|
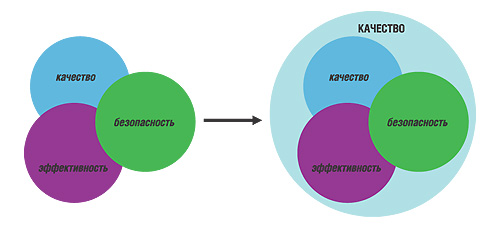
 В рекомендациях ВОЗ, которые посвящены разработке и внедрению национальной лекарственной политики в развивающихся государствах, отмечено, что ЛС должны быть качественными, безопасными и эффективными (How to develop and implement a national drug policy, 2001). Здесь под термином «качество» понимается формальное соответствие препартов их спецификациям, то есть специфическим стандартам. В то же время эти три характеристики объединены модулем КАЧЕСТВО, где под ним в широком смысле понимается именно способность удовлетворять потребности пациента. Схематично это представлено на рис. 1.
В рекомендациях ВОЗ, которые посвящены разработке и внедрению национальной лекарственной политики в развивающихся государствах, отмечено, что ЛС должны быть качественными, безопасными и эффективными (How to develop and implement a national drug policy, 2001). Здесь под термином «качество» понимается формальное соответствие препартов их спецификациям, то есть специфическим стандартам. В то же время эти три характеристики объединены модулем КАЧЕСТВО, где под ним в широком смысле понимается именно способность удовлетворять потребности пациента. Схематично это представлено на рис. 1.
Как видно из данного рисунка, категории качества (как соответствия спецификации), безопасности и эффективности взаимно «проникают» друг в друга. Это вполне естественно, так как при несоответствии, например, количественного содержания или содержания примесей в ЛП, пределам, установленным аналитической документацией на этот препарат, нельзя говорить о его безопасности и эффективности и т.д. В то же время КАЧЕСТВО ЛС в широком смысле охватывает все три категории и именно об этом КАЧЕСТВЕ мы будем вести речь в процессе обсуждения. Во избежание терминологической путаницы, да и просто из уважения к этому интегрированному понятию в контексте нашего обзора, слово КАЧЕСТВО мы будем писать именно таким образом.
|
Важность рассматриваемого вопроса не подлежит сомнению, так как весьма очевидна следующая взаимосвязь:
|
КАЧЕСТВО ЛС |
→ |
Качество |
→ |
Качество жизни |
Чем же и каким именно образом обеспечивается КАЧЕСТВО ЛС? Общепризнанными в мире являются так называемые GXP, то есть надлежащие практики или «правила надлежащей…», соблюдение которых обязательно на всех этапах жизненного цикла ЛС. На этапе разработки препарата, его доклинического изучения, клинических исследований, производства, дистрибьюции и розничной реализации существуют свои правила, соблюдение которых, фактически, и обеспечивает КАЧЕСТВО ЛС.
Такая цепочка надлежащих практик на всех этапах жизненного цикла ЛС (от его разработки до применения) представлена на рис. 2.
|
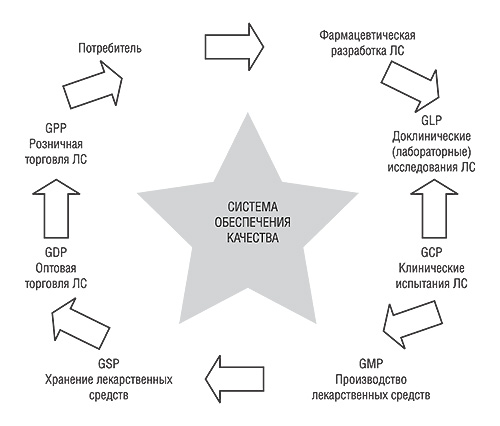
Так, Надлежащая лабораторная практика (GLP), Надлежащая клиническая практика (GCP), Надлежащая производственная практика (GMP), Надлежащая практика хранения (GSP), Надлежащая практика дистрибьюции (GDP) и Надлежащая фармацевтическая практика (GPP) — звенья одной цепи, поскольку каждая занимает свой сегмент жизненного цикла продукции. Например, продукция, изготовленная в соответствии с правилами GMP, может испортиться в период дистрибьюции, если не будут соблюдены соответствующие требования (стандарты GDP). То же самое произойдет при несоблюдении любой из надлежащих практик — прервется вся цепочка и фактически КАЧЕСТВО ЛС станет сомнительным, поскольку никто не сможет его гарантировать и обеспечивать.
Для того чтобы нагляднее проиллюстрировать обязательность исполнения надлежащих практик и соответствующие государственные регулирующие процедуры, которые при этом применяются во всем цивилизованном мире, представим рис. 3 (Левашова И.Г., Мурашко А.Н., Подпружников Ю.В., 2006).
|
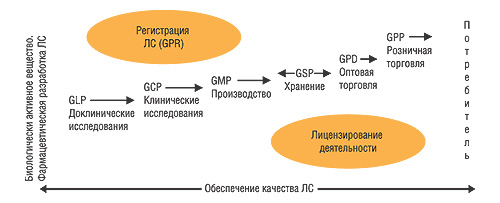
Таким образом, единственной общепризнанной в мире системой обеспечения КАЧЕСТВА ЛС является система надлежащих практик на всех этапах жизненного цикла препаратов. Это необходимо было понять для того, чтобы в дальнейшем можно было профессионально взвесить все «за» и «против» для оригинальных и генерических ЛС.
При этом мы проанализируем, какова ситуация в Украине с установлением и соблюдением принятых и используемых в ЕС надлежащих практик и самих регуляторных систем, а также как эта ситуация отражается на отечественном фармацевтическом рынке.
КРАТКАЯ ХАРАКТЕРИСТИКА УКРАИНСКОГО ФАРМАЦЕВТИЧЕСКОГО РЫНКА
Объем фармацевтического рынка Украины в 2006 г. в стоимостном выражении составил 1,86 млрд дол. США (Еженедельник «Аптека», № 6 (577) от 12 февраля 2007 г.). Величина рынка рассчитывалась исходя из формулы: производство+импорт–экспорт. Отечественный фармрынок уже не менее 5 лет демонстрирует стойкую тенденцию к росту в денежном выражении на уровне не менее 20% в год. Если в 2002–2003 гг. считалось, что пределом мечтаний может быть объем рынка Украины, равный 1 млрд дол., то в 2007 г. он достиг двухмиллиардного (в долларах США) рубежа.
Объем импортных ЛС за 9 мес 2007 г. в денежном выражении составил 5,8 млрд грн. (+39,6% по сравнению с аналогичным периодом предыдущего года), объем отечественного производства медикаментов за тот же период — 2,5 млрд грн. (+8,4%). Объем фармрынка за 9 мес 2007 г., рассчитанный по приведенной выше формуле, составил в ценах производителя 7,9 млрд грн., увеличившись на 28,1% по сравнению с аналогичным периодом предыдущего года («Еженедельник АПТЕКА», № 45 (616) от 19 ноября 2007 г.).
По состоянию на 1.12.2007 г. в Украине зарегистрирован 15 591 препарат. Под препаратами мы в данном случае понимаем медикаменты, различающиеся торговым названием, производителем, лекарственной формой, дозой и первичной упаковкой. То есть, упакован ли он в блистеры № 10 или № 30, в пачку вложен 1 или, например, 3 блистера — в нашем обзоре мы будем считать его одним препаратом. В то же время, если таблетки или капсулы упакованы, например, в блистер или баночку — это будет 2 разных препарата (помня предыдущие определения, в дальнейших рассуждениях нами будет сделан акцент именно на обеспечении КАЧЕСТВА препаратов).
В выступлениях руководителей Государственного фармакологического центра констатировалось, что Украина является генерической страной, когда процент оригинальных препаратов составляет около 7–8%, а генерических — около 90%.
ТЕРМИНОЛОГИЯ
Во избежание путаницы вначале определимся с терминологией, то есть с тем, что мы будем понимать под оригинальными и генерическими ЛС.
Оригинальным мы будем считать препарат, который был зарегистрирован в Украине в установленном порядке на основе экспертизы полного регистрационного досье, согласно приказу Министерства здравоохранения (МЗ) Украины от 26.08.2005 г. № 426. Если с оригинальным препаратом сравнивают генерический в процессе государственной регистрации последнего по сокращенной процедуре (сокращенному досье), тогда он выполняет функцию референтного.
Наличие полного досье означает, что препарат прошел все стадии доклинического изучения и все (как правило — 4) фазы клинических исследований, материалы всех этих исследований представлены в досье. Другими словами — это эталон, с которым должны сравнивать все копии (генерические препараты), выходящие на рынок по истечении срока патентной защиты на оригинатор.
Определение оригинального препарата, приведенное в приказе МЗ Украины от 11.09.2006 г. № 536: «ЛС, защищенное патентом, которое поступает в обращение под собственным зарегистрированным названием» — нельзя признать корректным. Во-первых, оригинатор не всегда защищен патентом, а во-вторых, он остается оригинальным и референтным и после истечения срока патента. Норма о собственном зарегистрированном названии вообще не выдерживает критики, так как под эту норму подпадают практически все зарегистрированные брэндированные генерики, когда патентом защищается торговое название. Поэтому под оригинальным препаратом мы будем понимать именно препарат, который был зарегистрирован по полному досье. Насколько это принципиально? Дело в том, что на разработку и полное исследование препарата уходит не менее 5 лет и стоит это не менее 1 млрд дол. Поэтому ситуацию, когда генерическая компания подает полное досье на препарат для признания его оригинальным (референтным), представить себе невозможно.
Под генерическим (многоисточниковым, по сути аналогичным) ЛС мы будем понимать фармацевтически эквивалентный (содержит то же количество того же действующего вещества и в той же лекарственной форме, что и референтный), либо фармацевтически альтернативный (отличается от референтного химической формой субстанции, или лекарственной формой, или силой действия). Генерический препарат может быть терапевтически эквивалентным, либо неэквивалентным оригинальному (EMEA, 2001; Приказ МЗ Украины от 17.04.2007 г. № 190).
Для более обоснованного анализа, который мы проведем далее, из всех зарегистрированных препаратов необходимо вычленить еще 2 большие группы. В первую очередь, это так называемые традиционные препараты или grandfather’s drugs, то есть «дедушкины» (или, как у нас почему-то говорят, «бабушкины») лекарства. Речь идет о препаратах, которые могут регистрироваться по сокращенной процедуре (по сокращенному досье), если их применение в медицинской практике было очень длительным (не менее 30 лет).
Действительно, кому придет в голову при регистрации либо перерегистрации цветков календулы либо настойки валерианы требовать от заявителя данных по токсичности или исследования этих препаратов в клинике? А до недавнего времени приходило! Однако Европейская директива (Directive 2004/24/EC, 2004) узаконила процедуру сокращенной регистрации для традиционных препаратов растительного происхождения. При этом существует ряд оговорок: препараты должны быть безрецептурными, применяемыми в определенных концентрациях, предназначенными для орального, наружного или ингаляционного применения и т.д.
В Украине мы стремимся к гармонизации нормативных документов, поэтому в приказе МЗ Украины от 11.09.2006 г. № 536 также появилось определение традиционных ЛС. Однако так называемую гармонизацию мы иногда проводим с учетом национальной специфики (об этом еще неоднократно будет упомянуто в данном обзоре). В частности, в этом же приказе появилась вроде бы невинная приставка о том, что традиционный — это препарат прежде всего растительного происхождения. В результате под понятие «прежде всего» можно подвести все что угодно и зарегистрировать по совершенно формальным критериям.
Мы уже не говорим, что данная норма, существовавшая в более раннем приказе МЗ Украины от 26.08.2005 г. № 426, вообще почему-то запрещала использовать для традиционных препаратов сокращенную процедуру регистрации. Отметим, что отсутствие до настоящего времени официального списка традиционных ЛС в Украине делает такую процедуру непрозрачной и переводит ее в режим «ручного управления».
Из списка зарегистрированных в Украине препаратов мы попытались вычленить препараты растительного происхождения, которые использовались в медицинской практике не менее 30 лет, из них не менее 15 лет — в Украине. Кроме того, к данной категории были отнесены такие препараты, как йод, бриллиантовый зеленый, раствор аммиака, мазь серная и пр. При сопоставлении оригинальных препаратов и генериков обсуждать традиционные ЛС смысла нет — у них в принципе не может быть полного досье, соответственно, оригинатора.
Сложнее ситуация с теми препаратами, которые формально подпадают под указанный выше «возрастной ценз», то есть те, которые применялись еще со времен Советского Союза, но по другим характеристикам, прежде всего — безопасности (парентеральная форма введения, возможные побочные реакции и т.д.) не могут быть отнесены к традиционным. С другой стороны, к генерическим их тоже отнести нельзя, поскольку референтных препаратов для них не существует в принципе. Например, если какой-то производитель сегодня регистрирует и начинает выпуск таблеток анальгина, либо инъекционного раствора натрия хлорида во флаконах, то с каким препаратом как референтным можно его сравнить и нужно ли это делать? Для такой категории ЛС наиболее удачным мы посчитали название, приведенное в приказе МЗ Украины от 26.08.2005 г. № 426 — «препараты с хорошо изученным медицинским применением».
Итак, анализ зарегистрированных в Украине препаратов с использованием приведенных выше категорий приводит нас к следующим показателям (таблица).
Таблица
Количество зарегистрированных в Украине по состоянию на 1.12.2007 г. ЛС с учетом торгового названия, производителя, лекарственной формы, дозировки и первичной упаковки в разрезе категорий ЛС
| №п/п | Категория ЛС | Количество ЛС, зарегистрированных в Украине | Доля зарегистрированных ЛС, % |
| 1. | Оригинальные | 1284 | 8,2 |
| 2. | Генерические | 8528 | 54,7 |
| 3. | Традиционные | 2050 | 13,1 |
| 4. | С хорошо изученным медицинским применением | 2552 | 16,4 |
| 5. | Субстанции и in bulk | 1177 | 7,6 |
| Всего | 15 591 | 100,0 | |
Поскольку в Украине пока не существует официально утвержденного списка оригинальных ЛС, к этой категории были отнесены препараты, которые рекомендуют в качестве оригинальных ВОЗ, Управление по контролю за пищевыми продуктами и лекарственными средствами США (FDA) и Британский национальный формуляр (ВNF). Отметим, что все они в свое время проходили процедуру регистрации по полному досье и являются действительно оригинальными в соответствии с их определением, приведенным выше.
Украина действительно является генерической страной, при этом доля генериков составляет 55% всех зарегистрированных в стране ЛС (см. таблицу).
|
Более половины всех зарегистрированных в Украине ЛС — генерики |
Почему же у нас так много генериков? Ответ на этот вопрос лежит в экономической плоскости. Средневзвешенная розничная стоимость упаковки генерического препарата составляет 7,5 грн., тогда как оригинального — 26 грн., то есть генерический препарат в среднем на 70% дешевле оригинального.
Это происходит в результате того, что компания, которая разработала и вывела на рынок оригинальный препарат, затратила на это сотни миллионов, а то и миллиарды долларов. Генерическая же компания сделала свою копию и зарегистрировала ее по сокращенному досье — без результатов доклинических испытаний и нескольких фаз клиники. Очень редко регистрационное досье может содержать материалы исследований биоэквивалентности (об этом подробно речь пойдет ниже). Выходящая таким образом на рынок Украины копия, конечно же, гораздо дешевле, чем оригинальный препарат, но будет ли при этом объективно обеспечен и доказан эквивалентный уровень качества, безопасности и эффективности генерика, то есть, будет ли он КАЧЕСТВЕННЫМ? Доказана ли объективно терапевтическая эквивалентность или взаимозаменяемость с оригинальными препаратами для генерических препаратов, используемых в лечебном процессе?
Для того чтобы дать ответ на этот вопрос, постараемся проанализировать движение препаратов в Украине по всем стадиям их жизненного цикла. При этом оговоримся сразу, что препараты не имеют национальности, то есть соответствие общепринятым надлежащим практикам должно быть обязательным для ЛС, производимых как в США, так и в Украине, как в Великобритании, так и в Индии.
Вот здесь уже мы наталкиваемся на первое «но». Это «НО» можно написать заглавными буквами, поскольку оно связано с безнадежной отсталостью нашей законодательной базы от динамики развития фармацевтического сектора Украины и развитых странах. Дело в том, что еще в 2001 г. были подготовлены изменения и дополнения к Закону Украины «О лекарственных средствах», который с момента его принятия в 1996 г. устарел, не отражает реалии фармацевтического сектора и не способствует гармонизации. Это, по сути, даже не изменения и дополнения, а, фактически, новая редакция закона. На него были получены положительные отзывы специалистов фармацевтического права ЕС, он был поддержан Европейской штаб-квартирой ВОЗ и центральным аппаратом в Женеве. В этом документе содержатся требования по соответствию всех этапов оборота ЛС надлежащим практикам (GLP, GCP, GMP, GDP), по фармацевтической разработке и розничной реализации. По терминологии и сути он максимально приближен к основной в ЕС Директиве (Directive 2001/83/EC, 2001), регулирующей сферу обращения ЛС. Самое интересное, что этот проект закона был принят в первом чтении еще в мае 2002 г. Верховной Радой Украины! Однако прошло больше 5 лет, но до второго чтения и принятия этого Закона в целом у наших парламентариев дело не дошло.
В этом кроется корень всех дальнейших проблем с обязательным введением в Украине общепринятых стандартов обеспечения качества ЛС на протяжении всего их жизненного цикла. Так, Руководства (укр. — Настанови) по GMP (42-01-2001), GDP (42-01-2002), GCP (42-7.0:2005), утвержденные приказами МЗ Украины, соответственно, в 2001, 2002 и 2005 г., имеют рекомендательный, а не обязательный характер, поскольку таких обязательных норм в законодательстве Украины нет. Следует отметить, однако, что постановлением Кабинета Министров Украины от 28 октября 2004 г. № 1419 МЗ Украины поручено начиная с 1.01.2009 г. обеспечить обращение ЛС в соответствии с требованиями GLP, GCP, GMP и GDP, гармонизованными с соответствующими директивами ЕС и ВОЗ.
Ниже мы коснемся каждого из упомянутых стандартов и проанализируем их внедрение в Украине. А то, что несоответствие этим стандартам (или хотя бы одному из них) ставит под угрозу и не гарантирует КАЧЕСТВО ЛС, нет смысла доказывать — это очевидно.
ФАРМАЦЕВТИЧЕСКАЯ РАЗРАБОТКА
Итак, начнем обсуждать стандарты качества на всех этапах оборота ЛС (см. рис. 2) и состояние с их внедрением в Украине с первого шага — фармацевтической разработки. Европейским агентством по ЛС (ЕМЕА) в 1996 г. было принято руководство, содержащее требование к фармацевтической разработке препаратов (СРМР/QWP/155/96). Исполнение этого руководства стало обязательным при подаче на регистрацию досье на ЛС в страну ЕС. В Украине перевод этого документа появился в виде Руководства МЗ Украины 42-3.1-2004, которое было утверждено соответствующим приказом МЗ Украины.
Позже, в 2005 г., в рамках работы Международной конференции по гармонизации (задачей этой организации является глобальная гармонизация требований к медикаментам в рамках рынков США, ЕС и Японии) появился документ Q8 «Фармацевтическая разработка». Он был принят и по условиям этой организации уже имплементирован в законодательства государств-членов. Этот документ фактически стал продолжением и развитием документа СРМР/QWP/155/96. Он связан с требованиями к модулю «качество» в регистрационном досье, которое в этих странах подается в формате общего технического документа (CTD).
Документ Q8 впервые вводит понятие жизненного цикла ЛС, под которым подразумевают все стадии существования препарата от начальной разработки, выхода на рынок и до прекращения пребывания на нем. Отмечается, что КАЧЕСТВО должно быть заложено в продукт уже на стадии его разработки, то есть оно «встраивается» в продукт. Документ детально описывает требования к фармацевтической разработке, в том числе к разработке состава, включая выбор компонентов и обоснование этого выбора, к технологии производства, включая обоснование оптимальных режимов ведения технологического процесса, системы закрытия контейнеров (первичной упаковки) и т.д.
Еще дальше и системнее, чем Q8, продвинулся в вопросах непрерывного обеспечения качества ЛС документ Q10 Международной конференции по гармонизации (). Он, кстати, так и называется: «Фармацевтическая система качества». Его проект, так же, как и все другие документы ICH, находится в свободном доступе на официальном веб-сайте этой организации. Под фармацевтической разработкой, которая охватывается глобальной системой качества, понимается:
- разработка субстанций;
- разработка нового наполнителя;
- разработка лекарственной формы (включая тару/систему упаковки);
- разработка системы доставки (если это применимо);
- разработка технологического процесса и масштабирование;
- разработка аналитического метода.
В Украине документы, соответствующие ICH Q8 и ICH Q10, не приняты ни в виде руководств, ни в виде рекомендаций. Сегодня ведущие предприятия Украины самостоятельно переводят эти документы и пытаются внедрить требования, содержащиеся в них. Определенные сведения об этих документах можно получить лишь на семинарах, которые проводят в Украине немногочисленные специалисты, никак не связанные с регуляторными органами.
Но если бы все проблемы в области отечественной фармацевтической разработки ограничивались лишь незнанием наиболее современных документов, это было бы еще не так страшно. Самое ужасное то, что в Украине на сегодня можно пересчитать по пальцам предприятия, которые технически и организационно готовы соответствовать данным требованиям и стремятся к этому. Это, в частности, «Дарница», Борщаговский химико-фармацевтический завод, «Фармак», «Артериум», «ФармаСтарт», «ИнтерХим» (если я случайно кого-то не упомянул, прошу меня простить). На каждом из этих предприятий есть фактически собственный опытно-внедренческий центр, укомплектованный высококлассными специалистами, которые целенаправленно занимаются фармацевтической разработкой. Пилотное оборудование, которым оснащены данные центры, позволяет проводить масштабирование технологии производства разработанных ЛС, поскольку является уменьшенной копией промышленного оборудования, которое установлено на производственных линиях.
|
Количество фармпредприятий в Украине, которые способны обеспечить фармацевтическую разработку, ограничено |
Относительно же других более чем 150 лицензированных производителей ЛС в Украине этого сказать нельзя. Я утверждаю это как человек, стоявший у истоков создания Инспектората GMP и до 2006 г. возглавлявший его. Я и мои сотрудники инспектировали абсолютно все украинские предприятия, имевшие в то время лицензию на производство ЛС. Для того чтобы снять возможные обвинения в непатриотизме, повторю ранее изложенный тезис о том, что препараты национальности не имеют. Для пациента и врача не важно, где препарат разработан и произведен, главное, чтобы он лечил, то есть был КАЧЕСТВЕННЫМ. А это, в свою очередь, возможно лишь при соблюдении системы обеспечения качества по всему жизненному циклу препарата. Оговорюсь при этом, что страны ЕС для гарантии еще и правовой ответственности за КАЧЕСТВО ЛС, импортируемых из третьих стран, нашли очень простой выход.
В Директиве 2001/83/EC от 6 ноября 2001 г. указано, что торговая лицензия (по нашему — регистрационное свидетельство) на препарат может быть выдана только заявителю, зарегистрированному на территории ЕС. Таким образом, если с КАЧЕСТВОМ препарата в ЕС возникнут проблемы, наступит юридическая ответственность заявителя, который находится в ЕС.
В Украине, к сожалению, такой правовой нормы нет, то есть мы не защищены от возможных больших неприятностей в случае возникновения серьезных проблем с КАЧЕСТВОМ импортных медикаментов. При этом если отечественные препараты исследуют на этапе доклиники и клиники, то большинство импортных — нет. Одно дело — досье компании на оригинальный препарат, которая провела огромные по объему и доказательности исследования, производство соответствует требованиям GMP ЕС или FDA США, и совсем другое — когда компания находится в третьих странах, где о КАЧЕСТВЕ знают понаслышке. В таких случаях необходимо в процессе регистрации на все 100% убеждаться в наличии у данной компании и ее препаратов всей цепочки обеспечения КАЧЕСТВА и ее эффективности.
Одной из самых больших проблем, оставшихся в наследство от фармацевтической отрасли Советского Союза, было и остается то, что разрабатывается в лаборатории один препарат, исследуется на животных — другой, в клинике испытывается третий, а поступает в продажу — четвертый, и при этом речь вроде бы должна идти об одном и том же препарате! Почему так происходит? Да потому, что невозможно без внесения изменений воспроизвести в промышленных масштабах технологию производства препарата, полученного изначально в пробирке или ступке в лаборатории.
При изменении технологии неизбежно меняются свойства препарата, в том числе не только технологические и физико-химические, но и фармакотерапевтические. Не зря в Приложении 13 GMP ЕС (ему соответствует Приложение L украинских GMP (Руководство МЗ Украины 42-01-2001)) «Производство исследуемых лекарственных средств» содержится требование о том, что клинические испытания должны проводиться для препаратов, полученных в промышленных условиях и, естественно, с соблюдением требований GMP. То же соответствие требованиям GMP для препаратов, которые используются в процессе клинических испытаний, содержится в Директиве ЕС (DIRECTIVE 2001/20/EC, 4 April 2001), то есть является нормой Закона.
Для того чтобы разрабатывался, испытывался и затем поступал на рынок один и тот же препарат, в руководстве EMEA 2001 г. содержится норма о том, что серия препарата, биоэквивалентность которого исследуется, должна составлять не менее 1/10 объема промышленной серии или 100 тыс. ед. продукции (выбирается большая цифра из двух).
Чтобы завершить анализ состояния фармацевтической разработки в Украине, необходимо остановиться на отечественной фармацевтической науке. Традиционно из всех научных учреждений Украины больше всего препаратов разрабатывается в Государственном научном центре лекарственных средств (ГНЦЛС). Это и понятно, ведь ГНЦЛС был в свое время головным в СССР по готовым ЛС. В нем в начале 90-х годов ХХ в. работало более 500 научных сотрудников, он по праву мог называться флагманом отечественной фармацевтической науки.
К сожалению, времена изменились радикально, и сейчас там трудится чуть более ста научных сотрудников соответствующего возраста. Оборудование, которым мог гордиться институт во времена Брежневского застоя, сейчас безнадежно устарело морально и технически. В настоящее время на базе ГНЦЛС невозможно провести фармацевтическую разработку в соответствии с требованиями ICH Q8 и ICH Q10 (я проработал в ГНЦЛС более 10 лет и поэтому могу утверждать это с полной ответственностью). Какой же выход? Его нашли ведущие отечественные заводы, создавшие в течение последнего десятилетия на собственной базе исследовательские центры, укомплектованные специалистами и оснащенные самым современным технологическим и аналитическим оборудованием.
Следует отметить, что зарубежные фармацевтические фирмы изначально двигались по этому пути, поскольку никто не надеялся и не надеется, что государство в лице какого-то института подарит частной фирме свежеразработанный препарат. Фирмы сами занимаются фармацевтической разработкой, дальнейшим исследованием и продвижением препарата на рынке. Например, компания «Novartis» имеет 4 исследовательских института, один из них с несколькими отделениями в разных странах, общее количество ученых, которые занимаются исследованиями и разработкой новых ЛС в этой фирме, превышает 4,5 тыс. ( ).
В крупнейшей в мире фармацевтической корпорации «Pfizer» работают более 13 тыс. ученых, которые проводят исследования в 19 научных центрах корпорации ( ).
Еще раз подчеркнем, что ни в коем случае никто не собирается обижать отечественных фармпроизводителей. Мы анализируем систему обеспечения ЛС и реальное положение дел в Украине с КАЧЕСТВОМ препаратов — оригинальных и генерических.
ДОКЛИНИЧЕСКОЕ ИССЛЕДОВАНИЕ
Следующий за фармацевтической разработкой этап обращения ЛС — их доклиническое изучение. Первый пункт Хельсинской декларации Всемирной медицинской ассоциации (WHA), содержащей рекомендации по проведению биомедицинских исследований с участием людей, гласит: «Биомедицинские исследования на людях должны проводиться в соответствии с общепринятыми научными принципами и основываться на адекватной постановке лабораторных экспериментов, использовании животных и изучении научной литературы».
Таким образом, обязательным требованием, принятым практически во всех странах мира, является проведение перед клиническими испытаниями нового (оригинального) ЛС доклинических исследований in vitro и на животных — на моделях in vivo с соблюдением правил GLP.
Концепция GLP зародилась в США в 70-е годы ХХ в., когда сотрудники FDA, не удовлетворенные качеством выполнения и обработки результатов экспериментальных исследований, начали создание ее основных принципов. Впервые правила GLP были сформулированы в США в 1976 г. и вступили в силу с 1979 г.
Наиболее широкое международное признание получили правила GLP Организации экономического сотрудничества и развития (Organization for Economic Cooperation and Development — OECD). Она объединяет 30 индустриально развитых стран Северной Америки, ЕС и бассейна Тихого океана. Членов этой организации называют еще золотой миллиард, поскольку в странах OECD производят более 2/3 ВВП всей Земли и в них проживает около 1 млрд человек. Украина в этом году начала предпринимать интенсивные попытки для вступления в данную организацию.
Правила GLP OECD были изданы впервые в 1981 г., пересмотрены в 1997 г. В ЕС правила GLP приняты в виде Директивы Европарламента и Совета Европы 2004/10 от 11.02.2004 г. «О гармонизации законов, постановлений и административных условий, касающихся применения принципов надлежащей лабораторной практики…». Часть 2 этого документа эквивалентна принципам GLP OECD.
Не будем детально останавливаться на содержании этих достаточно подробных правил, эффективность которых была доказана многолетней практикой. Приведем лишь определение: «GLP — это система качества, охватывающая организационный процесс и условия, при которых выполняются неклинические исследования, связанные со здоровьем и экологической безопасностью: планируются, проводятся, проверяются, регистрируются, архивируются и оформляются в виде отчета».
Отметим, что биоаналитические лаборатории, которые участвуют в исследовании биоэквивалентности генерических препаратов (о них речь пойдет далее), также должны соблюдать требования GLP и соответствовать им. Это многолетняя практика развитых стран, кроме того, на необходимость соблюдения GLP в биоаналитических лабораториях однозначно указывает пункт 20.1 WHO TRS 937 (2006), ann. 9. Процедура проверки соблюдения GLP детально изложена в Директиве ЕС 2004/9.
Какова же ситуация с GLP в Украине? К сожалению, самих правил GLP, соответствующих GLP ЕС, в Украине хотя бы на уровне утвержденного МЗ руководства для добровольного применения просто не существует. Есть приказ МЗ Украины от 01.11.2001 г. № 441, зарегистрированный в Минюсте, который излагает порядок проведения доклинического изучения ЛС. В том же документе содержится порядок определения организаций, которые проводят доклинические исследования. В тексте документа есть упоминание о том, что «при проведении доклинических исследований Исполнитель руководствуется правилами GLP, …, с учетом требований, применяемых в международной практике, в частности, в директиве ЕС 75/318».
Естественно, трудно ожидать, что кто-либо всерьез может воспринять эту «обязательную» норму приказа при отсутствии как минимум самих официально утвержденных в Украине правил GLP. С другой стороны, при отсутствии такой обязательной нормы в законе даже то, что GLP хотя бы упоминается в минздравовском приказе — это уже прогресс, который, все же ограничивается лишь благими намерениями.
|
Правил GLP, соответствующих GLP ЕС в Украине хотя бы на уровне утвержденного МЗ руководства, для добровольного применения не существует |
Напомним, что требования GLP для доклинического изучения касаются только оригинальных препаратов. Никому за рубежом и в голову не придет испытывать на животных генерический препарат, токсичность, показания к применению и дозы которого известны из полного досье оригинального препарата, находящегося много лет на рынке (при соблюдении в стране патентных прав — 6–10, иногда до 20 лет патентной защиты). Более того, ст. 10 основной регулятоной Директивы ЕС (Directive 2001/83/EC, 2001) прямо указывает на то, что от заявителя при регистрации генерического препарата не требуется предоставление данных о доклиническом изучении и клинических исследованиях.
За рубежом для генерического препарата требуется доказать его терапевтическую эквивалентность или взаимозаменяемость с оригинальным. Поскольку наши регуляторные органы целенаправленно начали работать в направлении биоэквивалентности всего 2–3 года назад, общепринятой у нас практикой регистрации генериков до недавнего времени была следующая. Генерический препарат должен был пройти доклинические испытания на животных, а затем — ограниченные клинические испытания на пациентах (больных), чтобы получить допуск на рынок. При этом в первую очередь торжествовал принцип «бей своих, чтоб чужие боялись». То есть, и доклинические, и ограниченные клинические испытания требовались в обязательном порядке для отечественных генериков с мотивировкой, что, мол, досье ваше подано не в CTD-формате. При этом масса импортных препаратов проходила одобрение списком, особенно это касалось in bulk (нерасфасованных) препаратов индийского производства.
Что же лаборатории, которые исследовали и исследуют препараты на доклиническом этапе? В доступных нам источниках их список невозможно найти, но полагаем, что в Украине их не менее 30 (НФаУ, мединституты, профильные институты АМН, некоторые институты НАН Украины и научные центры). Из них лишь 1 лаборатория по результатам зарубежных аудитов и инспекций подтвердила свое соответствие требованиям GLP.
Это лаборатория с образцово-показательным мини-виварием Института фармакологии и токсикологии АМН Украины. Отметим это как знаменательное событие, ведь для соответствия GLP необходимы и виварий с современной воздухоподготовкой и поточностью, и чистые линии животных, и стандартизованные и контролируемые корма, и протоколирование каждого шага в исследованиях, и пр., и пр.
Что же остальные лаборатории? По-прежнему продолжают делать десятикратно повторяющиеся испытания токсичности генериков для того, чтобы отечественный производитель мог зарегистрировать препарат в Украине. После этого отчет о доклинике можно благополучно положить в корзину, поскольку за рубежом он все равно никому не нужен (может быть, до поры нужен пока еще в некоторых странах СНГ, стоящих в процессе гармонизации с ЕС еще дальше Украины). Что же до исследования оригинальных препаратов — как мы уже говорили, без соответствия правилам GLP ни одно исследование, сделанное в такой лаборатории, не будет признано в цивилизованных странах.
|
По результатам международного аудита в Украине только 1 лаборатория, которая исследует препараты на этапе доклиники, подтвердила свое соответствие требованиям GLP |
Что касается биоаналитических лабораторий, их в Украине в настоящее время 3. Пока только одна из них имеет аккредитацию со стороны Госстандарта. Все 3 лаборатории декларируют свое соответствие требованиям GLP. Однако, к сожалению, не существует органа, который мог бы проинспектировать лабораторию и выдать по результатам инспекции документ о соответствии правилам GLP в Украине, как нет, собственно, официально принятых и самих правил. В этих условиях тем лабораториям, которые хотят работать на уровне, признаваемом во всем мире, остается одно — обращаться в зарубежные инспекции и проходить аудит там.
КЛИНИЧЕСКИЕ ИСПЫТАНИЯ
Следующий этап жизненного цикла ЛС (см. рис. 2) — клинические испытания.
Начнем с цитаты. «Надлежащая клиническая практика (GCP) — международный этический и научный стандарт качества планирования и проведения клинических исследований ЛС на человеке, а также документального оформления и представления их результатов. Соблюдение правил GCP для общества является гарантией достоверности результатов клинических испытаний, безопасности субъектов испытаний, охраны их прав и здоровья в соответствии с основополагающими принципами Хельсинкской декларации» (Руководство МЗ Украины 42-7.0:2005). Лучше о сути GCP сказать невозможно.
В ЕС стандарты GCP, в соответствии с которыми проводятся клинические испытания, изложены в Директиве (Directive 2001/20/EC, 2001) и Руководстве (CPMP/ICH/135/95 (E6)). Последнее, кстати, полностью гармонизовано с документом ICH Е6, поэтому имеет двойной номер. Следует отметить, что Государственный фармакологический центр (ГФЦ) МЗ Украины давно и целенаправленно ведет работу по гармонизации требований к проведению клинических испытаний, чтобы правила GCP прижились на украинской земле. И все это в условиях, когда норма обязательности GCP до сих пор, к сожалению, не содержится в Законе Украины «О лекарственных средствах», о чем шла речь выше.
В Украине переведено, утверждено приказом МЗ Украины как рекомендательное и издано Руководство по GCP (Руководство МЗ Украины 42-7.0:2005), являющееся квалифицированным переводом Руководства ЕС и ICH. Кроме того, в нашей стране были подготовлены и изданы монографии, популяризующие GCP (Мальцев В.И., Ефимцева Т.К., Белоусова Ю.В., Коваленко В.Н., 2002; 2006).
Была создана также соответствующая нормативно-правовая база, в которой оговорены требования к проведению клинических испытаний и приведено Типовое положение о Комиссии по этике (приказ МЗ Украины от 13.02.2006 г. № 66), изложен порядок определения лечебно-профилактических учреждений, в которых могут проводиться клинические испытания (приказ МЗ Украины от 17.05.2007 р. № 245).
В разделе 3 приказа МЗ Украины от 13.02.2006 г. № 66 «Общие принципы проведения клинических испытаний» содержится правовая норма о том, что планирование, проведение и отчетность всех фаз клинических испытаний, в том числе испытаний биодоступности и биоэквивалентности, осуществляются с соблюдением требований надлежащей клинической практики, утвержденных МЗ Украины. Там же содержится указание о том, что производство и хранение исследуемого ЛС должно проводиться в соответствии с требованиями GMP, что совпадает с требованиями GCP ЕС.
В приказе МЗ Украины от 17.05.2007 г. № 245 отмечено, что лаборатория для проведения фармакокинетических исследований, участвующая в клинических испытаниях, должна отвечать требованиям GLP. Поскольку эти документы зарегистрированы в Минюсте Украины, следовательно, и они сами, и данные нормы являются обязательными (де-юре) в Украине. Мы должны снять шляпу перед сотрудниками ГФЦ, которые смогли отстоять GCP и GLP в упомянутых документах, не имея подобных правовых норм в законе (в свое время автор этих строк готовил, согласовывал и регистрировал в Минюсте не менее 10 приказов МЗ Украины, поэтому поверьте мне, что это очень и очень непросто).
В приказе МЗ Украины от 17.05.2007 г. № 245 помимо прочих требований к клиническим базам указано, что «врачи, которые будут принимать участие в клинических испытаниях, должны… быть знакомы с международными требованиями надлежащей клинической практики». Это, безусловно, также является положительным моментом для дальнейшего внедрения GCP в Украине.
Во многом благодаря прогрессу во внедрении стандартов GCP в Украине отмечено ежегодное увеличение количества международных многоцентровых клинических испытаний. Если в 2000 г. таких испытаний было проведено 36, то в 2006 — 158, а за 10 мес 2007 г. — уже 146. Эта динамика свидетельствует о доверии иностранных фирм, которые являются спонсорами данных испытаний, к регуляторной системе и самим клиническим базам, об их соответствии требованиям GCP, что подтверждается проведенными аудитами и инспекциями. Повторимся, что при несоблюдении в клиническом исследовании требований GCP его результаты не могут быть признаны ни в одном регуляторном органе цивилизованных стран.
|
Количество ежегодно проводимых в Украине международных многоцентровых клинических испытаний значительно увеличивается. За первые 10 мес 2007 г. было проведено 146 таких испытаний |
Но не все так гладко с внедрением GCP в Украине, на очереди — ложка дегтя к описанной бочке меда. Существует ряд важных вопросов обеспечения КАЧЕСТВА препаратов, касающихся клинических испытаний (в частности, изучения фармакокинетики и биоэквивалентности) и следующих этапов обращения ЛС, таких как производство, хранение, дистрибьюция и розничная реализация. Подробнее о них мы расскажем в следующем номере «Еженедельника АПТЕКА». n
Редакция «Еженедельника АПТЕКА» благодарит компанию «Pfizer» за содействие в подготовке материала. Полный список использованной для подготовки обзора литературы находится в редакции.
Ю.В. Подпружников,
доктор фармацевтических наук,
профессор кафедры управления качеством
Национального фармацевтического университета,
сертифицированный специалист/инспектор/преподаватель GMP и GDP
|
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим