.
– Люмінофорні пластини. |
Примітка: Це відноситься лише до засобів запису, таких як рентгенівські знімки, а не до засобів для подальшого відображення.
Правило/пункт 25 — Вироби, що використовують нежиттєздатні тваринні тканини або похідні
Загальне пояснення правила
Це правило поширюється на вироби, що містять або зроблені з тканин тваринного походження, що були визначені нежиттєздатними, тобто такими, в яких більше немає ніякої можливості для клітинної метаболічної активності. Вироби, що містять живі тваринні тканини та/або будь-які людські тканини чи похідні, виключені зі сфери застосування ТР щодо МВ.
При виробництві деяких виробів може використовуватися промислова сировина, що містить малі кількості жиру або жирових похідних (наприклад, стеарат в полімерах). Такі речовини не вважаються похідними тваринних тканин в рамках цього правила.
| Правило/пункт 25 | ПРИКЛАДИ |
|---|---|
| Усі медичні вироби, виготовлені з використанням тканин тварин або нежиттєздатних тканин тварин1, відносяться до класу III, крім випадків, коли такі медичні вироби призначені для контакту з непошкодженою2 шкірою. | – Біологічні клапани серця.
– Свинячі ксенотрансплантатні перев’язувальні матеріали. – Імплантати та перев’язувальні матеріали, виготовлені з колагену. – Вироби, що використовують гіалуронову кислоту тваринного походження. |
Практичні проблеми класифікації
Вироби, виготовлені з нежиттєздатної тваринної тканини, що прямо контактують тільки з неушкодженою шкірою (наприклад, шкіряні компоненти ортопедичних пристосувань), відносяться до класу I відповідно до Правила 1.
Примітка 1: Похідні — продукти, що обробляються з тваринної тканини і включають такі речовини як молоко, шовк, бджолиний віск, волосся, ланолін.
Примітка 2: Неушкоджена шкіра включає шкіру навколо встановленої стоми, якщо шкіра непошкоджена.
Правило/пункт 26 — Мішки для крові
Загальне пояснення правила
Це спеціальне правило, що поширюється тільки на мішки для крові.
| Правило/пункт 26 | ПРИКЛАДИ |
| Мішки для крові відносяться до класу IIб. | – Мішки для крові (в тому числі такі, що містять або вкриті антикоагулянтом).
У випадках, коли мішки для крові виконують не тільки функцію зберігання та включають відмінні від антикоагулянтів системи зберігання (наприклад, правило пункту 21), можуть застосовуватися інші правила. |
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Медичні вироби. Допоміжні засоби. Виробник.»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.1/1, квітень 1994 та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753, Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 754 та Технічного регламенту щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
1. Медичні вироби
1) Медичний виріб — допоміжний засіб
Медичний виріб та допоміжний засіб не можна ототожнювати. Для їх розмежування визначальним є визначення термінів «медичний виріб» та «допоміжний засіб», які наведено у Технічному регламенті щодо медичних виробів, затвердженому постановою Кабінету Міністрів України від 02.10.2013 № 753 (далі — ТР щодо МВ) та у Технічному регламенті щодо активних медичних виробів, які імплантують, затвердженому постановою Кабінету Міністрів України від 02.10.2013 № 755 (далі — ТР щодо АМВІ). Допоміжні засоби в цілях ТР щодо АМВІ, розглядаються як медичні вироби, у той час як згідно з ТР щодо МВ, є відмінність між медичним виробом і допоміжним засобом. Допоміжні засоби класифікуються окремо від медичних виробів, з якими вони використовуються;
2) Медичне призначення
Медичне призначення кожного виробу визначається виробником. Відповідно до вимог технічних регламентів виробник для кожного свого виробу зазначає медичне призначення на етикетці, в інструкції із застосування та рекламних матеріалах. В технічних регламентах обумовлено захист пацієнтів і користувачів. Тому медичне призначення відноситься у загальному розумінні до готових виробів, незалежно від того, чи призначені вони для використання окремо, чи разом. Отже, сировина, компоненти або проміжні продукти, зазвичай не є медичними виробами. Перераховане повинно мати властивості або характеристики, які є визначальними для безпеки готових виробів. Тому, виробник готових виробів забезпечує відповідний відбір і контроль його сировини або проміжних продуктів відповідно до пункту 1, розділу ІІ, додатку 1 ТР щодо МВ.
Комплектувальні частини, що постачаються для заміни існуючих компонентів готового виробу, щодо якого вже була здійснена оцінка відповідності вимогам технічних регламентів, не є медичними виробами. У разі якщо комплектувальні частини суттєво змінюють характеристики або показники ефективності готового виробу, такі комплектувальні частини рекомендовано вважати окремими медичними виробами;
3) Налаштування
Після доставки кінцевому користувачеві або споживачеві готовий виріб може не бути готовим для використання чи застосування. Перед використанням може знадобитись додаткова обробка, підготовка, налаштування, установка, монтаж, адаптація або пристосування до потреб користувача або пацієнта. Наприклад, стерилізація медичних виробів, які постачаються нестерильними, монтаж систем, виготовлення суміші для зубних пломб, адаптація протеза до потреб пацієнта.
Вищезазначені дії, як правило, не виконуються виробниками, якщо вони здійснюються кінцевим користувачем під час використання або підготовки до використання. У цьому контексті необхідно розрізняти типову професійну діяльність, що здійснюється медичним працівником, та діяльність з обробки та складання, що здійснюється відповідним фахівцем.
В останньому випадку такою діяльністю може стати діяльність з виробництва або складання систем медичних виробів, процедурних наборів та процедур стерилізації, як це передбачено пунктами 27-30 ТР щодо МВ.
Особливу увагу в даному контексті слід приділяти виготовленню виробів на замовлення. Вироби на замовлення (наприклад, протези, індивідуальні слухові апарати), в більшості випадків є виробами індивідуального використання. Перед введенням в обіг медичного виробу, виготовленого на замовлення, виробник повинен провести процедуру, зазначену в додатку 9 і скласти заяву, зазначену в додатку 9 ТР щодо МВ.
У цих випадках проміжні продукти, спеціально призначені для виготовлення медичних виробів на замовлення, рекомендується вважати медичними виробами. Це відноситься, в значній мірі, до стоматологічних сплавів, модульних компонентів для протезування, якщо передбачене використання таких виробів безпосередньо пов’язане з медичними виробами;
4) Медично-гігієнічне призначення
Згідно визначення «медичний виріб», яке наведено у ТР щодо МВ, такі вироби, загалом призначені для використання у медичних цілях. Тому, вироби гігієнічного або косметичного призначення не є медичними виробами, навіть у тому випадку, коли вони можуть використовуватись для профілактики захворювання.
Приклади виробів, які, зазвичай, не можуть визначатись як медичні вироби:
– зубні щітки, зубні палички, зубна нитка;
– дитячі підгузки, гігієнічні тампони;
– контактні лінзи без коригувальних функцій, що призначені для зміни кольору очей;
– продукти для відбілювання зубів;
– інструменти для нанесення татуювань.
Приклади виробів, які можуть бути медичними виробами:
– вироби, що вбирають рідини при нетриманні;
5) Допомога особам з інвалідністю
У випадку, якщо обладнання призначене для полегшення або компенсації інвалідності, необхідно встановлювати прямий зв’язок між коригувальною функцією та відповідною особою. Тому, якщо цього не досягнуто, обладнання не вважається медичним виробом, наприклад, акустичні сигнали на світлофорах, туалетне обладнання для інвалідів.
6) Програмне забезпечення
Відмінність: програмне забезпечення, що впливає на належне функціонування виробу і програмне забезпечення, що використовується разом з немедичним обладнанням.
Програмне забезпечення, пов’язане з функціонуванням медичного виробу, може бути частиною виробу або окремим медичним виробом, якщо воно реалізується на ринку збуту окремо від відповідного медичного виробу.
У випадку якщо програмне забезпечення призначене для використання з багатофункціональним інформаційним обладнанням, необхідно встановити відмінність між програмним забезпеченням, що забезпечує належні діагностичні або терапевтичні властивості пристрою та програмним забезпеченням для обробки загальних даних пацієнтів.
Тільки в першому випадку можна визначити медичне призначення.
Приклади медичних виробів: програмне забезпечення для обчислення анатомічних частин тіла, програмне забезпечення для покращення зображення, що призначене для діагностичних цілей, програмне забезпечення для програмування медичного виробу.
Медичне призначення виробу відсутнє у випадку, коли програмне забезпечення використовується для обробки загальних даних пацієнта;
7) Багатоцільові вироби
Вироби багатоцільового призначення, які іноді можуть використовуватись в медичному середовищі, зазвичай, не є медичними виробами, за винятком виробів, для яких буде встановлено спеціальне медичне призначення, наприклад, багатоцільовий ПК, принтер, сканер, магнітоскоп, монітор.
2. Допоміжні засоби
Питання, чи є продукт «медичним виробом» або «допоміжним засобом», не має практичної значущості. Згідно із ТР щодо МВ допоміжні засоби слід розглядати як медичні вироби.
Визначення «допоміжний засіб», яке наведено у ТР щодо МВ, вимагає, щоб допоміжний засіб був спеціально призначений виробником для використання разом з медичним виробом. Передбачене використання допоміжного засобу повинно бути таким, щоб забезпечувати застосування медичного виробу відповідно до його передбаченого використання. Тому продукт може стати допоміжним засобом до медичного виробу тільки тоді, коли виробник такого продукту встановив його передбачуване використання разом з одним або декількома медичними виробами.
Приклади допоміжних засобів в залежності від визначених обставин використання медичного виробу:
– стерилізатори для використання в медичному середовищі;
– мішки для упаковки повторно стерилізованих медичних виробів;
– спеціальні зарядні пристрої для електромедичних виробів з живленням від акумуляторної батареї;
– засоби по догляду за контактними лінзами, дезінфікуючі засоби, які спеціально призначені для медичних виробів;
– спеціальний пристрій для очистки води, призначений для використання разом з апаратами для діалізу,
– газові балони/пристрої зменшення тиску для використання разом з апаратами для анестезії.
3. Виробник
Законодавство визначає виробника як фізичну або юридичну особу, яка відповідає за визначену виробничу діяльність, пов’язану з медичним виробом з метою його реалізації на ринку під власною назвою.
Причиною цього зв’язку з реалізацією на ринку є те, що технічні регламенти спрямовані на дотримання вимог щодо захисту виробу під час переходу з області відповідальності виробника в область відповідальності користувачів. ТР щодо МВ не розповсюджується на вироби у випадку, коли медичний виріб виготовлений користувачем (наприклад, лікарнею) без передачі іншій особі. Проте, це стосується виключно такої власної виробничої діяльності, коли медичний виріб залишається разом з користувачами в установі, і не стосується тих випадків, коли, наприклад, лікарня виготовляє ортопедичні вироби для використання пацієнтами.
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Однорідні партії»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.5/6 версія 1, лютий 1998 та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753, Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 754 та Технічного регламенту щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
Однорідність виробничих партій
Деякі статистичні процедури оцінки відповідності мають значення тільки для партій (lots), які є однорідними.
Партія вважається однорідною, якщо еквівалентні частини або матеріали виготовлені та/або протестовані у єдиний спосіб, без перерви, як правило, в той же день або в той же період часу, і виготовляються тією ж особою, або з такими ж налаштуваннями обладнання і відповідають тій же специфікації.
Для медичних виробів для діагностики in vitro і деяких інших медичних виробів, таких як стоматологічні матеріали, додатково застосовується наступне визначення: Суміші речовин, такі як реагенти аліквоти з тієї ж об’ємної суміші, вважаються однорідними, якщо процес змішування та аліквоти підтверджено1.
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Субпідряд — зв’язок з системою управління якістю»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.5/3, редакція 2, червень 1998 та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753, Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 754 та Технічного регламенту щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
Розглянемо для прикладу Технічний регламент щодо медичних виробів, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 753 (далі — ТР щодо МВ).
Відповідно до пункту 6 додатку 3, пункту 6 додатку 6, пункту 4 додатку 7 до ТР щодо МВ процедура оцінювання в разі потреби має включати інспектування постачальників виробника з метою перевірки виробничих процесів.
Органу з оцінки відповідності слід звернути увагу на два основних питання при розгляді субпідрядників:
1) чи субпідрядник бере істотну участь у розробці та/або виробництві виробу?
2) чи субпідрядник постачає деталі, матеріали або послуги, які можуть вплинути на відповідність виробу основним вимогам технічних регламентів?
Якщо відповідь на обидва питання 1) і 2) «НІ», ніяких подальших дій не потрібно здійснювати.
Якщо відповідь на вищенаведені питання 1) або 2) «ТАК», то орган з оцінки відповідності повинен оцінити чи достатньо доказів компетентності субпідрядника для постачання деталей, матеріалів або послуг пов’язаних з медичним виробом (-ами), що розглядаються. Оцінка буде стосуватися різних аспектів, включаючи контроль, що здійснюється виробником за діяльністю субпідрядника.
Орган з оцінки відповідності може не проводити аудит субпідрядника, але лише у тому випадку, якщо буде доведено, що інший орган з оцінки відповідності, компетентний стосовно оцінки частин, матеріалів або послуг, провів оцінку субпідрядника щодо частин, матеріалів чи послуг і засвідчив компетентність субпідрядника в цій області.
У всіх інших випадках органу з оцінки відповідності повинна бути надана можливість розглянути актуальність або критичність субпідрядника для медичного виробу, і, якщо надані виробником докази є незадовільними, орган з оцінки відповідності має здійснити аудит/оцінку субпідрядника або вимагати виробника провести повторну оцінку субпідрядника1.
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Дата «використати до»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.2/3 верс.3, червень 1998 та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753, Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 754 та Технічного регламенту щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
1. Мета рекомендацій
Метою рекомендацій є:
1) сприяння у прийнятті рішення виробником про те, що дата «використати до» необхідна для його конкретного медичного виробу, медичного виробу для діагностики in vitro та активних медичних виробів, які імплантують (далі — вироби);
2) визначення, яка інформація необхідна, для підтвердження рішення виробника;
3) визначення рекомендацій щодо поводження з виробами, що повністю або частково втратили свої властивості і не мають подальшого використання за місцем їх утворення чи виявлення і від яких їх власник позбувається, має намір або повинен позбутися шляхом утилізації чи видалення.
1. Мета цих рекомендацій
Мета цих рекомендацій полягає в тому, щоб:
1) сприяти у прийнятті рішення виробником про те, що дата «використати до» необхідна для його конкретного виробу;
2) вказують на те, яка інформація необхідна, для підтвердження його рішення.
2. Вимоги
Технічний регламент щодо медичних виробів, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 753 (далі — ТР щодо МВ), Технічний регламент щодо медичних виробів для діагностики in vitro, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 754 (далі — ТР щодо МВ in vitro), Технічний регламент щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755 (далі — ТР щодо АМВІ), вимагають вказувати на медичному виробі та/або на його упаковці, та/або у супровідній інформації, що супроводжує виріб, будь-які часові обмеження щодо безпечного використання виробу. Хоча формулювання відрізняються, технічні регламенти встановлюють необхідність надання цієї інформації:
| ТР щодо МВ, додаток 1 | ТР щодо МВ in vitro, додаток 1 | ТР щодо АМВІ, додаток 1 |
|---|---|---|
| 44. Етикетка медичного виробу повинна містити такі елементи:
5) в разі потреби — строк, до якого гарантовано безпечне застосування медичного виробу, із зазначенням року та місяця; |
40. Етикетка виробу повинна містити таку інформацію…
дату, до настання якої сам виріб чи його частина можуть безпечно використовуватися без погіршення експлуатаційних характеристик, у такому порядку: рік, місяць та в разі потреби — день; |
15. Кожен виріб повинен містити такі відомості…
зазначення строку придатності для безпечної імплантації виробу; |
Щодо виробів, на які поширюється дія ТР щодо АМВІ, дата придатності завжди повинна бути вказана.
У випадку виробів, охоплених ТР щодо МВ та ТР щодо МВ in vitro, обмеження за часом необхідне «в разі потреби» (або «необхідне»)1.
Якщо виріб багаторазовий, ТР щодо МВ окремо вимагає наводити інформацію щодо будь-яких обмежень кількості повторних застосувань.
Якщо виріб призначений для одноразового використання, але зберігається протягом тривалого періоду, будь-які обмеження на цей період та особливості використання потрібно зазначати як підпункти 10, 11 пункту 44 додатку 1 ТР щодо МВ:
інформацію про будь-які спеціальні інструкції з експлуатації;
інформацію про будь-які запобіжні заходи та/або застереження2.
3. Як вирішити, що дата «використати до» необхідна?
Наносити дату «використати до» потрібно, коли пов’язані з безпекою характеристики або заявлена дія можуть погіршитися з плином часу.
При прийнятті рішення, чи є таке погіршення «пов’язане з безпекою», виробник повинен звернутися до результатів аналізу ризиків та заходів, прийнятих для управління ризиками.
1) аналіз ризиків визначатиме дію і характеристики, що необхідні для безпечного використання конкретного виробу.
Наприклад, аналіз ризику може показати, що стерильність необхідна для безпечного використання. Водночас, аналіз ризику не буде охоплювати колір виробу, якщо це лише аспекти естетики, але може враховувати його, якщо цей колір нанесено з метою, пов’язаною з безпечним використанням виробу (наприклад, колір визначає розмір самого виробу);
2) аналіз ризиків і заходи з управління ризиками будуть також визначати рівень або ступінь ефективності або характеристики, але тільки в тій мірі, в якій вони впливають на безпечне використання виробу.
Наприклад, рівень опору потоку газу або швидкості витоку з дихальної системи, або ймовірність, що виріб стане нестерильним;
3) аналіз ризику і заходи з управління ризиками будуть також визначити період, протягом якого ефективність або характеристики залишаються сталими, щоб забезпечити безпечне використання, у тому числі щодо терміну придатності і передбачуваного періоду використання.
Наприклад, період, протягом якого батарея кардіостимулятора зберігає достатньо енергії для функціонування після імплантації, такий же, як визначений виробником.
4. Інформація, яка потрібна для підтримки рішення
1) Інформація, яка необхідна, якщо встановлено дату «використати до»
Виробник повинен продемонструвати, що відповідні дія і характеристики виробу зберігаються протягом заявленого терміну придатності, який відображається датою «використати до».
Це може бути досягнуто шляхом, та/або:
перспективних досліджень з використанням прискореного старіння, валідованих з кореляцією деградації реального часу;
ретроспективних досліджень, що використовують реальний досвід часу, наприклад, з використанням тестування збережених зразків, огляду історії скарг чи опублікованої літератури і т.д.
2) Інформація, яка необхідна, якщо не визначено дату «використати до»
Так як відсутність дати «використати до» являє собою неявне декларування нескінченного терміну придатності, виробник повинен продемонструвати, та/або:
відсутність пов’язаної з безпекою дії або характеристик, які б, ймовірно, погіршилися за тривалий час (підпункт 1 пункту 3);
у разі будь-якого ймовірного погіршення (підпункт 2 пункту 3) не являють собою неприйнятний ризик;
що період, протягом якого відбувається погіршення перевищує межу ймовірного часу першого використання виробу (підпункт 3 пункту 3), наприклад, 30 років.
При цьому, виробник повинен розглянути, серед іншого:
матеріали самого виробу, що використовуються у виробництві, у тому числі клеї, покриття, упаковки і т.д.;
методи виготовлення (наприклад, кріплення компонентів, процес герметизації упаковки);
методи захисту виробу або їх частини від псування (наприклад упаковку, інструкції для зберігання);
при необхідності, стан, в якому виріб має знаходитися до першого використання (наприклад, без встановленої батареї);
потенціальну деградацію з часом певних матеріалів (наприклад, деградація полімеру за рахунок довгострокових ефектів стерилізації вільними радикалами з гамма-опроміненням).
Якщо виробник не може виконати вищезазначені вимоги абзацу третього підпункту 2, або абзацу шостого підпункту 2, або абзацу восьмого підпункту 2, то дата «використати до» повинна бути вказана.
3) Поводження з виробами, що повністю або частково втратили свої властивості і не мають подальшого використання за місцем їх утворення чи виявлення і від яких їх власник позбувається, має намір або повинен позбутися шляхом утилізації чи видалення.
Вимогами ТР щодо МВ (пункт 47 Розділу II «Вимоги до розроблення і виготовлення медичних виробів» додатку 1 до ТР щодо МВ) встановлено, що у разі потреби в інструкції із застосування зазначаються, зокрема, запобіжні заходи, що мають бути вжиті для усунення будь-яких спеціальних, незвичайних ризиків, пов’язаних з утилізацією медичних виробів.
Вимогами ТР щодо МВ in vitro (пункт 43 додатку 1 до ТР щодо МВ in vitro) встановлено, що інструкції із застосування в разі доцільності містять, зокрема, інформацію про заходи безпеки, яких необхідно вжити для запобігання будь-яким специфічним, незвичайним ризикам, пов’язаним з використанням чи утилізацією виробів, включаючи спеціальні захисні заходи (якщо виріб містить речовини людського або тваринного походження, слід звертати увагу на їх потенційну інфекційну природу (інфектогенність).
Загальні вимоги до поводження з медичними відходами в закладах охорони здоров’я з метою попередження їх негативного впливу на життя, здоров’я населення та довкілля і визначають порядок збирання, перевезення, зберігання, сортування, оброблення (перероблення), утилізації, видалення, знезараження, захоронення, знищення медичних відходів встановлено Державними санітарно-протиепідемічними правилами і нормами щодо поводження з медичними відходами, затвердженими наказом Міністерства охорони здоров’я України від 08 червня 2015 року № 325, зареєстрованими в Міністерстві юстиції України 07 серпня 2015 року за № 959/27404.
У інших випадках, якщо власник виробів позбувається, має намір або повинен позбутися шляхом утилізації чи видалення виробів, що повністю або частково втратили свої властивості і не мають подальшого використання за місцем їх утворення чи виявлення застосовуються положення Закону України «Про відходи».
5. Приклади:
1) Серцевий катетер з балоном із латексу3
Серцевий катетер включає в себе балон з латексу, що розташований на його кінці і тимчасово закриває кровоносну судину. Здатність балона витримувати певний тиск необхідна для безпечного використання. Якість латексу балона, однак, з часом погіршується. Упаковка та правила зберігання захищають виріб від світла, зменшують швидкість руйнування, але не запобігають йому. Тому необхідно вказувати дату «використати до».
Виробник повинен продемонструвати, що балон з латексу залишається в змозі витримати заявлений тиск впродовж відповідного терміну придатності, якщо виріб зберігається у відповідності до вказівок виробника.
2) Ортопедичний імплантат тазостегнового суглоба (постачається стерильним)4
Метало-керамічний ортопедичний імплантат постачається стерильним в композитному контейнері з пластику/паперу. Здатність упаковки підтримувати стерильність є необхідною для безпечного використання. У той час як підтримка стерильності залежить від обставин (тобто в залежності від фактичних умов зберігання та поводження), стерильність також залежить і від часу, в результаті, наприклад, зниження гнучкості та міцності, щільності пакувального матеріалу, що робить його більш сприйнятливим до подій, які можуть вплинути на стерильність.
Окрім того, оскільки такі імплантати доступні в різних розмірах конкретний виріб може залишатися в його упаковці протягом тривалого часу, поки він не буде потрібний для імплантації.
Тому, необхідно вказувати дату «використати до». Щодо підтримки стерильності, дата «використати до» буде відображати:
а) пов’язане з часом погіршення характеристик паковання, наприклад ізоляцію, цілісність і стійкість до проникнення часток, що несуть мікроорганізми;
б) ймовірність пошкодження під час транспортування і зберігання, що призводять до порушення стерильності, але не є очевидними, а, отже, попередження про невикористання виробу при пошкодженні паковання не допоможе.
Виробник повинен продемонструвати, що:
– пакувальний матеріал здатний зберігати стерильність виробу довше ніж заявлений термін придатності, за умови зберігання відповідно до інструкцій виробника.
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Сфера застосування Технічного регламенту щодо активних медичних виробів, які імплантують, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 755»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.1/2rev2 April 1994 та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753 та Технічного регламенту щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
Дані рекомендації необхідно застосовувати разом з Технічним регламентом щодо медичних виробів, затвердженим постановою Кабінету Міністрів України від 02.10.2013 № 753 (далі — ТР щодо МВ) та Технічним регламентом щодо активних медичних виробів, які імплантують, затвердженим постановою Кабінету Міністрів України від 02.10.2013 № 755 (далі — ТР щодо АМВІ), що, в свою чергу, забезпечує практичне підтвердження однакового застосування вказаних технічних регламентів.
1. Активний медичний виріб, який імплантують
1) ТР щодо АМВІ застосовується під час введення в обіг та/або введення в експлуатацію активних медичних виробів, які імплантують.
Дія ТР щодо АМВІ поширюється на активні медичні вироби, які імплантують, якщо вони відповідають визначенню, наведеному в цьому Технічному регламенті. Це означає, що вироби повинні бути за визначенням «медичними виробами», які є одночасно «активними» та «виробами, які імплантують».
Станом на квітень 2019 року чинна версія Технічного регламенту щодо активних медичних виробів, які імплантують, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 755 не містить визначення «медичний виріб», як це визначено Директивою Ради ЄС від 20 червня 1990 р. № 90/385/ЄЕС щодо наближення законодавства держав-членів в частині активних медичних виробів, які імплантують. Тому, слід керуватися визначенням «медичний виріб, визначеного Директивою Ради ЄС від 20 червня 1990 р. № 90/385/ЄЕС в частині визначення «медичного виробу» до внесення відповідних змін.
2) Під визначенням «медичний виріб», згідно з ТР щодо АМВІ, слід розуміти будь-який інструмент, апарат, прилад, матеріал або інший виріб, незалежно від того, використовується він окремо або в комбінації разом з будь-якими допоміжними засобами або програмним забезпеченням для його належного функціонування, призначений виробником з метою використання для людей в цілях:
– діагностики, профілактики, моніторингу, лікування або полегшення захворювання або травми,
– дослідження, заміни або зміни анатомії або фізіологічного процесу,
– контролю процесу зачаття,
та основна передбачувана дія яких в організмі або на організм людини не досягається за допомогою фармакологічних, імунологічних або метаболічних засобів, але функціонуванню яких такі засоби можуть сприяти.
Під визначенням «медичні вироби» розуміють вироби, призначені для використання в лікувальних цілях, «які застосовуються окремо або разом з будь-якими допоміжними засобами, або програмним забезпеченням для їх належного функціонування». Медичне призначення може бути досягнуто шляхом застосування «одиночного виробу» або декількох виробів, що діють одночасно один з одним, утворюючи систему. Якщо медичне призначення досягається системним шляхом, кожен елемент системи можна розглядати як медичний виріб. Визначення медичного виробу може застосовуватися стосовно системи, як такої, або до взаємозамінних частин, призначених для формування системи разом з іншими виробами, тому, для цілей ТР щодо АМВІ, кожна частина якої належить до такої системи, охоплюється ТР щодо АМВІ незалежно від того, чи є така частина сама по собі «активною», «активно імплантованою», чи ні.
Приклади активних медичних виробів, які імплантують:
– електричний кардіостимулятор, який імплантують;
– електричний кардіостимулятор, який імплантують без електрода;
– електрод;
– виріб введення лікарських засобів з/без катетера, який імплантують;
– катетер виробу для введення лікарських засобів, який імплантують;
3) Згідно з метою ТР щодо АМВІ медичний виріб є активним, якщо його робота залежить від наявності джерела електричної енергії або будь-якого іншого джерела енергії, крім безпосередньо генерованої людським організмом або силою тяжіння (гравітації). Це включає в себе, наприклад, вироби, що активуються за допомогою тиску, за винятком того випадку, коли ефект досягається енергією, яка надходить з організму пацієнта. Під визначенням розуміється, що функція виробу включає використання джерела енергії для здійснення корисної роботи. Проста передача тепла, світла, тиску або вібрації не означає, що виріб є активним.
Приклади:
– зниження тиску при гідроцефалії дозволяє випустити спинномозкову рідину, коли пружина ослаблена виріб не є «активним». Навіть якщо положення пружини може регулюватись електромагнітними засобами, виріб залишається неактивним, оскільки медична функція виробу полягає в зменшенні тиску, а не в регулюванні;
– виріб введення лікарського засобу, в якому лікарський засіб рухається від резервуара за допомогою збереженого джерела енергії (пружина, рідина, газ тощо…), є «активним»;
– внутрішньосудинний катетер, що містить волокно-оптичний джгут, під’єднаний до зовнішнього джерела світла, може використовуватись для вимірювання тиску або інших параметрів крові, якщо показники променю світла будуть змінюватись в залежності від параметрів крові та будуть реєструватись виробом. Хоча система в цілому залежить від джерела живлення, для досягнення своєї медичної функції (вимірювання параметрів крові) інвазійний елемент не є «активним», оскільки він тільки передає світло;
– кохлеарний імплантат, що активується за допомогою зовнішнього передавача енергії вважається «активним», оскільки імплантований компонент, безумовно, залежить від джерела живлення для його функціонування і його мета полягає в перетворенні енергії, яку він отримує, в електричні сигнали, які є тригером відповідних каналів сенсорного сприйняття в головному мозку, тобто він виконує корисну роботу.
4) Активний медичний виріб є «виробом, який імплантують», якщо він призначений для повного або часткового введення в тіло пацієнта хірургічним чи іншим медичним шляхом або через природний отвір, що повинен залишатися в тілі після закінчення процедури введення.
ТР щодо АМВІ був розроблений для активних медичних виробів, які можуть характеризуватись потенційно високим ризиком внаслідок того, що вони повністю або частково імплантуються в організм. Такі вироби можуть становити небезпеку, зокрема, у зв’язку з неможливістю їх обслуговування, калібрування чи контролю, та внаслідок проблем, пов’язаних із старінням матеріалів, як зазначено в декількох основних вимогах Додатку 1 до ТР щодо АМВІ. Тому, властивість «виріб, який імплантують» повинна інтерпретуватися з урахуванням того, що ці небезпеки є типовими для імплантованих медичних виробів.
З вищезазначених причин, зовнішній насос введення лікарських засобів, як для довготривалого, так і для постійного використання, який з’єднаний з катетером шляхом «часткового введення» в організм, не вважається активним імплантованим медичним виробом.
Однією з важливих характеристик імплантованого виробу є його відносно довготривале використання. Необхідно вказати відмінність в передбаченому використанні виробу, який може в довготривалому порядку використовуватись протягом декількох місяців, у порівнянні з тимчасовим користуванням протягом зазначеного медичного втручання. Зовнішній електрокардіостимулятор, в тому числі його електрод, використовується тимчасово, отже, не вважається виробом, що «залишається в організмі людини після завершення процедури». Те ж саме відноситься до використання внутрішньоаортального балонного насоса. Згідно з метою ТР щодо АМВІ, термін «процедура» повинен інтерпретуватись як процес діагностики, моніторингу та лікування, який може тривати протягом декількох днів, як правило, в лікарні, і не обов’язково стосуватись виключно хірургічної операції, в ході якої виріб був розміщений всередині організму.
2. Допоміжний засіб до активного медичного виробу, який імплантують
1) «Допоміжний засіб» до активного медичного виробу, який імплантують за визначенням є «активним медичним виробом, який імплантують» та, на підставі цього, на них поширюється дія ТР щодо АМВІ. Вказане не передбачає, що властивості «активний» та «виріб, який імплантують» обов’язково повинні відповідати вимогам до визначення виробу, як «допоміжні засоби». Достатньою умовою є те, що продукт за його передбаченим використанням є допоміжним для цілей застосування активного медичного виробу, який імплантують таким чином, що це дозволяє використовувати медичний виріб у відповідності з його передбаченим використанням, або для посилення цілей застосування виробу, визначених його виробником. Згідно з цим пристрій програмування або зовнішній передавач, призначений для активації або контролювання частини виробу, яка імплантована, підпадає під дію визначення «активний імплантований медичний виріб».
2) Репрезентативний перелік активних медичних виробів, які імплантують:
– електрокардіостимулятори, які імплантують;
– дефібрилятори, які імплантують;
– проводи, електроди, адаптери для виробів;
– нейростимулятори, які імплантують;
– стимулятори сечового міхура;
– стимулятори сфінктера;
– стимулятори діафрагми;
– кохлеарні імплантати;
– активний виріб для введення лікарського засобу, який імплантують;
– катетери, датчики для виробів;
– активні вироби моніторингу, які імплантують;
– вироби програмування, програмне забезпечення, передавачі.
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 22.01.2020 р. № 142
МЕТОДИЧНІ РЕКОМЕНДАЦІЇ
«Уповноважений представник»
ВСТУП
Методичні рекомендації розроблено на основі Керівного документу Європейської Комісії MEDDEV 2.5/10, січень 2012 року та не є юридично обов’язковими. Методичні рекомендації розроблено задля однозначного тлумачення норм Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753, Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 754 та Технічного регламенту щодо активних медичних виробів, які імплантують, затверджений постановою Кабінету Міністрів України від 02.10.2013 № 755.
Обов’язковими є лише тексти технічних регламентів:
– Технічного регламенту щодо медичних виробів, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 753 (далі — ТР щодо МВ);
– Технічного регламенту щодо медичних виробів для діагностики in vitro, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 754 (далі — ТР щодо МВ in vitro);
– Технічного регламенту щодо активних медичних виробів, які імплантують, затвердженого постановою Кабінету Міністрів України від 02.10.2013 № 755 (далі — ТР щодо АМВІ).
Текст технічних регламентів застосовується там, де існують розбіжності між положеннями технічних регламентів та змістом даних рекомендацій.
Протягом кількох років висловлювались занепокоєння щодо відсутності чіткого роз’яснення ролі уповноваженого представника. Особлива плутанина була викликана тим, що деякі виробники передавали певні повноваження своїм уповноваженим представникам. Також плутанину викликала відсутність чіткого переліку даних, які повинні/можуть надавати уповноважені представники.
1. Підсумки поточних положень технічних регламентів про медичні вироби
Виробник, а не уповноважений представник, несе відповідальність за зобов’язання відповідно до технічних регламентів.
Якщо виробник, який вводить вироби в обіг під власною назвою, є нерезидентом України, він повинен призначити уповноваженого представника.
Виробник може мати більш ніж одного уповноваженого представника, за умови, що один виріб (мається на увазі один тип/модель виробу) пов’язаний тільки з одним уповноваженим представником. Намір такого положення — відсутність обмеження одним представником для всього спектру продукції.
В інформації на зовнішній упаковці або інструкції із застосування має міститися адреса уповноваженого представника, якщо виробник є нерезидентом.
Мета цього примусового призначення — надання органам влади можливості контакту з особою, яка несе відповідальність за розміщення виробу на ринку України, особливо в надзвичайних випадках.
Уповноважений представник має певні обов’язки, визначені відповідними технічними регламентами, такі як:
– інформування компетентних органів влади про зареєстроване місцезнаходження (ТР щодо МВ: клас І, системи медичних виробів та процедурні набори, медичні вироби, виготовлені на замовлення; ТР щодо АМВІ: медичні вироби виготовлені на замовлення; ТР щодо МВ in vitro: про вироби та сертифікати;
– зберігання певної інформації для подальшого використання компетентними органами, такої як декларації про відповідність та технічної документації.
Виробник може доручити своєму уповноваженому представникові ініціювати проведення процедур, передбачених у додатках 4, 5, 8 і 9 ТР щодо МВ.
Виробник може уповноважувати свого представника розпочати процедури оцінки відповідності, наведені в додатках 3, 5, 6 і 8 ТР щодо МВ in vitro.
Виробник може уповноважити свого представника ініціювати процедури, наведені у додатках 3, 4, і 6 ТР щодо АМВІ.
1) Роль уповноваженого представника
Загальні дані
Так як технічні регламенти не включають детального опису ролі та зобов’язань уповноваженого представника, важливим для виробника та уповноваженого представника є укладення контракту із зазначенням обов’язків та повноважень, які виробник делегуватиме уповноваженому представникові, також і у випадках, коли уповноважений представник є дочірньою компанією/представництвом виробника.
Призначення уповноваженого представника не змінює відповідальності виробника. Уповноважений представник повинен бути призначеним та знаходитися під наглядом виробника.
Уповноважений представник є представником виробника і буде безпосередньо нести відповідальність. Наприклад, його відповідальністю може бути забезпечення проведення відповідної процедури оцінки відповідності та підтвердження того, що виріб пройшов оцінку відповідності. Ще один приклад — встановлення системи пост-маркетингового нагляду, сумісною з такою ж системою виробника. Уповноважений представник таким чином буде повністю поінформованим про свої зобов’язання. Такі зобов’язання повинні бути відображені у зазначеному вище договорі з виробником.
Призначення уповноваженого представника
Якщо виробник, який під власним іменем вводить в обіг медичні вироби, зазначені в абзаці першому цього пункту, не є резидентом України, він зобов’язаний призначити одного уповноваженого представника, відповідального за введення в обіг цих виробів на ринку України. Те саме стосується ТР щодо МВ in vitro без зазначення «один».
Уповноважений представник повинен бути єдиним уповноваженим представником у межах України для мінімум всіх виробів того ж самого типу. Виробник може мати різних уповноважених представників для різних виробів (типів).
Вимога мати уповноваженого представника застосовується для всіх медичних виробів, розміщених на ринку України, виробник яких знаходиться поза межами України.
Вимога мати уповноваженого представника також застосовується до виробів, призначених для клінічних досліджень (ТР щодо МВ, ТР щодо АМВІ), або оцінки характеристик (ТР щодо МВ in vitro), де виробник-нерезидент України.
Реєстрація
ТР щодо МВ та ТР щодо АМВІ
Реєстрація уповноважених представників, виробників та виробів
Уповноважені представники призначені для виробу щодо якого існує зобов’язання повідомити Державну службу України з лікарських засобів та контролю за наркотиками (далі — Держлікслужба) (ТР щодо МВ: клас І, системи медичних виробів та процедурні набори, медичні вироби, виготовлені на замовлення; ТР щодо АМВІ: медичні вироби, виготовлені на замовлення), зобов’язані зареєструвати у Держлікслужбі своє місцезнаходження та виробника, а також надати перелік і опис відповідних виробів.
Реєстрація Клінічних Досліджень
Виробник або уповноважений представник повинні проінформувати Держлікслужбу про наміри провести клінічні дослідження. Вони також інформують про закінчення таких досліджень та надають письмовий звіт про клінічні дослідження. Виробник може передоручити такі завдання повністю або частково уповноваженим представникам.
ТР щодо МВ in vitro
Реєстрація уповноважених представників, виробників, виробів та сертифікатів
Уповноважений представник, призначений для виробів, які підпадають під ТР щодо МВ in vitro, зобов’язаний зареєструвати у Держлікслужбі своє місцезнаходження та виробника, інформацію щодо виробів та сертифікатів.
Реєстрація оцінки характеристик
Виробник-нерезидент України, який бажає здійснити оцінювання характеристику діагностичного виробу, повинен призначити уповноваженого представника. Уповноважений представник передає інформацію про виробника та такий виріб до Держлікслужби. Заявка, необхідна для оцінювання характеристик виробу, видається виробником уповноваженому представникові.
Оцінювання відповідності
Технічний регламент дозволяє виробникові передати виконання деяких вимог Технічних регламентів призначеному уповноваженому представникові. Це повинно бути окремо вказано у договорі між виробником та уповноваженим представником.
ТР щодо МВ
Виробник може доручити своєму уповноваженому представникові ініціювати проведення процедур, передбачених у додатках 4, 5, 8 і 9.
– Подати заявку для оцінки відповідності для перевірки типу (додаток 4),
– Скласти декларацію відповідності, включаючи додаток 8 пункт 5 у випадку з виробами, розміщеними на ринку в стерильних умовах та виробів класу І з функцією вимірювання,
– Скласти заяву для виготовлених на замовлення виробів (додаток 9 пункт 2.1).
ТР щодо АМВІ
Виробник може уповноважити свого представника ініціювати процедури, наведені у додатках 3, 4, і 6.
– Подати заявку для оцінки відповідності для перевірки типу (додаток 3),
– Скласти декларацію відповідності типу, вказаному у сертифікаті перевірки типу (додаток 4),
– Скласти заяву для виготовлених на замовлення виробів (додаток 6).
ТР щодо МВ in vitro
Виробник може уповноважувати свого представника розпочати процедури оцінки відповідності, наведені в додатках 3, 5, 6 і 8.
– Виконати вимоги додатку 3.
– Подати заявку для оцінки відповідності для перевірки типу (додаток 5).
– Скласти декларацію відповідності типу, вказаному у сертифікаті перевірки типу (додаток 6).
Уповноважений представник буде контактною особою між органами влади та іншими органами, замість виробника, стосовно зобов’язань останнього згідно технічних регламентів
Інформація яка вимагається від виробника
Уповноважений представник зобов’язується надавати певну інформацію до компетентних органів, таку як декларації відповідності та технічну документацію.
Уповноважений представник повинен мати можливість надавати всю документацію та інформацію, яку можуть запитувати компетентні органи, з метою нагляду за ринком.
Інформація може зберігатися в уповноважених представників, які мають повноваження передавати таку інформацію безпосередньо до компетентних органів. У такому випадку, в договорі будуть зазначені зобов’язання від виробника для постійного оновлення інформації.
Якщо виробник обирає не зберігати інформацію в уповноважених представників, він надаватиме уповноваженим представникам всю документацію та інформацію, якої можуть потребувати органи ринкового нагляду, з метою спостереження за ринком при отриманні запиту, переданого уповноваженими представниками до виробника. Уповноважені представники повинні мати доступ до всієї документації та інформації.
У цьому випадку договір повинен містити дані про те, що виробник надає інформацію, на яку здійснено запит уповноваженим представникам завчасно, а також включати зобов’язання від виробника про своєчасне надання уповноваженим представникам інформації про будь-які зміни.
Уповноважені представники повинні розірвати договір з виробником, якщо він не забезпечує надання йому доступу до необхідної інформації.
Інформація, на яку здійснюється запит, може включати:
декларацію відповідності;
копії етикетки, упаковки та інструкцій із застосування;
сертифікати;
дані пост-маркетингового нагляду за ринком, звіти та скарги;
технічну документацію;
відповідні клінічні дані/повідомлення;
деталі про дистриб’юторів/постачальників, які розміщують вироби з маркуванням національним знаком на ринку;
звіти про інциденти та здійснені корегувальні заходи.
Виробник повинен інформувати своїх уповноважених представників щодо всіх питань, пов’язаних з виробами, розміщеними на ринку України.
Інформація, яку можна отримати від уповноважених представників
Уповноважений представник повинен інформувати виробника щодо всіх питань, які можуть бути пов’язані з виробами, що розміщуються на ринку України.
Значні негативні результати під час клінічних досліджень, тобто перед введенням на ринок
Усі побічні ефекти повинні бути повністю задокументовані і негайно доведені до відома Держлікслужби відповідно до законодавства.
Забезпечення виконання обов’язків
У разі встановлення Держлікслужбою або органами доходів і зборів факту нанесення маркування знаком відповідності технічним регламентам з порушенням вимог законодавства або відсутності такого маркування, уповноважений представник повинен привести медичні вироби у відповідність.
Врегулювання потребують також випадки, коли маркування знаком відповідності технічним регламентам було нанесене на вироби, на які не поширюється їх дія.
2. Договір між виробником та уповноваженим представником
Виробники передоручатимуть свої права безпосередньо уповноваженим представникам і це бажано зазначати в письмовому договорі. Вони визначатимуть завдання уповноважених представників та межі їхніх повноважень.
Для того щоб переконатися, що ролі представника та виробника є чіткими, рекомендовано, щоб їх зобов’язання були викладені в письмовій формі у договорі між виробником та його уповноваженим представником.
Договір між виробником та уповноваженим представником повинен зазначати, що уповноважений представник зобов’язується інформувати виробника про рішення компетентного органу відносно відмови або заборони допуску виробу на ринок або будь-які дії, пов’язані з введенням в експлуатацію виробу.
Договір між виробником та уповноваженим представником зазначає те, що уповноважений представник зобов’язується інформувати виробника про відомі йому несприятливі ситуації.
Договір між виробником та уповноваженим представником зазначатиме, що виробник зобов’язується інформувати уповноваженого представника з усіх питань, пов’язаних з виробами, які були розміщеними на ринку України.
Орган з оцінки відповідності перевіряє, щоб виробник-нерезидент, призначив уповноваженого представника та уклав договір, в якому зазначив передоручення відповідних обов’язків.
Нижче перелічені очікування компетентних органів влади. Це, в ідеалі, повинно бути зазначено у договорі між виробником та уповноваженим представником.
– Уповноважений представник повинен вміти оцінити здатність виробника виконувати свої регуляторні зобов’язання. Для того щоб виконувати оцінювання, уповноважений представник повинен мати доступ до технічної документації.
– Уповноважені представники повинні мати змогу перевірити існування необхідної інформації/документації у виробника та виконання ним відповідних процесів (наприклад, пост-ринковий нагляд).
– Уповноважені представники повинні мати відповідні знання, навички та ресурси для оцінювання та перевірки зазначеного.
– Компетентні органи повинні мати змогу отримати на запит наступну інформацію від уповноваженого представника:
декларацію відповідності;
копію маркування, упаковки та інструкцію з користування;
сертифікати органу з оцінки відповідності (де необхідно);
дані про процеси пост-ринкового нагляду, звіти та скарги, дані про інші процеси;
технічну документацію, яка необхідна під час нагляду за ринком;
відповідні клінічні дані/повідомлення;
дані про дистриб’юторів/постачальників, які постачають вироби на ринок;
звіти про інциденти та здійснені корегувальні дії.
У випадку виникнення суперечностей, де уповноважені представники вважатимуть, що виробник не відповідає вимогам технічних регламентів, його обов’язком є здійснення переговорів стосовно таких суперечностей з виробником. Якщо суперечність триває, питання повинне бути передано до компетентних органів для отримання рішення. Уповноважений представник має право розірвати договір.
У випадку наявності недотримання виробником умов, які стосуються відповідальності уповноваженого представника та які виробник відмовляється виправити, уповноважений представник має право розірвати договір з виробником. Уповноважений представник має право розірвати договір, якщо невиконання обов’язків виробника викликає порушення державного законодавства з боку уповноваженого представника. Уповноважені представники повинні проінформувати про це компетентні органи та орган з оцінки відповідності виробника.
3. Нормативні вимоги
В таблиці зазначено статті та розділи додатків до технічних регламентів в сфері медичних виробів, які містять вимоги стосовно уповноважених представників.
| ТР щодо МВ | ТР щодо МВ in vitro | ТР щодо АМВІ |
|---|---|---|
| Призначення | ||
| Пункт 31 абзац 2.
Якщо виробник, який під власним іменем вводить в обіг медичні вироби, зазначені в абзаці першому цього пункту, не є резидентом України, він зобов’язаний призначити одного уповноваженого представника, відповідального за введення в обіг цих виробів на ринку України. |
Пункт 16 абзац 5.
У разі коли виробник не є резидентом України, обов’язок щодо надання доступу до зазначеної документації для уповноважених органів державної влади покладається на його уповноваженого представника. |
Пункт 34.
Якщо виробники, не є резидентами України, вони повинні призначити уповноваженого представника в Україні. |
| Реєстрація | ||
| Пункт 31 абзац 2.
Такий представник зобов’язаний повідомити Держлікслужбі щодо свого місцезнаходження і надати перелік та опис відповідних виробів. |
Пункт 23 абзац 10.
У разі коли виробник, який вводить в обіг вироби під своїм найменуванням, не є резидентом України, інформація подається Держлікслужбі уповноваженим представником, якого він призначає. |
Пункт 33 абзац 1.
Держлікслужба може запитувати у виробників зазначених виробів або їх уповноважених представників усі дані щодо ідентифікації таких виробів, зокрема за етикеткою, інструкцією із застосування тощо. |
| Пункт 31 абзац 1.
Будь-який виробник, який під власним іменем вводить медичні вироби в обіг, згідно з процедурами, зазначеними у пунктах 18 і 19 цього Технічного регламенту, а також будь-яка інша фізична особа — підприємець або юридична особа, яка провадить діяльність, зазначену в пунктах 27–30 цього Технічного регламенту, зобов’язана повідомити Держлікслужбі щодо свого місцезнаходження і надати перелік та опис відповідних виробів. |
Пункт 23.
Виробник, який під своїм найменуванням вводить в обіг вироби, повідомляє Держлікслужбі: – своє місцезнаходження; – відомості про реагенти, реагентні продукти, калібратори, контрольні матеріали та про будь-які істотні зміни, включаючи припинення введення в обіг; – щодо виробів, зазначених у додатку 2, та виробів для самоконтролю — дані, що необхідні для ідентифікації таких виробів, аналітичні та в разі потреби діагностичні параметри відповідно до пункту 3 додатка 1, результати оцінки характеристик згідно з додатком 8, інформацію про видані сертифікати та про будь-які істотні зміни, включаючи припинення введення в обіг; – інформацію про виріб, який є новим виробом, із зазначенням інформації про маркування виробу знаком відповідності технічним регламентам. |
Пункт 33 абзац 1.
Виробник, що є резидентом України та під власним найменуванням вводить в обіг вироби, виготовлені на замовлення відповідно до процедури, зазначеної в пункті 20 цього Технічного регламенту, повідомляє Держлікслужбі про своє місцезнаходження, а також надає перелік і опис відповідних виробів. |
| Процедура оцінювання відповідності | ||
| Пункт 21.
Виробник може доручити своєму уповноваженому представникові ініціювати проведення процедур, передбачених у додатках 4, 5, 8 і 9. Дивись також: Додаток 4 пункт 2 Додаток 5 пункт 1 Додаток 8 пункт 1 Додаток 9 пункт 1 |
Пункт 15.
Виробник може уповноважувати свого представника розпочати процедури оцінки відповідності, наведені в додатках 3, 5, 6 і 8. Дивись також: Додаток 3 пункт 1 Додаток 5 пункт 2 Додаток 6 пункт 1 Додаток 8 пункт 1 |
Пункт 21.
Виробник може уповноважити свого представника ініціювати процедури, наведені у додатках 3, 4, і 6. Дивись також: Додаток 2 пункт 2 Додаток 3 пункт 2 Додаток 4 пункти 1 та 2 Додаток 5 пункт 2 Додаток 6 пункт 1 |
| Документація оцінювання відповідності | ||
| Додаток 4 пункт 7.
Інші органи з оцінки відповідності можуть отримати копії сертифікатів перевірки типу та/або додатків до них. Іншим органам з оцінки відповідності надається доступ до додатків сертифікатів на їх обґрунтований запит після інформування виробника про такий запит. Додаток 5 пункт 11. Виробник або його уповноважений представник не менш як п’ять років, а для медичних виробів, які імплантують, — не менш як п’ятнадцять років з моменту виготовлення останнього медичного виробу повинен зберігати для надання на запит уповноважених органів державної влади такі документи:- декларацію про відповідність; – документацію, зазначену в пункті 2 цього додатка; – сертифікати, зазначені в пунктах 6 і 10 цього додатка; – сертифікат перевірки типу, зазначений в додатку 4 до Технічного регламенту щодо медичних виробів. |
Пункт 16.
Виробник протягом п’яти років після виготовлення останнього виробу зберігає: – декларацію про відповідність; – технічну документацію, зазначену в додатках 3–8; – рішення та сертифікати органів з оцінки відповідності. У разі коли виробник не є резидентом України, обов’язок щодо надання доступу до зазначеної документації для уповноважених органів державної влади покладається на його уповноваженого представника. |
Додаток 2 пункт 16.
Виробник або його уповноважений представник протягом не менш як 15 років з моменту виготовлення останнього виробу повинен зберігати для надання в разі потреби Держлікслужбі: – декларацію про відповідність; – документацію, зазначену пункті 3 цього додатка, а також документацію, дані і записи, зазначені в пункті 4 цього додатка; – зміни, зазначені в пунктах 7 і 9 цього додатка; – рішення та звіти органів з оцінки відповідності, зазначені в пунктах 7, 10, 14 і 15 цього додатка. |
| Додаток 6 пункт 12.
Виробник або його уповноважений представник не менш як п’ять років, а для медичних виробів, які імплантують, — не менш як п’ятнадцять років з моменту виготовлення останнього медичного виробу повинен зберігати для надання на запит Держлікслужби: – декларацію про відповідність; – документацію щодо систем управління якістю, зазначену в пункті 3 цього додатка; – зміни, зазначені в пункті 7 цього додатка; – рішення та звіти органу з оцінки відповідності, про які йдеться в пунктах 10 і 11 цього додатка; – технічну документацію на затверджені типові зразки та копії сертифікатів перевірки типу, зазначених в додатку 4 до Технічного регламенту щодо медичних виробів. |
Додаток 3 пункт 9.
Виробник або його уповноважений представник протягом не менш як 15 років з моменту виготовлення останнього виробу повинен зберігати разом з технічною документацією копію сертифіката перевірки типу та додатки до нього для надання на запит Держлікслужби. Додаток 4 пункт 10. Виробник або його уповноважений представник забезпечують можливість надання сертифікатів відповідності, виданих органом з оцінки відповідності, на вимогу інших органів з оцінки відповідності та уповноважених органів державної влади. |
|
| Додаток 7 пункт 8.
Виробник або його уповноважений представник не менш як п’ять років, а для медичних виробів, які імплантують, — не менш як п’ятнадцять років з моменту виготовлення останнього медичного виробу повинен зберігати для надання на запит Держлікслужби такі документи: – декларацію про відповідність; – технічну документацію на затверджені типові зразки та копії сертифікатів перевірки типу; – зміни, зазначені в пункті 5 цього додатка; – рішення та звіти органу з оцінки відповідності, про які йдеться в пунктах 5 і 7 цього додатка; – в разі потреби — сертифікат перевірки типу, зазначений у додатку 4 до Технічного регламенту щодо медичних виробів. |
Додаток 6 розділ 3.
Виробник зобов’язаний зберігати для Держлікслужби такі документи: 1) для виробів, виготовлених на замовлення, — документацію, в якій наводиться опис виробничих ділянок, а також інформацію про розроблення, виробництво та експлуатаційні характеристики виробів, зокрема очікувані експлуатаційні показники, для забезпечення можливості оцінки виконання вимог Технічного регламенту. Виробник зобов’язаний вжити всіх необхідних заходів для забезпечення виготовлення виробів відповідно до зазначеної документації; 2) для виробів, призначених для клінічних досліджень, — документацію, в якій наводиться: – загальний опис виробу та його призначення; – проектні креслення, інформацію про методи виробництва, зокрема методи стерилізації, а також схеми компонентів, вузлів, ланцюгів тощо; – описи та пояснення, необхідні для розуміння зазначених креслень, схем, а також принципів роботи виробу; |
|
| Додаток 8 розділ 2.
Виробник повинен підготувати технічну документацію, зазначену в пункті 3 цього додатка. Виробник або його уповноважений представник протягом п’яти років з моменту виготовлення останнього медичного виробу повинен зберігати для надання на запит Держлікслужби технічну документацію, зокрема декларацію про відповідність, з метою її перевірки. У випадку з імплантованими виробами, такий термін складатиме 15 років. Додаток 9 пункт 4. Інформація, що міститься в заявах, передбачених цим додатком, зберігається не менш ніж п’ять років. Строк зберігання інформації про медичні вироби, які імплантують, становить не менш ніж п’ятнадцять років. |
– результати аналізу ризиків, інформація про стандарти, що застосовуються повністю або частково, з числа тих, що включені до переліку національних стандартів, які відповідають європейським гармонізованим стандартам та добровільне застосування яких може сприйматися як доказ відповідності виробів вимогам Технічного регламенту, а також опис рішень, прийнятих з метою виконання встановлених вимог Технічного регламенту у разі часткового застосування зазначених стандартів;
– якщо виріб містить як невід’ємну частину речовину або похідні крові людини, зазначені в пункті 11 додатка 1 до Технічного регламенту, — дані про випробування, проведені у зв’язку з цим, необхідні для оцінювання безпеки, якості та ефективності даної речовини або похідних крові людини з урахуванням цільового призначення цього виробу; – результати проектних розрахунків та проведених перевірок і технічних випробувань тощо.Виробник зобов’язаний вжити всіх необхідних заходів для забезпечення виготовлення виробів відповідно до документації, зазначеної у підпункті 1 пункту 3 цього додатка та абзаці першому цього підпункту. Виробник повинен забезпечити проведення оцінки за необхідності шляхом проведення перевірки ефективності зазначених заходів. |
|
| Маркування | ||
| Додаток 1 пункт 44.
Етикетка медичного виробу повинна містити такі елементи: 1) найменування або торгову марку і місцезнаходження виробника. Для медичних виробів, що імпортуються з метою введення в обіг, на етикетці, або зовнішньому пакуванні, або в інструкції із застосування додатково зазначається найменування та місцезнаходження уповноваженого представника, якщо виробник не є резидентом України. |
Додаток 1 пункт 40.
Етикетка виробу повинна містити таку інформацію, що може наводитися у формі символів: 1) найменування або торгову марку та місцезнаходження виробника. Для виробів, що імпортуються з метою введення в обіг, на етикетці або зовнішньому пакуванні, або в інструкції із застосування додатково зазначається найменування та місцезнаходження уповноваженого представника, якщо виробник не є резидентом України. |
Додаток 1 пункт 15.
Кожен виріб повинен містити такі відомості (в разі можливості — у вигляді символів), нанесені чітко і розбірливо: 1) на стерильній упаковці: метод стерилізації; позначення, що дозволяє розпізнати упаковку; найменування та місцезнаходження виробника; опис виробу; напис: «виключно для клінічних досліджень», якщо виріб призначений для клінічних досліджень; напис: «виріб, виготовлений на замовлення», якщо виріб виготовлений на замовлення; повідомлення, що виріб є стерильним; місяць і рік виробництва; зазначення строку придатності для безпечної імплантації виробу; |
| 2) на зовнішній упаковці:
найменування та місцезнаходження виробника і найменування та місцезнаходження уповноваженого представника, якщо виробник не є резидентом України; опис виробу; призначення виробу; характеристики для використання; напис: «виключно для клінічних досліджень», якщо виріб призначений для клінічних досліджень; напис: «виріб, виготовлений на замовлення», якщо виріб був виготовлений на замовлення; повідомлення, що виріб є стерильним; місяць і рік виробництва; зазначення строку придатності для безпечної імплантації виробу; умови транспортування і зберігання виробу; для виробу, зазначеного у пункті 5 Технічного регламенту, зазначається, що виріб містить речовину, отриману з крові або плазми крові людини. |
||
| Клінічні дослідження/оцінка характеристик | ||
| Пункт 32 абзац 1.
У разі виготовлення медичних виробів, призначених для клінічних досліджень, виробник або його уповноважений представник, зареєстрований в Україні, повинні дотримуватися процедури, зазначеної в додатку 10, і повідомити про це Держлікслужбі шляхом подання заяви, вимоги щодо заповнення якої зазначені в пункті 2 додатка 10. |
Додаток 8 пункт 1.
Для введення в обіг виробів, призначених для оцінки характеристик, виробник або його уповноважений представник повинен скласти заявку, що містить інформацію, зазначену в пункті 2 цих Процедур, і забезпечити виконання відповідних положень Технічного регламенту щодо медичних виробів для діагностики in vitro. |
Пункт 28.
У разі виготовлення виробів, призначених для клінічних досліджень, виробник або його уповноважений представник за 60 календарних днів до початку досліджень повинен подати Держлікслужбі заяву щодо виробів особливого призначення, вимоги до якої наведені у додатку 6. |
| Застосування прав | ||
| Пункт 46.
У разі встановлення Держлікслужбою або органами доходів і зборів факту нанесення маркування знаком відповідності технічним регламентам з порушенням вимог законодавства або відсутності такого маркування, що є порушенням цього Технічного регламенту, виробник або його уповноважений представник повинні привести медичні вироби у відповідність з вимогами цього Технічного регламенту. |
Пункт 32.
У разі встановлення Держлікслужбою або органами доходів і зборів факту нанесення маркування знаком відповідності технічним регламентам з порушенням вимог законодавства або відсутності такого маркування, що є порушенням вимог цього Технічного регламенту, виробник або його уповноважений представник повинні привести вироби у відповідність з вимогами цього Технічного регламенту. |
Пункт 44.
У разі встановлення Держлікслужбою або органами доходів і зборів факту нанесення маркування знаком відповідності технічним регламентам з порушенням вимог законодавства або відсутності такого маркування, що є порушенням вимог цього Технічного регламенту, виробник або його уповноважений представник повинен привести вироби у відповідність з вимогами цього Технічного регламенту. |
| Рішення відносно заборони або відмови | ||
| Пункт 47.
Якщо порушення не усунуто, Держлікслужба або органи доходів і зборів вживають заходів щодо обмеження або заборони введення в обіг медичного виробу або мають переконатися, що він виведений з обігу, в установленому законодавством порядку. |
Пункт 33.
Якщо порушення залишається, Держлікслужба або органи доходів і зборів вживають заходів до обмеження чи заборони введення в обіг виробу або повинні переконатися, що він виведений з обігу в установленому законодавством порядку. |
Пункт 45.
Якщо порушення залишається, Держлікслужба або органи доходів і зборів вживають заходів обмеження або заборони введення в обіг виробу або повинні переконатися, що він виведений з обігу в установленому законодавством порядку. |
Генеральний директор Фармацевтичного директоратуОлександр Комаріда
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим