Повідомлення про оприлюднення проекту Наказу Міністерства охорони здоров’я України
«Про затвердження змін до наказу МОЗ
від 26.08.2005 № 426»
Міністерством охорони здоров’я України на громадське обговорення пропонується проект Наказу Міністерства охорони здоров’я України «»Про затвердження змін до наказу МОЗ від 26.08.2005 № 426», який розроблено з метою:
- удосконалення процедури державної реєстрації (перереєстрації) лікарських засобів та внесення змін до реєстраційних матеріалів у відповідності із директивами та іншими документами ЄС, рекомендаціями Всесвітньої організації охорони здоров’я;
- запровадження рівних умов для вітчизняних та іноземних виробників лікарських засобів;
- підсилення контролю за тривалістю, прозорістю та дотриманням процедури державної реєстрації (перереєстрації), внесенням змін до реєстраційних матеріалів лікарських засобів;
- оптимізації системи реєстрації лікарських засобів, реалізація якої гарантуватиме забезпечення населення України якісними, безпечними та ефективними лікарськими засобами.
Пропозиції та зауваження стосовно проекту від фізичних та юридичних осіб у письмовій та електронній формі просимо надавати протягом місяця з дня опублікування цього повідомлення за адресою:
01601, м. Київ, вул. М. Грушевського, 7, Управління розвитку фармацевтичного сектору галузі охорони здоров’я, Державний експертний центр МОЗ, тел.: (044) 498-43-01, e-mail: zinс[email protected].
01011, м. Київ, вул. Арсенальна, 9/11, Державний комітет України з питань регуляторної політики та підприємництва, e-mail: [email protected].
Проект наказу МОЗ
«Про затвердження змін до наказу МОЗ
від 26.08.2005 № 426»
Відповідно до постанови Кабінету Міністрів України від 26.05.2005 № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)» (із змінами) та з метою удосконалення процедури державної реєстрації лікарських засобів та забезпечення поетапного впровадження в Україні світових вимог щодо проведення експертизи матеріалів на лікарські засоби, що подаються на державну реєстрацію
НАКАЗУЮ :
1. Затвердити Зміни до Порядку проведення експертизи матеріалів на лікарських засобів, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення, затвердженого наказом Міністерства охорони здоров’я України від 26.08.2005 № 426, зареєстрованого в Міністерстві юстиції України 19.09.2005 № 1069/11349 (додаються).
2. Заступнику директора Департаменту контролю якості медичних послуг, регуляторної політики та санітарно-епідемічного благополуччя — начальнику Управління розвитку фармацевтичного сектору галузі охорони здоров’я (Стеців В. В.) забезпечити подання цього наказу в установленому порядку на державну реєстрацію до Міністерства юстиції України.
3. Генеральному директору Державного експертного центру МОЗ (Бліхар В. Є.) забезпечити організацію дотримання вимог Порядку, зазначеного у п.1 цього наказу, при проведенні експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію) та при внесенні змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення.
4. Цей наказ набирає чинності з дня його офіційного опублікування.
5. Контроль за виконанням цього наказу покласти на Першого заступника Міністра Моісеєнко Р. О.
| Міністр | О.В. Аніщенко |
ПОЯСНЮВАЛЬНА ЗАПИСКА
до проекту наказу МОЗ України
«Про затвердження змін до наказу МОЗ
від 26.08.2005 № 426»
1. Обґрунтування необхідності прийняття акта
Проект наказу «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» розроблений Міністерством охорони здоров’я з метою удосконалення системи державної реєстрації (перереєстрації) лікарських засобів в Україні. Проектом передбачено внесення змін до чинного Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення у відповідності із директивами та іншими документами ЄС, рекомендаціями Всесвітньої організації охорони здоров’я тощо.
2. Цілі і завдання прийняття акта
Як визначено Загальнодержавною програмою адаптації законодавства України до законодавства Європейського Союзу, затвердженою Законом України від 18 березня 2004 року № 1629-IV. метою такої адаптації є досягнення відповідності правової системи України acquis communautaire з урахуванням критеріїв, що висуваються ЄС до держав, які мають намір вступити до нього, тобто гармонізація нормативно-правових актів України щодо якості, безпечності та ефективності лікарських засобів з Директивою Європейського Парламенту та Ради, що стосується реєстрації лікарських засобів:
DIRECTIVE 2001/83/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 6 November 2001
on the Community code relating to medicinal products for human use
(OJ L 311, 28.11.2001, p. 67)
ДИРЕКТИВА 2001/83/ЄС ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ ТА РАДИ
від 6 листопада 2001 року
про Кодекс спільноти відносно лікарських препаратів, призначених для споживання людьми
(OВ № L 311, 28.11.2001, С. 67),
а також Директиви та Постанови (Регламенти), що вносять зміни у Директиву 2001/83/ЄС:
DIRECTIVE 2002/98/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 27 January 2003
setting standards of quality and safety for the collection, testing, processing, storage and distribution
of human blood and blood componentsand amending Directive 2001/83/EC
ДИРЕКТИВА 2002/98/ЄС ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ ТА РАДИ
від 27 січня 2003 року,
що встановлює стандарти якості та безпеки збору, тестування, обробки, зберігання та розподілу людської крові та її складових, а також вносить зміни до Директиви 2001/83/ЄС
COMMISSION DIRECTIVE 2003/63/EC
of 25 June 2003
amending Directive 2001/83/EC of the European Parliament and of the Council on the Community
code relating to medicinal products for human use
(Text with EEA relevance)
Директива Комісії 2003/63/ЄС
від 25 червня 2003р.,
що вносить зміни до директиви 2001/83/ЄС Європейського парламенту та Ради про Кодекс спільноти відносно лікарських препаратів, призначених для споживання людьми (текст в рамках ЄЕЗ)
DIRECTIVE 2004/24/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 31 March 2004
amending, as regards traditional herbal medicinal products, Directive 2001/83/EC on the
Community code relating to medicinal products for human use
Директива 2004/24/ЄС ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ ТА РАДИ
від 31 березня 2004р.,
що вносить зміни відносно традиційних рослинних лікарських засобів до Директиви 2001/83/ЄС про Кодекс спільноти відносно лікарських препаратів, призначених для споживання людьми
DIRECTIVE 2004/27/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 31 March 2004
amending Directive 2001/83/EC on the Community code relating to medicinal products for human use
(Text with EEA relevance)
Директива 2004/27/ЄС ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ ТА РАДИ
від 31 березня 2004р.,
що вносить зміни до директиви 2001/83/ЄС про Кодекс спільноти відносно лікарських препаратів, призначених для споживання людьми
текст в рамках ЄЕЗ)
REGULATION (EC) No 1901/2006 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 12 December 2006
on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive
2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004
(Text with EEA relevance)
Регламент (ЄС) № 1901/2006 ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ ТА РАДИ
від 12 грудня 2006р.
про лікарські засоби для педіатричного використання та що вносить зміни у постанову (ЕЄС) № 1768/92, директиву 2001/20/ЄС, директиву 2001/83/ЄС та постанову (ЕЄС) №726/2004
(текст в рамках ЄЕЗ)
REGULATION (EC) No 1394/2007 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 13 November 2007
on advanced therapy medicinal products and amending Directive 2001/83/EC
and Regulation (EC) No 726/2004
(Text with EEA relevance)
РЕГЛАМЕНТ (ЄС) № 1394/2007 ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ І РАДИ
від 13 листопада 2007 року
про лікувальні препарати прогресивної терапії, що вносить зміни до Директиви 2001/83/ЄС
і до Регламенту (ЄС) № 726/2004
(Текст стосується ЄЕП)
DIRECTIVE 2008/29/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 11 March 2008
amending Directive 2001/83/EC on the Community code relating to medicinal products for human
use, as regards the implementing powers conferred on the Commission
Директива 2008/29/ЄС ЄВРОПЕЙСЬКОГО ПАРЛАМЕНТУ І РАДИ
Від 11 березня 2008р.
Що вносить зміни до директиви 2001/83/ЄС про Кодекс спільноти відносно лікарських препаратів, призначених для споживання людьми, відносно впровадження повноважень, що надаються Комісії
DIRECTIVE 2009/53/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
of 18 June 2009
amending Directive 2001/82/EC and Directive 2001/83/EC, as regards variations to the terms of marketing authorisations for medicinal products
(Text with EEA relevance)
Директива 2009/53/ЄС Європейського парламенту та Ради
від 18 червня 2009р.,
що вносить зміни до директиви 2001/82/ЄС та директиви 2001/83/ЄС відносно змін в умови видачі торгових ліцензій на лікарські засоби
(текст в рамках ЄЕЗ)
COMMISSION DIRECTIVE 2009/120/EC
of 14 September 2009
amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards advanced therapy medicinal products
(Text with EEA relevance)
Директива Комісії 2009/120/ЄС
від 14 вересня 2009р.,
що вносить зміни до директиви 2001/83/ЄС Європейського парламенту та Ради про Кодекс спільноти відносно лікарських препаратів, призначених для споживання людьми, стосовно лікувальних препаратів прогресивної терапії (текст в рамках ЄЕЗ).
В Україні створена і функціонує 3-х рівнева система контролю якості лікарських засобів, яка є однією з кращих серед країн СНД, а саме:
1. Державний контроль при їх ввезенні на територію України.
2. Контроль уповноваженими особами суб’єктів господарювання.
3. Контроль інспекторами територіальних органів Держлікслужби Україні при здійсненні планових та позапланових перевірок суб’єктів господарювання.
Україна стала першою країною серед колишніх республік Радянського Союзу, в якій стандарти GMP були визнані обов’язковими, а регуляторна система визнана міжнародним співтовариством. З 2011 року регуляторний орган Україні у сфері забезпечення якості лікарських засобів — Державна служба України з лікарських засобів стала членом міжнародної Системи співробітництва фармацевтичних інспекцій — Pharmaceutical Inspection Cooperation Scheme (PIC/S).
Однак протягом останніх трьох років іноземні виробники лікарських засобів є безперечними «лідерами» за кількістю приписів за показниками якості або побічних дій. Одна з причин — нерівні умови при допуску на ринок вітчизняних підприємств і закордонних. Відповідність виробництва вимогам GMP зараз не є обов’язковою для зарубіжних виробників при постачанні лікарських засобів в Україну. У минулому році, при проведенні сертифікації зарубіжних підприємств, продукція яких реалізується в Україну, в 30% випадків перевірок виявлено критичні невідповідності виробництв, тобто існує потенційна можливість випуску препаратів, що загрожують життю пацієнта. Таким чином, для захисту українського фармацевтичного ринку і населення країни від неякісних лікарських засобів назріла необхідність в інспектуванні іноземних виробників згідно вимог GMP на етапі реєстрації лікарських засобів.
Вступ України до Світової організації торгівлі (СОТ) обумовив визначення нових правових норм у сфері обігу лікарських засобів. Базовими засадами документів СОТ визначено, що торговельна система має бути відкритою для чесної конкуренції. Отже, виникла потреба в імплементації в чинне законодавство положень Угоди про торгові аспекти прав інтелектуальної власності ТРИПС (TRIPS — Agreement on Trade Related Aspects of Intellectual Property Rights), яка встановлює необхідні стандарти в сфері інтелектуальної власності і зобов’язує країни учасниці СОТ забезпечити повну патентну охорону (як виробничих процесів, так і самих продуктів, у тому числі до фармацевтичної продукції).
Прийняття цього наказу сприятиме:
- удосконаленню процедури державної реєстрації (перереєстрації) лікарських засобів та внесення змін до реєстраційних матеріалів у відповідності із директивами та іншими документами ЄС, рекомендаціями Всесвітньої організації охорони здоров’я;
- запровадженню рівних умов для вітчизняних та іноземних виробників лікарських засобів;
- підсиленню контролю за тривалістю, прозорістю та дотриманням процедури державної реєстрації (перереєстрації) та внесення змін до реєстраційних матеріалів лікарських засобів;
- оптимізація системи реєстрації лікарських засобів, реалізація якої гарантуватиме забезпечення населення України якісними, безпечними та ефективними лікарськими засобами.
3. Загальна характеристика і основні положення проекту акта
Проектом акту передбачено викладення деяких термінів відповідно до європейських та міжнародних стандартів, зокрема, термінів «побічна реакція», «післяреєстраційне дослідження з безпеки», «реєстраційні матеріали», «інструкція для медичного застосування лікарського засобу», «ризик, пов’язаний із застосуванням лікарського засобу», «експертиза матеріалів на лікарський засіб», «реєстраційне посвідчення на лікарський засіб», «ефективність лікарського засобу», «якість лікарського засобу», «повна і незалежна заява (автономна заява)», «діючі речовини (субстанції)», «комбінований препарат-генерик», «заява на лікарський засіб, який має добре вивчене медичне застосування», «неінтервенційне дослідження», «фармацевтична еквівалентність», «фармацевтично альтернативні лікарські засоби», «біовейвер», «біофармацевтична система класифікації (БСК)», «дослідження еквівалентності in vitro», «дослідження еквівалентності, «заява на продукцію із in bulk».
Проектом наказу МОЗ України «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» пропонується викласти у новій редакції деякі терміни, внести зміни до Порядку проведення експертизи лікарського засобу, що подається на державну реєстрацію (перереєстрацію) та Порядку експертизи матеріалів про внесення змін до реєстраційних матеріалів на лікарський засіб, а також Доповнити Порядок проведення експертизи розділом 5 «Порядок проведення контролю якості лікарського засобу, який подається на державну реєстрацію (перереєстрацію)» тощо.
Відповідно до норм постанови Кабінету Міністрів України від 26 травня 2005 р. № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)» (із змінами) передбачено подання копі] документу, що підтверджує відповідність умов виробництва чинним в Україні вимогам належної виробничої практики (GMP), виданого Держлікслужбою (для вітчизняних виробників — копія чинної ліцензії на виробництво лікарських засобів, зазначена у підпункті и) цього пункту) або копія офіційного документу, виданого уповноваженим органом, який входить до міжнародної системи співробітництва фармацевтичних інспекцій (PIC(S), про відповідність виробництва вимогам GMP.
З метою забезпечення виконання вимог Угоди про торгові аспекти прав інтелектуальної власності ТРИПС щодо захисту прав інтелектуальної власності в Україні, пропонується внести наступну зміну: «якщо після проведення розгляду заяви про порушення прав суд установив, що внаслідок такої реєстрації можуть бути порушені захищені патентом України майнові права інтелектуальної власності, у тому числі в процесі виробництва, використання, продажу лікарських засобів».
4. Стан нормативно-правової бази у цій сфері правового регулювання
На сьогодні у цій сфері правового регулювання діє Закон України «Про лікарські засоби» та постанова Кабінету Міністрів України від 26 травня 2005 р. № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)», наказ Міністерства охорони здоров’я України від 26 серпня 2005 року № 426 «Про затвердження Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення». З метою забезпечення гармонізації вищезазначених документів з директивами ЄС та рекомендаціями ВООЗ потребує внесення змін вищезазначений наказ МОЗ від 26.08.2005 № 426, оскільки адаптація законодавства України до законодавства ЄС є пріоритетною складовою процесу інтеграції України до ЄС та пріоритетним напрямом її зовнішньої політики. Реалізація нормативно-правового акта не передбачає визнання актів такими, що втратили чинність.
5. Фінансово-економічне обґрунтування
Реалізація проекту акта не потребує додаткових фінансових та інших витрат з Державного та місцевих бюджетів та від суб’єктів господарювання.
6. Позиція заінтересованих органів
Проект наказу необхідно погодити з Державною службою України з лікарських засобів та Державним комітетом України з питань регуляторної політики та підприємництва. Підлягає державній реєстрації у Міністерстві юстиції України.
7. Регіональний аспект
Проект наказу «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» не стосується розвитку адміністративно-територіальних одиниць України.
8. Запобігання корупції
У проекті акта не має правил і процедур, які можуть містити ризики вчинення корупційних правопорушень
9. Громадське обговорення.
Проект наказу «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» розміщений на офіційному веб — сайті МОЗ України www.moz.gov.ua для громадського обговорення.
10. Позиція соціальних партнерів
Проект наказу «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» не стосується соціально-трудової сфери, тому не потребує погодження з уповноваженими представниками від всеукраїнських профспілок, їх об’єднань та всеукраїнськими об’єднаннями організацій роботодавців.
11. Прогноз результатів
Прийняття змін до наказу МОЗ від 26.08.2005 № 426 сприятиме удосконаленню процедури державної реєстрації (перереєстрації) лікарських засобів в Україні, запровадженню рівних умов для вітчизняних та іноземних виробників при виробництві лікарських засобів відповідно до вимог належної виробничої практики (GMP), оптимізації системи реєстрації лікарських засобів, реалізація якої гарантуватиме забезпечення населення України якісними, безпечними та ефективними лікарськими засобами відповідно до сучасних міжнародних правил і норм, зокрема, щодо захисту прав інтелектуальної власності, раціональному медичному застосуванню лікарських засобів згідно потреб сучасної медичної допомоги.
| Перший заступник Міністра | Р.О.Моісеєнко |
АНАЛІЗ РЕГУЛЯТОРНОГО ВПЛИВУ
проекту наказу МОЗ України
«Про затвердження змін до наказу МОЗ
від 26.08.2005 № 426»
1. Аналіз проблеми, яку буде розв’язано шляхом державного регулювання
Державна політика у сфері обігу лікарських засобів визначає необхідність забезпечення населення України ефективними, безпечними та якісними лікарськими засобами. Експертиза матеріалів реєстраційного досьє є важливою процедурою, яка дає можливість підтвердити (або спростувати) ефективність, безпечність та якість лікарських засобів.
Предметом правового регулювання запропонованого проекту акта є удосконалення процедури державної реєстрації лікарських засобів.
Проект наказу «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» розроблений Міністерством охорони здоров’я.
Проектом передбачено внесення змін до чинного Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення у відповідності із директивами та іншими документами ЄС, рекомендаціями Всесвітньої організації охорони здоров’я тощо.
При постачанні лікарських засобів в Україну від зарубіжних виробників проблемою стала відповідність виробництва цих засобів вимогам належної виробничої практики (GMP), оскільки в даний час національними нормативно-правовими актами не передбачена їх обов’язковість.
У минулому році, при проведенні сертифікації зарубіжних підприємств, продукція яких реалізується в Україну, в 30% випадків перевірок виявлено критичні невідповідності виробництв, тобто існує потенційна можливість випуску препаратів, що загрожують життю пацієнта. Таким чином, для захисту українського фармацевтичного ринку і населення країни від неякісних лікарських засобів назріла необхідність в інспектуванні іноземних виробників згідно вимог GMP на етапі реєстрації лікарських засобів.
2. Цілі державного регулювання
Відповідно до статті 51 Угоди про партнерство і співробітництво між Європейським Співтовариством і Україною важливою умовою для зміцнення економічних зв’язків між Україною та Співтовариством є зближення існуючого і майбутнього законодавства України з законодавством Співтовариства. Тобто, Україна має вжити заходів для забезпечення того, щоб її законодавство поступово було приведене у відповідність до законодавства Співтовариства,
Україна стала першою країною серед колишніх республік Радянського Союзу, в якій стандарти GMP були визнані обов’язковими, а регуляторна система визнана міжнародним співтовариством. З 2011 року регуляторний орган Україні у сфері забезпечення якості лікарських засобів — Державна служба України з лікарських засобів стала членом міжнародної Системи співробітництва фармацевтичних інспекцій — Pharmaceutical Inspection Cooperation Scheme (PIC/S). Членство в PIC/S — це своєрідна конвенція з питань фармацевтичного інспектування, основні завдання якої — міжнародний розвиток, впровадження і дотримання стандартів GMP. Для досягнення вищезазначених цілей PIC/S розробляє стандарти GMP, гармонізовані з нормативними документами ЄС, та інші настанови, сприяє їх впровадженню на фармацевтичних виробництвах. Також PIC/S сприяє оптимізації співробітництва між національними регуляторними органами і міжнародними організаціями. Переваги для країни від її членства в PIC/S полягають у забезпеченні захищеності споживачів від неякісної фармацевтичної продукції. Безперечною перевагою для фармацевтичних підприємств є зменшення кількості дублюючих перевірок, з чого випливає раціональне використання фінансових ресурсів; спрощення процедур експорту-імпорту продукції на міжнародних ринках. Управління якістю — одна з основних функцій управління на виробництві. Система управління якістю дає регуляторним органам більше впевненості в тому, що виробництво є послідовним і надійним з точки зору виробничих стандартів. Основний елемент механізму управління якістю на виробництві — правильна інфраструктура системи якості, яка включає необхідні процедури, процеси і ресурси. Система забезпечення якості передбачає дотримання вимог GMP, а також розробку і дотримання при виробництві належного дизайну продукції та упаковки. На підставі вищевикладеного, Міністерством охорони здоров’я України розроблений проект наказу, приписами якого передбачено наявність в реєстраційних матеріалах документів, які підтверджують відповідність умов виробництва чинним в Україні вимогам належної виробничої практики (GMP).
Вступ України до Світової організації торгівлі (СОТ) обумовив визначення нових правових норм у сфері обігу лікарських засобів. Базовими засадами документів СОТ визначено, що торговельна система має бути відкритою для чесної конкуренції. Отже виникла потреба в імплементації в чинне законодавство положень Угоди про торгові аспекти прав інтелектуальної власності ТРИПС (TRIPS — Agreement on Trade Related Aspects of Intellectual Property Rights), яка встановлює необхідні стандарти в сфері інтелектуальної власності і зобов’язує країни учасниці СОТ забезпечити повну патентну охорону (як виробничих процесів, так і самих продуктів, у тому числі до фармацевтичної продукції.
Прийняття цього наказу сприятиме:
- удосконаленню процедури державної реєстрації (перереєстрації) лікарських засобів та внесення змін до реєстраційних матеріалів у відповідності із директивами та іншими документами ЄС, рекомендаціями Всесвітньої організації охорони здоров’я;
- запровадженню рівних умов для вітчизняних та іноземних виробників лікарських засобів;
- підсиленню контролю за тривалістю, прозорістю та дотриманням процедури державної реєстрації (перереєстрації) та внесення змін до реєстраційних матеріалів лікарських засобів;
- оптимізація системи реєстрації лікарських засобів, реалізація якої гарантуватиме забезпечення населення України якісними, безпечними та ефективними лікарськими засобами.
- Оцінка альтернативних способів досягнення зазначених цілей
Як альтернативний спосіб досягнення цілей була розглянута ситуація, коли діючі умови залишаться без змін, тобто залишаться наявні розбіжності положень різних нормативно-правових актів.
Слід зазначити, що результатом такого підходу можуть стати:
- неналежне виконання положень Законів України «Про лікарські засоби» та «Про загальнодержавну адаптацію законодавства України до законодавства Європейського Союзу»;
- відсутність можливості координування та контролю впровадження міжнародних стандартів у фармацевтичній галузі;
- гальмування розвитку вітчизняного виробництва лікарських засобів, зокрема, генериків;
- подальшому застосуванню нерівних умов для вітчизняних та іноземних виробників лікарських засобів;
- неможливість здійснення належної оцінки якості, безпечності та ефективності високотехнологічних лікарських засобів та надходження таких препаратів на фармацевтичний ринок України.
Оскільки основним нормативним документом, що регулює процедуру державної реєстрації лікарських засобів в Україні та вимоги до реєстраційних документів є наказ № 426, то з метою забезпечення гармонізації вищезазначеного документу з директивами ЄС та вимогами СОТ, він потребує змін.
Адаптація законодавства України до законодавства ЄС є пріоритетною складовою процесу інтеграції України до ЄС та пріоритетним напрямом її зовнішньої політики.
Запропонований проект акта відповідає правилам та нормам ЄС, та законодавству, яке прийнято в Україні на вимоги СОТ.
З огляду на вищевикладене, альтернативи запропонованому регуляторному акта немає.
Механізм, який застосовується для розв’язання проблеми, і відповідні заходи
Прийняття наказу удосконалить систему реєстрації лікарських засобів, посилить контроль за обігом лікарських засобів в частині державної реєстрації, реалізація якої гарантуватиме забезпечення населення України якісними, безпечними та ефективними лікарськими засобами. Окрім того, будуть усунені розбіжності з нормативними актами, що мають вищу юридичну силу, а саме: Законом України «Про лікарські засоби» та постановою Кабінету Міністрів України від 26 травня 2005 р. № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)» (із змінами).
Проектом акту передбачено викладення деяких термінів відповідно до європейських та міжнародних стандартів, зокрема, термінів «побічна реакція», «післяреєстраційне дослідження з безпеки», «реєстраційні матеріали», «інструкція для медичного застосування лікарського засобу», «ризик, пов’язаний із застосуванням лікарського засобу», «експертиза матеріалів на лікарський засіб», «реєстраційне посвідчення на лікарський засіб», «ефективність лікарського засобу», «якість лікарського засобу», «повна і незалежна заява (автономна заява)», «діючі речовини (субстанції)», «комбінований препарат-генерик», «заява на лікарський засіб, який має добре вивчене медичне застосування», «неінтервенційне дослідження», «фармацевтична еквівалентність», «фармацевтично альтернативні лікарські засоби», «біовейвер», «біофармацевтична система класифікації (БСК)», «дослідження еквівалентності in vitro», «дослідження еквівалентності, «заява на продукцію із in bulk».
Проектом наказу МОЗ України «Про затвердження змін до наказу МОЗ від 26.08.2005 № 426» пропонується викласти у новій редакції деякі терміни, внести зміни до Порядку проведення експертизи лікарського засобу, що подається на державну реєстрацію (перереєстрацію) та Порядку експертизи матеріалів про внесення змін до реєстраційних матеріалів на лікарський засіб, а також доповнити Порядок проведення експертизи розділом 5 «Порядок проведення контролю якості лікарського засобу, який подається на державну реєстрацію (перереєстрацію)» тощо.
Відповідно до норм постанови Кабінету Міністрів України від 26 травня 2005 р. № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)» (із змінами) передбачено подання копі] документу, що підтверджує відповідність умов виробництва чинним в Україні вимогам належної виробничої практики (GMP), виданого Держлікслужбою (для вітчизняних виробників — копія чинної ліцензії на виробництво лікарських засобів, зазначена у підпункті и) цього пункту) або копія офіційного документу, виданого уповноваженим органом, який входить до міжнародної системи співробітництва фармацевтичних інспекцій (PIC(S), про відповідність виробництва вимогам GMP.
З метою забезпечення виконання вимог Угоди про торгові аспекти прав інтелектуальної власності ТРИПС щодо захисту прав інтелектуальної власності в Україні, пропонується внести наступну зміну: «якщо після проведення розгляду заяви про порушення прав суд установив, що внаслідок такої реєстрації можуть бути порушені захищені патентом України майнові права інтелектуальної власності, у тому числі в процесі виробництва, використання, продажу лікарських засобів».
3. Обґрунтування можливостей досягнення визначених цілей у разі прийняття регуляторного акта
Прийняття наказу сприятиме удосконаленню нормативно-правового поля для реалізації державної політики у сфері обігу лікарських засобів шляхом удосконалення порядку експертизи, що сприятиме введенню на фармацевтичний ринок України якісних, ефективних та безпечних лікарських засобів шляхом подальшої розбудови системи реєстрації лікарських засобів у відповідності до європейських норм, посилить контроль за обігом лікарських засобів в частині державної реєстрації, запобігатиме появі неякісних та неефективних препаратів на ринку України. Прийняттям наказу будуть усунені розбіжності з нормативними актами, що мають вищу юридичну силу, а саме: Законом України «Про лікарські засоби» та постановою Кабінету Міністрів України від 26 травня 2005 р. № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)» (із змінами).
Соціально-економічні та інші результати впровадження запропонованого акта, у разі його прийняття, пов’язані із запобіганням застосування неякісних, неефективних та небезпечних лікарських засобів, з метою охорони здоров’я громадян і захисту їх життя.
4. Очікувані результати прийняття акта
| Вигоди | Витрати |
|---|---|
| Сфера інтересів держави | |
| 1. Реалізація державної політики у сфері обігу лікарських засобів. 2. Обіг на ринку України безпечних, якісних та ефективних лікарських засобів. 3. Удосконалення процедури державної реєстрації (перереєстрації) лікарських засобів. 4. Запровадження прозорої процедури державної реєстрації (перереєстрації), внесення змін до реєстраційних матеріалів лікарських засобів. |
Відсутні. |
| Сфера інтересів суб’єктів господарювання | |
| 1. Створення прозорої системи експертизи реєстраційних матеріалів. 2. Створення конкурентних умов ведення бізнесу в частині виведення на ринок лікарських засобів. |
Відсутні. |
| Сфера інтересів громадян | |
| 1. Створення сприятливих умов для поліпшення забезпечення населення і закладів охорони здоров’я якісними, високоефективними, безпечними та доступними лікарськими засобами. 3. Покращення стану здоров’я населення, отримання доступу до якісних, ефективних та безпечних лікарських засобів. |
Відсутні. |
5. Обґрунтування терміну дії регуляторного акта
Акт набирає чинності з дня опублікування. Строк дії регуляторного акта встановлюється на необмежений термін. Перегляд документу буде здійснюватись за потребою.
– Показники результативності акта
Прогнозовані показники результативності регуляторного акта:
а) Розмір надходження до державного та місцевих бюджетів цільових фондів — реалізація акта не передбачає додаткових надходжень та втрат до державного та місцевих бюджетів.
б) Кількість суб’єктів господарювання, та/або фізичних осіб, на які поширюватиметься дія акта — дія акта поширюється на усіх суб’єктів господарювання (резидентів та нерезидентів), які є заявниками та/або виробниками лікарських засобів, і відповідають за їх ефективність, безпечність та якість.
в) Розмір коштів, що витрачатиметься суб’єктами господарювання та/або фізичними особами, пов’язаний з виконанням вимог акта — запропонований акт не вимагає від суб’єктів господарювання додаткових витрат.
г) Час, що витрачатиметься суб’єктами господарювання та/або фізичними особами, пов’язаний з виконанням вимог акта — прийняття запропонованого акту не збільшить витрати часу суб’єктами господарювання при проведенні державної реєстрації (перереєстрації) лікарських засобів, та внесення змін до матеріалів реєстраційного досьє протягом дії реєстраційного посвідчення.
д) Рівень поінформованості суб’єктів господарювання та/або фізичних осіб оцінюється як середній за рахунок публікації на офіційному сайті Міністерства охорони здоров’я України. (www.moz.gov.ua).
6. Заходи, за допомогою яких здійснюватиметься відстеження результативності акта
Відстеження результативності зазначеного регуляторного акта буде здійснюватися шляхом обробки статистичної інформації щодо кількості лікарських засобів, що зареєстровані в Україні.
Базове відстеження результативності регуляторного акта буде здійснюватися після набуття чинності цим регуляторним актом.
Повторне відстеження буде здійснюватися через рік після набрання чинності цим регуляторним актом за результатами якого можливо здійснити порівняння показників базового та повторного відстеження.
Відстеження результативності акта буде проводитись шляхом аналізу даних щодо ефективності, безпечності та якості лікарських засобів, опитування суб’єктів господарювання, зайнятих у сфері обігу лікарських засобів.
Аналіз регуляторного впливу підготовлено Міністерством охорони здоров’я України.
| Перший заступник Міністра |
ЗАТВЕРДЖЕНО
наказом Міністерства охорони здоров’я України
від ___ _________2011 р. №__
Зареєстровано
в Міністерстві юстиції України
__ _______ 2011 р. за №_________
Зміни до Порядку проведення експертизи матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
У тексті Порядку проведення експертизи матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення, затвердженого наказом Міністерства охорони здоров’я України від 26.08.2005 № 426, зареєстрованого в Міністерстві юстиції України 19.09.2005 за № 069/11349 (далі — Порядок) та додатків до нього слова «Державний фармакологічний центр МОЗ України» в усіх відмінках замінити словами «Державний експертний центр МОЗ» у відповідних відмінках.
По тексту Порядку та додатках до нього слова «активна субстанція» в усіх відмінках та числах замінити словом «субстанція» у відповідних відмінках та числах.
Абзац перший пункту 2.19 Порядку викласти у такій редакції:
«2.19 Побічна реакція — будь-яка небажана негативна реакція, включаючи відсутність ефективності, яка виникає при застосуванні лікарських засобів.».
Підпункт 2.19.3. пункту 2.19. Порядку викласти в такій редакції:
«2.19.3. Післяреєстраційне дослідження з безпеки — будь-яке дослідження зареєстрованого лікарського засобу, що проводиться з метою визначення та вивчення ступеню ризиків/небезпеки при його застосуванні, підтвердження профілю безпеки лікарського засобу або оцінки ефективності заходів по управлінню ризиками.».
Пункт 2.21 Порядку після слів «в Україні» доповнити словами «а також несе відповідальність за достовірність інформації, що міститься у наданих ним реєстраційних та додаткових матеріалах».
Пункт 2.22. Порядку викласти у такі редакції:
«2.22. Реєстраційні матеріали (реєстраційне досьє) — комплект документів, що додаються до заяви про державну реєстрацію (перереєстрацію, внесення змін до реєстраційних матеріалів) лікарського засобу на підставі яких можна зробити обґрунтований висновок щодо його ефективності, безпеки та якості».
Пункт 2.30 Порядку викласти в такій редакції:
«2.30. Інструкція для медичного застосування лікарського засобу (інструкція про застосування лікарського засобу) — офіційно затверджена інформація про застосування лікарського засобу, яка супроводжує лікарський засіб.».
Пункт 2.31. Порядку викласти у такій редакції:
«2.31. Ризик, пов’язаний із застосуванням лікарського засобу — сукупність даних із підтвердженим причинно-наслідковим зв‘язком про розвиток побічних реакцій лікарського засобу, зокрема, внаслідок прояву небезпечних властивостей лікарського засобу та небезпечних факторів у групі осіб, які його застосовували.
Співвідношення ризик/користь — співвідношення кількісної та якісної оцінки виявлених факторів небезпечних властивостей лікарського засобу при його медичному застосуванні, які погіршують перебіг захворювання або є причиною розвитку нових шкідливих впливів лікарського засобу на організм та якість життя споживача лікарського засобу до позитивного впливу лікарського засобу на серйозність та тяжкість перебігу захворювання.».
Пункт 2.32. Порядку викласти у такій редакції:
«Експертиза матеріалів на лікарський засіб — це перевірка, аналіз та спеціалізована експертиза реєстраційних матеріалів та матеріалів додаткових експертиз (випробувань) лікарського засобу, що здійснюється на підставі відповідної заяви, з метою підготовки вмотивованих висновків та рекомендацій щодо прийняття рішення про можливість його державної реєстрації (перереєстрації, внесення змін до реєстраційних матеріалів).».
Пункт 2.36 Порядку викласти у такій редакції:
«2.36. Реєстраційне посвідчення на лікарській засіб — документ, у якому міститься інформація про лікарській засіб, зареєстрований в Україні, внесений до Державного реєстру лікарських засобів України, та дозволений до медичного застосування в Україні.».
Абзац 2 пункту 2.37. Порядку вилучити.
Пункт 2.40. Порядку викласти у такій редакції:
«2.40. Ефективність лікарського засобу — сприятлива лікувальна, профілактична чи діагностична дія лікарського засобу на перебіг і тривалість захворювання чи корекцію стану і фізіологічних функцій організму людини».
У пункті 2.41 Порядку слова «або листка-вкладиша» вилучити.
Пункт 2.45 викласти у такій редакції:
«2.45. Якість лікарського засобу — сукупність властивостей, які надають лікарському засобу здатність задовольняти споживачів відповідно до свого призначення і відповідають вимогам, встановленим законодавством.».
Пункт 2.47. Порядку викласти у такій редакції:
«2.47. Повна і незалежна заява (автономна заява) — заява на державну реєстрацію лікарського засобу, до складу якого входить нова активна субстанція або відома активна субстанція — компонент лікарського засобу, зареєстрованого в Україні в іншій лікарській формі.».
У пункті 2.48. Порядку слова «комплект реєстраційних документів на новий лікарський засіб» замінити словами «заява про державну реєстрацію лікарського засобу».
У пункті 2.49. Порядку слова «Скорочена заява» замінити словом «Заява».
У пункті 2.50. Порядку слова «комплект реєстраційних документів на лікарський засіб» замінити словами «заява про державну реєстрацію лікарського засобу».
Доповнити розділ 2 Порядку пунктами 2.59 — 2.69 у такій редакції:
«2.59. Діючі речовини (субстанції) — біологічно активні речовини у вигляді розчинів, порошків, гранул, пелет тощо, в тому числі біологічні агенти (мікроорганізми, віруси, клітини та тканини людини, тварин та їх компоненти), які можуть змінювати стан і функції організму або мають профілактичну, діагностичну чи лікувальну дію та використовуються для виробництва готових лікарських засобів.
2.60. Комбінований препарат-генерик — багатокомпонентний лікарський засіб, який за складом та кількістю активних речовин є аналогічним до зареєстрованого багатокомпонентного лікарського засобу, що містить відомі активні субстанції, які використовувались раніше в цій комбінації або раніше використовувались у медичній практиці у цій комбінації як окремі лікарські засоби у тій же лікарській формі, дозах та відповідному співвідношенні
2.61. Заява на лікарський засіб, який має добре вивчене медичне застосування — заява про державну реєстрацію лікарського засобу, який містить відому діючу речовину та у тій самій лікарській формі, що й подібні лікарські засоби, зареєстровані в Україні протягом щонайменше 10 років (до заяви додається комплект реєстраційних матеріалів, у яких наведені повні адміністративні, фармацевтичні та бібліографічні дані, що свідчать про ефективність та безпеку лікарського засобу), який немає системної дії на організм.
2.62. Неінтервенційне дослідження — дослідження, у якому лікарські засоби призначаються звичайним способом відповідно до затвердженої інструкції з медичного застосування. Залучення пацієнта в групу з визначеним методом лікування в протоколі клінічного дослідження заздалегідь не передбачено, а призначення лікарського засобу диктується сучасною практикою і не залежить від рішення включити пацієнта у випробування. Не застосовують додаткових діагностичних або моніторингових процедур щодо пацієнтів, а для аналізу зібраних даних використовують епідеміологічні методи.
2.63. Фармацевтична еквівалентність — лікарські препарати є фармацевтично еквівалентними, якщо вони вводяться тим самим шляхом, містять ту саму кількість тієї самої діючої речовини (тих самих діючих речовин) у тих самих дозованих формах, відповідають вимогам тих самих або порівнюваних стандартів. Фармацевтична еквівалентність не обов’язково передбачає терапевтичну еквівалентність, оскільки відмінності у допоміжних речовинах та/або у процесі виробництва, або інші коливання можуть призвести до швидшого або повільнішого розчинення та/або до швидшої або повільнішої абсорбції.
2.64. Фармацевтично альтернативні лікарські засоби — препарати, що містять однакову молярну кількість тієї самої діючої речовини, але відрізняються за лікарською формою (наприклад, таблетки у порівнянні з капсулами) та/або хімічною формою (наприклад, інші солі, інші ефіри). Альтернативні лікарські засоби доставляють ту саму діючу речовину тим самим шляхом введення, але не є фармацевтично еквівалентними. Вони можуть бути або можуть не бути біоеквівалентними або терапевтично еквівалентними референтному препарату.
2.65. Біовейвер — це процедура, за якої проводиться державна реєстрація генеричного лікарського засобу на основі біофармацевтичної системи класифікації та результатів порівняльних досліджень in vitro.
2.66. Біофармацевтична система класифікації (БСК) — це наукова система класифікації діючих речовин на основі їх розчинності у водних розчинах та ступеня проникнення/повної абсорбції у людини.
2.67. Дослідження еквівалентності in vitro — це комплексні дослідження, які базуються на класифікації діючої речовини згідно з БСК та розчиненні препарату, а також включають порівняння профілів розчинення генеричного та референтного препаратів у трьох середовищах зі значеннями рН 1,2, рН 4,5 і рН 6,8.
2.68. Дослідження еквівалентності — дослідження, яке визначає еквівалентність між генеричним та референтним препаратами при використані досліджень in vivo та/або in vitro.
2.69. Заява на продукцію із in bulk — заява на державну реєстрацію готового лікарського засобу, виробництво якого здійснюється шляхом фасування та/або кінцевого пакування і маркування.».
Розділ 2 Порядку доповнити останнім абзацом у такій редакції:
«Зміст інших термінів, що використовуються у цьому Порядку, відповідає визначеним законодавством України та прийнятим у світовій практиці.».
Пункт 3.1. Порядку доповнити абзацом у такій редакції:
««Після отримання заяви Центр встановлює можливість реєстрації (перереєстрації) лікарського засобу з точки зору його належності до заборонених до застосування в Україні, та, у разі державної реєстрації — здійснює кваліфікацію типу такої заяви і визначає обсяг необхідних реєстраційних матеріалів, у разі перереєстрації — можливість подовження терміну дії реєстраційного посвідчення.».
Пункт 3.2. Порядку після слів та символів «державну реєстрацію (перереєстрацію),» доповнити словами «починається з дати отримання комплекту відповідно до типу заяви реєстраційних матеріалів та».
Підпункт 3.2.1. пункту 3.2. Порядку вилучити, у зв’язку із чим вважати підпункти 3.2.2 та 3.2.3 підпунктами 3.2.1 та 3.2.2. відповідно.
У абзаці 3 пункт 3.2.1. слова «щодо доведення безпечності, ефективності та якості лікарського засобу» замінити словами «у відповідності до типу заяви».
Абзац 2 пункту 3.3. Порядку після слів «згідно з додатком 1» доповнити словами та символами «(у разі надання заяви на комбіновані лікарські засоби в декількох варіаціях доз, які збільшуються непропорційно, заява розглядається як на два різних лікарські засоби).».
У абзаці 4 пункту 3.3. Порядку слова, цифри та символи «або заяву про державну реєстрацію (перереєстрацію) лікарського засобу згідно з додатком 17 (за бажанням заявника).» замінити словами «, а у випадку традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами, згідно з додатком 17.»
Абзац 5 пункту 3.3. Порядку викласти у такій редакції:
«Комплект реєстраційних матеріалів надається з урахуванням проведеної Центром кваліфікації типу заяви та здійснення заявником оплати державного збору за реєстрацію (перереєстрацію) лікарського засобу та вартості експертних робіт або надання ним належним чином оформленого гарантійного листа щодо їх сплати.
Пункт 3.3. Порядку доповнити абзацом 6 у такій редакції:
«Якщо Заявник протягом 6 місяців з дати офіційного подання заяви про проведення експертизи матеріалів на лікарські засоби, що подаються для державної реєстрації (перереєстрації) (включаючи традиційні лікарські засоби та лікарські засоби, що виробляються згідно із затвердженими прописами), а також — на діючі речовини (активні субстанції), які подаються для державної реєстрації (перереєстрації) не подає до Центру реєстраційних матеріалів або листа з обґрунтуванням терміну, необхідного для їх підготовки, то заява знімається з розгляду. Надалі така заява може бути подана до Центру в установленому порядку.».
У зв’язку із цим вважати абзаци 6-8 абзацами 7-9.
У пункті 3.4. Порядку слова «первинної експертизи» замінити словами «кваліфікації типу заяви або розгляду можливості подовження терміну дії реєстраційного посвідчення», а слова «її позитивних висновків» — словами «встановлення можливості реєстрації (перереєстрації) заявленого лікарського засобу».
У пункті 3.6. Порядку слово «заяви» замінити словами «реєстраційних матеріалів відповідно до типу заяви».
Пункт 3.10. після слів «на державну реєстрацію» доповнити символами та словом «(перереєстрацію)».
Підпункт д) пункту 3.13. Порядку викласти у такій редакції:
«д) якщо після проведення розгляду заяви про порушення прав суд установив, що внаслідок такої реєстрації можуть бути порушені захищені патентом України майнові права інтелектуальної власності, у тому числі в процесі виробництва, використання, продажу лікарських засобів.».
Пункт 3.15. Порядку після слів «очікуваною користю» доповнити словами та символами «, надана Заявником інформація, що міститься в реєстраційних матеріалах, є недостовірною.».
У абзаці 1 пункту 4.1. Порядку слова «про зміни в технології виробництва та/або зміну обладнання, виробника діючих та допоміжних речовин» замінити словами «про будь-які зміни щодо зареєстрованого лікарського засобу».
Абзаци 2—5 пункту 4.1. Порядку вилучити.
Доповнити пункт 4.1. Порядку підпунктами 4.1.1. — 4.1.3. у такій редакції:
«4.1.1. За характером зміни класифікують на:
- зміни типу IА — незначні зміни, що виявляють незначний або не виявляють впливу на якість, безпеку та ефективність лікарського засобу, які стосуються внесення поправок до змісту реєстраційних документів, поданих на момент прийняття рішення про реєстрацію (перереєстрацію) лікарського засобу, і не потребують його нової реєстрації;
- зміни типу II — будь-які зміни до реєстраційних документів, що не потребують нової реєстрації лікарського засобу та можуть виявляти значний вплив його на якість, безпеку та ефективність, але не можуть розглядатися як зміни типу I;
- зміни типу ІБ — незначні зміни, які не можуть бути змінами типу ІА та типу ІІ, і не потребують нової реєстрації.
4.1.2. Реєстрація додаткової упаковки — зміни до матеріалів реєстраційного досьє, що передбачають появу на ринку України нової упаковки лікарського засобу. Реєстрація додаткової упаковки може супроводжуватись зміною первинної упаковки (матеріалу первинної упаковки, з яким готовий лікарський засіб безпосередньо вступає в контакт).
4.1.3. Термінові зміни, що стосуються безпеки лікарського засобу — термінові тимчасові обмеження, пов’язані з безпекою використання лікарського засобу, і які впроваджуються заявником у разі виявлення ризику для здоров’я людини при застосуванні зареєстрованого (перереєстрованого) лікарського засобу. Про кожну причину, характер і дату передбачуваного введення в дію тимчасових термінових обмежень заявник повідомляє Центр, який протягом 24 годин приймає рішення щодо їх запровадження. При позитивному рішенні Центру подальша експертиза матеріалів про внесення змін здійснюється у порядку, встановленому даним розділом.
У разі, якщо заявнику стало відомо про виявлені небезпечні властивості лікарського засобу для здоров’я або життя людини, що стали підставою для прийняття заявником рішення про введення обмежень до його застосування, заявник повідомляє про це Центр у будь-якій спосіб та подає заяву про зміни, пов’язані з виявленими проблемами з безпеки, у супроводі відповідної документації щодо цих виявлених проблем з безпеки негайно, але не пізніше ніж через 15 календарних днів від дати отримання цієї інформації. Запропоновані зміни затверджуються відповідним наказом МОЗ України, після чого лікарський засіб має застосовуватись відповідно до оновленої інформації.
У разі, якщо Центр отримав обґрунтовану інформацію про виявлені небезпечні властивості лікарського засобу для здоров’я або життя людини, що стали підставою для прийняття відповідного рішення про введення обмежень для застосування лікарського засобу в установленому законодавством порядку, Центр повідомляє про це заявника у будь-який спосіб негайно, але не пізніше, ніж через 15 календарних днів від дати прийняття такого рішення.
У разі згоди заявника з прийнятим рішенням він повинен негайно, але не пізніше ніж через 15 календарних днів з моменту отримання цієї інформації, подати до Центру заяву про внесення змін щодо безпеки лікарського засобу до реєстраційних матеріалів. Запропоновані зміни затверджуються відповідним наказом МОЗ України, після чого лікарський засіб повинен вироблятися та застосовуватись відповідно до оновленої інформації.
У разі незгоди заявника з прийнятим рішенням та його відмови від унесення змін щодо безпеки лікарського засобу до реєстраційних матеріалів без обґрунтованих підстав або відсутності його реагування стосовно цього рішення МОЗ України приймає рішення про повну або тимчасову заборону застосування лікарського засобу шляхом припинення дії реєстраційного посвідчення у порядку, передбаченому чинним законодавством.
У разі, якщо 15-й календарний день випадає на вихідний або святковий день, інформація повинна бути надана у перший після нього робочий день.».
Абзаци 3—7 пункту 4.3. Порядку викласти у такій редакції:
«для змін, що потребують нової реєстрації — додаток 1;
для реєстрації додаткової упаковки — додаток 17.
До заяви додаються:
копія діючого реєстраційного посвідчення;
підтвердження затвердження запропонованих змін уповноваженим органом країни виробника/заявника (за необхідності).
Доповнити пункт 4.3. Порядку абзацом 8 у наступній редакції:
«Заява дійсна протягом 3- місяців з дати офіційного подання заяви. У разі ненадання матеріалів реєстраційного досьє протягом зазначеного терміну або листа з обґрунтуванням терміну, необхідного для їх підготовки, то заява знімається з розгляду. Надалі така заява може бути подана до Центру в установленому порядку»
Викласти підпункт 4.3.3. пункту 4.3. Порядку у такій редакції:
«4.3.3. «4.3.3. У випадку відсутності у Додатку 5 до Порядку класифікації відповідних змін, що вносяться до матеріалів реєстраційного досьє під час дії реєстраційного посвідчення, дана зміна зазначається як підпункт «Інша зміна» у відповідному пункті Додатку 7.
Дані зміни за класифікацією можуть бути як зміни типу ІА, ІБ, так і зміни ІІ-го типу.
До змін типу ІА відносять:
а) зміни виключно адміністративного характеру:
- зміна назви та/або адреси заявника (власника реєстраційного посвідчення);
- зміна назви та/або адреси виробника готового лікарського засобу;
- зміна назви та/або адреси виробника активної субстанції;
б) виключення будь-якої виробничої дільниці для таких стадій виробництва готового лікарського засобу: виробництво активної субстанції, виробництво проміжного продукту або готового лікарського засобу, пакування, виробництво серії, контроль серії;
в) зміни у методах контролю якості активної субстанції або вихідної сировини/проміжних продуктів або реагентів, що використовуються у виробничому процесі, за умови, що не змінюється сама методика (наприклад, змінюється довжина хроматографічної колонки але не її тип), проведена валідація зміненого методу контролю, яка показує відповідність попередньому методу, та не змінюються межі, встановлені у специфікації;
г) зміни, що вносяться у зв’язку зі змінами у національній фармакопеї або Європейській фармакопеї;
ґ) зміни в упаковці лікарського засобу, що не контактує безпосередньо з лікарським засобом, та не впливає на доступність, застосування, безпечність або стабільність лікарського засобу;
д) зміни у специфікаціях на лікарський засіб, активну субстанцію, вихідні матеріали, проміжні продукти, допоміжні речовини, що приводять до звуження меж, за умови, що нові параметри залишаються у рамках заявленого діапазону та не є результатом непередбачених обставин у процесі виробництва.
До змін типу ІІ відносять:
а) зміни, пов’язані з введенням нового показання до застосування або змінами вже існуючого;
б) значні зміни у короткій характеристиці лікарського засобу, зокрема, на підставі нових відомостей щодо якості, результатів доклінічних досліджень та клінічних випробувань, нових повідомлень про безпечність лікарського засобу;
в) зміни, що стосуються розширення затверджених специфікацій, меж та критеріїв прийнятності;
г) зміни, що стосуються суттєвих змін у виробничому процесі, складі лікарського засобу, специфікаціях, складі домішок у активній субстанції або готового лікарського засобу, які можуть мати істотний вплив на його якість, безпечність та ефективність;
ґ) зміни пов’язані зі змінами у виробничому процесі або дільниці для виробництва активної субстанції біологічного походження;
д) зміни, пов’язані з введенням нового проектного простору (ICH Q8 (R2) або розширенням затвердженого, який був розроблений відповідно до Європейських та міжнародних наукових керівництв.
е) термінові тимчасові обмеження, які пов’язані з безпечністю використання лікарського засобу і які впроваджуються заявником у разі виявлення ризику для здоров’я людини при застосуванні зареєстрованого (перереєстрованого) лікарського засобу. Про кожну причину, характер і дату передбачуваного введення в дію тимчасових термінових обмежень заявник повідомляє Центр, який протягом 24 годин приймає рішення щодо їх запровадження. При позитивному рішенні Центру подальша експертиза матеріалів про внесення змін здійснюється у порядку, описаному в даному розділі.
є) зміни заявника, крім одночасних змін заявника та виробника або місця виробництва.
Підпункти 4.3.4. та 4.3.5. пункту 4.3. Порядку вилучити.
Доповнити пункт 4.4. абзацом у такій редакції:
«Допускається початок проведення експертизи Центром після надання гарантійного листа щодо сплати експертних робіт, передбачених абзацом першим цього пункту, але при цьому Заявнику не повідомляються результати експертизи, включаючи попередню, до виконання абзацу першого у повному обсязі.»
Доповнити розділ 4 пунктом 4.5. у такій редакції:
«4.5. При внесенні змін І-го типу до Центру на експертизу подаються матеріали згідно Додатку 5.
При внесенні змін ІІ-го типу та реєстрації додаткової упаковки заявник повинен надати Центру:
- матеріали, які обґрунтовують унесення змін;
- відповідні матеріали з поправками, унесеними відповідно до заяви;
- доповнені або оновлені існуючі експертні звіти (огляди, висновки) з урахуванням змін (за необхідності).».
У зв’язку із цим пункти 4.5. — 4.9. вважати пунктами 4.6. — 4.10.
У абзаці 2 пункту 4.6. слово «заяви» замінити словами «реєстраційних матеріалів».
Пункт 4.8. викласти у такій редакції:
««У разі негативних висновків за результатами попередньої експертизи Центр повідомляє про це заявнику письмово і надає відповідне обґрунтування.
Заявник має доопрацювати реєстраційне досьє згідно із зауваженнями Центру в термін до 15 календарних днів. Час, потрібний для доопрацювання, не входить до терміну проведення експертизи.
Якщо заявник протягом визначеного терміну не надає Центру доопрацьованих матеріалів або листа з обґрунтуванням термінів, необхідних для їх доопрацювання, а також якщо надані заявником додаткові або відсутні дані та/або інформація не забезпечують відповідності реєстраційного досьє встановленим вимогам, то зміни до матеріалів реєстраційного досьє на лікарський засіб знімаються з розгляду. Про прийняте рішення Центр письмово повідомляє заявнику.
При цьому вартість проведення експертних робіт заявникові не повертається. Надалі, на бажання заявника, матеріали про внесення змін до реєстраційних документів протягом дії реєстраційного посвідчення подаються в установленому порядку.».
Доповнити Порядок розділом 5 у такій редакції:
«5. Порядок проведення контролю якості лікарського засобу, який подається на державну реєстрацію (перереєстрацію)
5.1. Контроль якості лікарського засобу проводиться під час експертизи матеріалів, що додаються до заяви про його державну реєстрацію відповідно до наданих у них специфікації та методів контролю.
5.2. Уповноважена лабораторія проводить контроль якості після оплати його вартості, установленої договором між заявником та уповноваженою лабораторією. Звіти щодо проведеного контролю якості заявник надає до Центру для проведення експертизи та долучення до матеріалів реєстраційного досьє.
5.3. Контроль якості лікарського засобу не проводиться у випадках, коли подана заява про реєстрацію:
- у частині фасування та/або пакування з форми in bulk за умови, що форма in bulk вже зареєстрована та під час її реєстрації проводився контроль якості в уповноваженій лабораторії;
- після проведених клінічних випробувань (дослідження біоеквівалентності) за умови, що контроль якості вже проводився на досліджуваному лікарському засобі і специфікація та технологія виробництва лікарського засобу не зазнали змін;
- діючої речовини (субстанції);
- препарату-сироти за умови, що лікарський засіб є зареєстрованим у країні із строгою регуляторною системою;
- додаткових дозувань лікарського засобу до вже зареєстрованого дозування за умови, що склад дозувань пропорційно-подібний, виробництво лікарського засобу відбувається на одному й тому ж обладнанні в умовах GMP, проведене дослідження з валідації методів контролю та технологічного процесу (примітка: у випадку одночасної реєстрації таких пропорційно-подібних дозувань контроль якості проводять для одного дозування);
- лікарського засобу того ж виробника під іншою назвою;
- лікарського засобу за «заявою інформованої згоди» за наявності реєстраційного посвідчення того самого або іншого власника.
5.4. У тих випадках, коли у уповноважених лабораторій України відсутня акредитація на право виконання вимірювань за певними показниками, перевірку якості лікарського засобу можливо здійснити на основі повного комплекту первинних матеріалів виробника, які слугували підставою для надання дозволу про випуск певної серії готового лікарського засобу, або шляхом інспектування дільниці проведення контролю якості даного лікарського засобу у виробника.
5.5. Контроль якості лікарського засобу не проводиться у випадках, коли подана заява про перереєстрацію та протягом дії реєстраційного посвідчення специфікація та методи контролю якості не змінювались, або заявником вносились або вносяться зміни до матеріалів реєстраційного досьє у частині, що стосується специфікації та методів контролю якості, у повній відповідності до вимог чинного законодавства. У іншому випадку проводиться контроль лікарського засобу за показником або методикою, які зазнали змін.»
У зв’язку із цим розділи 5—8 уважати відповідно розділами 6—9. У тексті Порядку та додатках до нього посилання на розділи 5—8 замінити посиланнями відповідно на розділи 6—9.
У пункті 6.1. слова «прийняття заяви» замінити словами «надходження до Центру реєстраційних матеріалів».
У абзаці 1 пункту 6.2. слова «прийняття відповідної заяви» замінити словами «надходження до Центру реєстраційних матеріалів».
Доповнити пункт 6.2. абзацом 5 у такій редакції:
«змін, що потребують нової реєстрації.».
У абзаці 1 пункту 6.3 слова «відповідної заяви» замінити словами «відповідних реєстраційних матеріалів».
Доповнити розділ 6 пунктами 6.4 та 6.5. у такій редакції:
«6.4. Датою завершення експертизи реєстраційних матеріалів, зазначених у п. 5.1. — 5.3. вважається день прийняття Центром рішення про:
6.4.1 надання вмотивованих висновків щодо ефективності, безпечності та якості лікарського засобу та рекомендацій лікарського засобу до державної реєстрації (перереєстрації);
6.4.2. надання рекомендацій щодо відмови у державній реєстрації (перереєстрації);
6.4.3. надання рекомендацій щодо внесення змін до реєстраційних матеріалів або нової реєстрації лікарського засобу в установленому порядку;
6.4.4. відмову у внесенні змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення на лікарській засіб
6.4.5. зняття лікарського засобу з розгляду.
6.5. До термінів експертних робіт, перелічених у підпунктах 6.1—6.3 Порядку, не входить час, коли матеріали були на доопрацюванні в заявника, час, необхідний на отримання відповідей від третіх осіб (у т. ч. — уповноважених органів) на запити Центру, пов’язані із проведенням експертизи, а також час проведення додаткових експертиз (випробувань).»
Пункт 7.1. викласти у такій редакції:
«7.1. Реєстраційні матеріали, що надаються на експертизу, повинні містити:
7.1.1. для державної реєстрації — інформацію, яка зазначена у додатках 2 або 3 (готові лікарські форми, продукція in bulk, продукція із in bulk), або додатку 12 (традиційні лікарській засоби або лікарські засоби, які виробляються згідно із затвердженим прописом) або додатку 13 (діючі речовини (субстанція), пелети, гранули тощо);
7.1.2. для державної перереєстрації — інформацію, яка зазначена у додатку 15;
7.1.3. для внесення змін до реєстраційних матеріалів — інформацію, яка зазначена у додатку 5, або інформацію, необхідну для експертизи для внесення змін ІІ типу, реєстрації додаткової упаковки та змін, що потребують нової реєстрації.
Зазначені відомості необхідно регулярно поновлювати.
При підготовці реєстраційних матеріалів заявник може використовувати рекомендації, наведені в додатку 4 до Порядку.
При реєстрації (перереєстрації) традиційних лікарських засобів та лікарських засобів, що виробляються згідно із затвердженими прописами, та реєстрації додаткової упаковки заява подається згідно з додатком 17.
Абзац 1 пункту 7.3. Порядку після слів «не вимагається надання результатів» доповнити словом «власних».
Абзац 1пункту 7.3.3 доповнити реченням у такій редакції:
«Вимоги щодо надання доказів еквівалентності генеричного лікарського засобу до референтного лікарського засобу наведені у додатку 20».
Пункт 7.8. вилучити.
Пункт 7.10 Порядку викласти у такій редакції:
«7.10. Перелік документів для проведення експертизи матеріалів для державної перереєстрації лікарського засобу визначений у додатку 16 або за бажанням Заявника у форматі ЗТД (додаток 3).
У разі проведення експертизи реєстраційних матеріалів на традиційні лікарські засоби, що подаються для держаної перереєстрації (заява — додаток 17), реєстраційні матеріали надаються відповідно до додатку 16.
Перелік документів, для проведення експертизи реєстраційних матеріалів на лікарські засоби, що виробляються згідно із затвердженими прописами для державної перереєстрації (заява — додаток 17) визначений у додатку 22.
Для проведення експертизи реєстраційних матеріалів на діючі речовини (субстанції), які подаються для державної перереєстрації разом із заявою про проведення експертизи матеріалів на діючі речовини (активні субстанції), які подаються для державної перереєстрації (додаток 16) подаються оновлені методи контролю якості, оновлені дані про технологію виробництва та інформація щодо відсутності змін у методах контролю та технології виробництва діючих речовин.
Примітка. У разі необхідності державної реєстрації (перереєстрації) лікарських засобів, які відносяться до групи лікарських засобів — «Медичні гази», відповідна заява подається до Центру згідно додатку 17, а перелік документів — як на відтворені діючі речовини (субстанції), доповнені Інструкцією для медичного застосування.».
Пункт 7.11. викласти у наступній редакції:
«7.11. Реєстраційне досьє подається до Центру відповідно до типу реєстраційної процедури у 3-х примірниках. За погодженням з Центром заявник може подавати окремі частини реєстраційного досьє на електронному носії (додаток 2: розділ 2 G частини ІІ, розділ IVB1 частини ІV; додаток 3: модуль V).
Реєстраційні матеріали подаються українською, або російською, або англійською мовою. У разі подання матеріалів англійською, з перекладом на українську або російську мову додатково надаються матеріали, перелічені в частині I додатка 2 або в Модулі 1 додатка 3, а на вимогу Центру — інформація, передбачена Модулем 2 додатка 3, методи контролю якості готового лікарського засобу, технологічний регламент або відомості про технологію виробництва.
У разі реєстрації діючої речовини інформація, передбачена додатком 13, подається англійською мовою, з перекладом на українську або російську додатково надаються матеріали, перелічені в частині 1 додатка 13, методи контролю якості.
Переклад реєстраційних матеріалів має бути засвідчений нотаріально або підписом особи, яка його здійснила, із зазначенням прізвища, ім’я, по батькові та посади такої особи, а також скріплений печаткою юридичної особи, яка подає такі матеріали до Центру (за наявності).».
Доповнити Порядок розділом 8 у такій редакції:
«8. Порядок видачі реєстраційного посвідчення (вкладки).
8.1. У разі складення Центром позитивних висновків за результатами проведеної експертизи та сплати Заявником державного збору та вартості експертних робіт, передбачених абзацом першим пункту 3.5. та абзацом першим пункту 4.4. Порядку, Центр протягом 10 робочих днів готує та видає Заявнику для узгодження проекти наступних документів:
- реєстраційного посвідчення, інструкції для медичного застосування та короткої характеристики лікарського засобу (за ії наявності), методів контролю якості лікарського засобу — під час здійснення державної реєстрації (перереєстрації) лікарського засобу;
- вкладки до реєстраційного посвідчення, змін до інструкції для медичного застосування та короткої характеристики лікарського засобу, змін до методів контролю якості — під час внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення на лікарській засіб.
Час, необхідний для підготовки Центром та узгодження Заявником документів, зазначених у абзацах 1—3 цього пункту, не входить до терміну експертизи реєстраційних матеріалів.
8.2. Протягом 3-х робочих днів після прийняття МОЗ рішення про державну реєстрацію (перереєстрацію) лікарського засобу або про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення Центр повідомляє Заявника про прийняте рішення, оформляє та подає до МОЗ проект реєстраційного посвідчення (вкладки до реєстраційного посвідчення).
8.3. Реєстраційне посвідчення (вкладка до реєстраційного посвідчення) протягом трьох робочих днів з моменту отримання підписуються уповноваженою посадовою особою та засвідчується печаткою МОЗ України. Оформлені реєстраційні посвідчення (вкладка до реєстраційного посвідчення) передаються у Центр для видачі Заявнику.
8.4. Підписане уповноваженою посадовою особою та засвідчене печаткою МОЗ України реєстраційне посвідчення із зазначенням строку, протягом якого дозволяється застосування лікарського засобу в Україні (вкладка до реєстраційного посвідчення) видається Центром за наявності належним чином оформлених документів, що підтверджують відповідні повноваження у представника Заявника.»
У зв’язку із цим розділи 8 та 9 уважати відповідно розділами 9 та 10. У тексті Порядку та додатках до нього посилання на розділи 8 та 9 замінити посиланнями відповідно на розділи 9 та 10.
У пункті 1В додатку 2 до пункту 7.8 Порядку слова «та листок-вкладиш» вилучити.
У підпункті 1В2 додатку 2 до пункту 7.8 Порядку символ та слова «, листка-вкладиша» вилучити.
Абзац другий преамбули частини IV Додатку 4 до Порядку викласти в такій редакції:
«До клінічних випробувань належить науково-дослідницька робота, метою якої є будь-яке дослідження за участю людини як суб’єкта дослідження, призначене для виявлення або підтвердження клінічних, фармакологічних та/або інших фармакодинамічних ефектів одного або декількох досліджуваних лікарських засобів, та/або виявлення побічних реакцій на один або декілька досліджуваних лікарських засобів, та/або для вивчення усмоктування, розподілу, метаболізму та виведення одного або кількох лікарських засобів з метою підтвердження його (їх) безпечності та/або ефективності».
У тексті Порядку та додатків до нього слова «листок-вкладиш» у всіх відмінках замінити словами «інструкція для медичного застосування лікарського засобу» у відповідних відмінках.
У тексті додатків до Порядку слова «Торгова назва лікарського засобу» в усіх відмінках замінити словами «Назва лікарського засобу».
У абзаці п’ятому пункту 1А частини І «Резюме досьє» додатку 2 до Порядку після слів «три виробничі» доповнити словами та символами) «(для вітчизняного виробника — науково-дослідні)».
Пункт IB 3 частини І «Резюме досьє» додатку 2 та пункт 1.3.5. Модулю 1 додатку 3 до Порядку викласти у такій редакції:
«І В 3. Копія короткої характеристики лікарського засобу/інструкції для медичного застосування, затвердженої в країні заявника/виробника (за наявності).»
У абзаці 4 пункту 1.2. та підпункті 1.3.5. пункту 1.3. Модулю 1 частини 1 Вимог до матеріалів реєстраційного досьє додатку 3 до Порядку слова та символ «копію короткої характеристики лікарського засобу, розробленої та затвердженої в країні виробника/заявника» замінити словами та символом «копію короткої характеристики лікарського засобу/ інструкції для медичного застосування, розробленої та затвердженої в країні заявника/виробника (за наявності).»
Розділ 5 частини 3 Вимог до матеріалів реєстраційного досьє додатку 3 до Порядку — вилучити.
У абзаці першому підпункту в) абзацу 1 пункту 5.2. додатку 3 до Порядку слова «історій хвороби суб’єктів досліджень» замінити словами «медичних карт стаціонарних/ амбулаторних хворих (суб’єктів досліджень)».
Абзац п’ятий підпункту в) абзацу 1 пункту 5.2. додатку 3 до Порядку викласти у такій редакції:
«Медичні карти стаціонарних/амбулаторних хворих (суб’єктів досліджень) повинні зберігатися у відповідних умовах та протягом строку, передбачених чинним законодавством.»
Доповнити підпункт 5.3.1. пункту 5.3. модулю 5 частини 1 розділу «Вимоги до матеріалів реєстраційного досьє» додатку 3 до Порядку абзацом 3 у такій редакції:
«В разі проведення процедури біовейвер необхідно надати звіт про дослідження in vitro за БСК в розділі 5.3.1.2. Оцінка та проведення досліджень біоеквівалентності або обґрунтування того, чому дослідження не проводилися, має бути відповідно до вимог керівництва з дослідження біоеквівалентності та додатку 20.»
Підпункт 5.3.7 модуля 5 розділу «Структура реєстраційного досьє» додатку 3 до Порядку викласти в такій редакції:
«5.3.7. Зразки індивідуальних реєстраційних форм та списки пацієнтів із збереженням конфіденційності персональних даних суб’єктів дослідження».
Назву пункту 5.3.7 модулю 5 частини 1 розділу «Вимоги до матеріалів реєстраційного досьє» Додатку 3 до порядку викласти в такій редакції:
«5.3.7. Зразки індивідуальних реєстраційних форм та списки пацієнтів із збереженням конфіденційності персональних даних суб’єктів дослідження.».
Додатки 1, 5, 6, 7, 8, 12, 16 до Порядку викласти у новій редакції (додаються).
Доповнити додаток 14 до Порядку абзацом 4 у такій редакції: «Я гарантую достовірність інформації, що міститься у наданих реєстраційних матеріалах», у зв’язку з чим абзац 4 вважати абзацом 5.
Доповнити Порядок додатком 9 у редакції, що додається, у зв’язку із чим вважати додатки 9 — 18 відповідно додатками 10 — 19. У тексті Порядку та додатках до нього посилання на додатки 9 — 18 замінити посиланнями відповідно на додатки 10 — 19.
Абзац 1 додатку 17 до Порядку викласти у такій редакції:
«Цим підтверджується, що всі існуючі дані, що стосується якості діючої речовини (субстанції) представлені у реєстраційному досьє та є достовірними.»
Доповнити додаток 10 до Порядку пунктом 5 у такій редакції:
«5. На упаковці лікарських засобів, що містять один чи декілька наркотичних засобів або одну чи декілька психотропних речовин, що є такими відповідно до чинного законодавства, повинні міститись докладні відомості, вказані в пунктах 1.1 та 1.2 цього Додатка. Додатково первинна упаковка повинна бути позначена подвійною червоною смугою.»
У зв’язку із цим уважати пункти 5 та 6 додатку 10 пунктами 6 та 7 відповідно.
Абзац сьомий пункту 6 додатку 10 до Порядку викласти у такій редакції:
«На зовнішній упаковці лікарських засобів також шрифтом Брайля зазначаються назва лікарського засобу, доза діючої речовини та лікарська форма у порядку, визначеному МОЗ України.»
Додаток 11 викласти в новій редакції, що додається.
У додатку 15 до Порядку слова «інструкції/листку-вкладиші» замінити словом «інструкції».
Доповнити Порядок додатками 20, 21 у редакції, що додається.
| Директор Департаменту контролю якості медичних послуг, регуляторної політики та санітарно-епідемічногоблагополуччя — начальник Управління розвитку фармацевтичного сектору галузі охорони здоров`я |
Стеців В. В. |
«Додаток 1
до пункту 3.3 Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
ЗАЯВА
про державну реєстрацію лікарського засобу
Дата надходження заяви
«___» __________ 20__ року № ______________________
Назва лікарського засобу
Діюча(-і) речовина(-и) або
комбінований лікарський засіб
Сила(-и) дії (дозування)
Лікарська форма та упаковка
Заявник
Уповноважена особа, що виступає від
імені заявника
Цим гарантую, що всі дані реєстраційного досьє, представлені у відповідності до додатку_________
_______________________________________________________________________________
(зазначити)
є достовірними. Надаю згоду, що у разі не надання реєстраційного досьє протягом 6 місяців у відповідності до додатку
______________________________________________________________________
(зазначити)
дана заява буде анульована.
Усі дані одержані заявником в установленому законом порядку і не порушують права третьої сторони, захищені патентом, свідоцтвом про знак для товарів та послуг (пункт 4.14 цього додатка).
Також цим підтверджується, що всі передбачені збори сплачені/будуть сплачені відповідно до вимог законодавства.
Від імені заявника
________________________________
Підпис
________________________________
Ім’я та прізвище
________________________________
Посада
М.п.
1. ЗАГАЛЬНІ ПУНКТИ ЗАЯВИ ПРО ДЕРЖАВНУ РЕЄСТРАЦІЮ
1. Загальні пункти заяви про державну реєстрацію
1.1. Ця заява подається відповідно до такого:
Примітка. Розділ повинен бути заповнений для будь-якої заяви, уключаючи заяви, на які є посилання в розділі 1.1.
У разі надання заяви на відповідний тип вилучають перелік інших видів заяв, на які не поширюється дана заява.
![]() Повна і незалежна заява/автономна заява
Повна і незалежна заява/автономна заява
![]() Нова діюча речовина (далі — ДР)
Нова діюча речовина (далі — ДР)
Відома ДР
![]() Бібліографічна заява
Бібліографічна заява
(надається реєстраційне досьє з адміністративними, фармацевтичними та бібліографічні дані, що свідчать про ефективність та безпечність лікарського засобу)
Заява про лікарський засіб, який має добре вивчене медичне застосування
(надається реєстраційне досьє з адміністративними, фармацевтичними та літературним оглядом даних, що свідчать про ефективність та безпечність лікарського засобу)
![]() Заява інформованої згоди
Заява інформованої згоди
(надається реєстраційне досьє з адміністративними, фармацевтичними, доклінічними та клінічними даними разом зі згодою власника доклінічних та клінічних даних)
Примітка. Зареєстрований лікарський засіб і заява інформованої згоди можуть мати як одного, так і різного(их) власника(ів) реєстраційного посвідчення.
Зареєстрований в Україні лікарський засіб:
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
— Додайте письмову згоду власника реєстраційного посвідчення
— на зареєстрований лікарський засіб (пункт 4.2 цього додатка).
![]() Заява про генеричний лікарський засіб
Заява про генеричний лікарський засіб
(надається реєстраційне досьє з адміністративними, фармацевтичними даними, уключаючи еквівалентність до референтного лікарського засобу, доклінічними (літературний огляд) та клінічними (власними даними або літературний огляд) даними)
– Референтний лікарський засіб:
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
– Лікарський засіб, що використовувався у дослідженнях на біоеквівалентність (якщо такі проводилися):
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
![]() Заява на комбінований препарат-генерик
Заява на комбінований препарат-генерик
(надається реєстраційне досьє з адміністративними, фармацевтичними, даними, уключаючи еквівалентність до референтного лікарського засобу, доклінічними (літературний огляд) та клінічними (власними даними або літературний огляд) даними)
– Референтний лікарський засіб:
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
– Лікарський засіб, що використовувався у дослідженнях на біоеквівалентність (якщо такі проводилися):
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
![]() Заява про подібний біологічний лікарський засіб
Заява про подібний біологічний лікарський засіб
(надається реєстраційне досьє з адміністративними, фармацевтичними, хімічними, біологічними даними, уключаючи еквівалентність до референтного лікарського засобу. Крім того, мають надаватись
додаткові дані, що підтверджують належний рівень безпечності (токсикологічні чи інші доклінічні дані) та ефективності (клінічні дані).
– Референтний лікарський засіб:
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
– Лікарський засіб, що використовувався у дослідженнях на біоеквівалентність:
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
![]() Заява про лікарський засіб з відомою діючою речовиною у новій лікарські формі, іншим терапевтичним використанням тощо
Заява про лікарський засіб з відомою діючою речовиною у новій лікарські формі, іншим терапевтичним використанням тощо
(надається реєстраційне досьє з адміністративними, фармацевтичними, доклінічними та клінічними даними)
– Референтний лікарський засіб:
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
– Лікарський засіб, що використовувався у дослідженнях на біоеквівалентність (якщо такі проводилися):
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
– Відмінності порівняно з референтним лікарським засобом:
– інша лікарська форма;
– інша(і) сила(и) дії (кількісні зміни ДР);
– інший спосіб уведення;
– інша фармакокінетика (уключаючи іншу біодоступність);
– інше терапевтичне застосування;
– інші відмінності __________________________________________
___________________________________________________________
![]() Заява_на_фіксовану_комбінацію
Заява_на_фіксовану_комбінацію
(надається реєстраційне досьє з адміністративними, фармацевтичними, власними доклінічними та власними клінічними даними)
![]() Заява на продукцію із in bulk
Заява на продукцію із in bulk
(надається реєстраційне досьє з адміністративними, фармацевтичними доклінічними (літературний огляд) та клінічними (літературний огляд) даними)
Примітка. У разі наявної державної реєстрації разом із продукцією in bulk готової лікарської форми надаються лише адміністративні та фармацевтичні дані для цього типу заяви
![]() Заява на гомеопатичний лікарський засіб
Заява на гомеопатичний лікарський засіб
(надається реєстраційне досьє з адміністративними, фармацевтичними доклінічними (літературний огляд) та клінічними даними)
![]() Заява на зміни, що потребують нової реєстрації
Заява на зміни, що потребують нової реєстрації
(надаються відповідні розділи реєстраційних матеріалів, які обґрунтовують указані зміни і є достатніми для експертизи лікарського засобу)
Вибрати необхідне:
1. Зміни активних речовин:
а) заміна активної речовини на іншу сіль, ефір, похідну тощо за умови, що характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих або
– використання інших ізомерів, або зміна ізомерного складу, або
– незначні зміни молекулярної структури речовин біологічного походження за виключенням зміни у активній речовині протигрипозних вакцин;
б) зміна вектора, який використовується для виробництва антигена або вихідного матеріалу, включаючи новий головний банк клітин від іншого джерела, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
в) новий ліганд або механізм з’єднання для радіофармацевтичного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
г) застосування інших екстрагентів або співвідношення рослинна лікарська сировина/рослинний препарат для рослинного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих.
2. Зміни сили дії, лікарської форми та способу застосування:
а) зміна біодоступності;
б) зміна фармакокінетики;
в) зміна або введення додаткового дозування лікарського засобу (додаткова доза);
г) зміна або додання нової лікарської форми;
ґ) зміна або додання нового шляху введення (для парентеральних форм у зв’язку з відмінностями в ефективності та безпечності лікарського засобу при внутрішньоартеріальному, внутрішньовенному, внутрішньом’язовому та інших шляхах уведення).
– Лікарський засіб, що зареєстровано в Україні і до якого вносяться відповідні зміни
– назва лікарського засобу, сила дії, лікарська форма;
– власник реєстраційного посвідчення;
– номер(и) реєстраційного(их) посвідчення(нь).
2. ОСОБЛИВІ ПУНКТИ ЗАЯВИ ПРО ДЕРЖАВНУ РЕЄСТРАЦІЮ
2.1. Назва та код АТС
2.1.1. Назва лікарського засобу
2.1.2. Назва ДР або склад (указати)
Примітка. Тільки одна назва повинна бути наведена в такому порядку: МНН*, ДФУ, Європейська Фармакопея, загальноприйнята назва, наукова (хімічна) назва.
2.1.3. Фармакотерапевтична група (використовуйте діючий код АТС)
Код АТС Група
Укажіть, якщо заява на одержання коду АТС перебуває на розгляді:
2.2. Сила дії (дозування), лікарська форма та упаковка, шлях уведення, об’єм контейнера та упаковки
2.2.1. Сила дії (дозування) і лікарська форма та упаковка
(використовуйте діючий список стандартних термінів — ДФУ або Європейської Фармакопеї)
Лікарська форма та упаковка
Діюча(-і) речовина(-и) або комбінований лікарський засіб
Сила(-и) дії (дозування)
2.2.2. Шлях(-и) введення (використовуйте діючий список стандартних термінів — ДФУ або Європейської Фармакопеї)
* ДР повинна бути названа за рекомендованими МНН відповідно до форми солі або гідрату (якщо існує). 2.2.3. Контейнер, кришка та пристрої для введення, уключаючи опис матеріалу, з якого вони виготовлені (використовуйте діючий список стандартних термінів — ДФУ або Європейської Фармакопеї)
Для кожного типу упаковки вкажіть:
2.2.3.1. Розмір(-и) упаковки.
2.2.3.2. Пропонований термін придатності.
2.2.3.3. Пропонований термін придатності (після першого розкриття контейнера).
2.2.3.4. Пропонований термін придатності (після розчинення або розведення).
2.2.3.5. Пропоновані умови зберігання.
2.2.3.6. Пропоновані умови зберігання після першого розкриття упаковки.
Додайте перелік макетів графічного зображення упаковки, які надаються разом із заявою, якщо можливо (пункт 4.13 цього додатка).
2.3. Правовий статус
2.3.1. Пропонована категорія відпуску:
![]() для рецептурного застосування;
для рецептурного застосування;
![]() для безрецептурного застосування.
для безрецептурного застосування.
2.3.2. Для лікарських засобів, пропонованих для рецептурного застосування заявник надає свої пропозиції щодо категорії відпуску лікарського засобу, однак право визначати категорію відпуску залишається за МОЗ України.
2.4. Заявник (власник реєстраційного посвідчення/компанія)
2.4.1. Власник реєстраційного посвідчення:
назва (найменування);
місцезнаходження;
країна;
телефон;
факс;
e-mail.
2.4.2. Уповноважена особа/компанія, визначена для ведення переговорів від імені заявника під час процедури реєстрації в Україні:
прізвище, ім’я/ назва компанії
![]() Якщо відрізняється від пункту 2.4.1 цього додатка, додайте доручення (пункт 4.1 цього додатка);
Якщо відрізняється від пункту 2.4.1 цього додатка, додайте доручення (пункт 4.1 цього додатка);
Місцезнаходження;
телефон;
факс;
e-mail.
2.4.3. Уповноважена особа/компанія, затверджена для ведення переговорів між власником реєстраційного посвідчення та вповноваженими органами України після реєстрації, якщо вони відмінні від визначених у пункті 2.4.2:
прізвище, ім’я/ назва компанії.
![]() Якщо відрізняється від пункту 2.4.1 цього додатка, додайте доручення (пункт 4.1 цього додатка);
Якщо відрізняється від пункту 2.4.1 цього додатка, додайте доручення (пункт 4.1 цього додатка);
місцезнаходження;
телефон;
факс;
e-mail.
2.4.4. Уповноважена особа заявника в Україні для здійснення фармаконагляду:
прізвище, ім’я;
назва компанії;
місцезнаходження;
цілодобовий телефон;
факс;
e-mail.
![]() Додайте документальне підтвердження кваліфікації уповноваженої особи, відповідальної за фармаконагляд (пункт 4.3 цього додатка), гарантійний лист заявника.
Додайте документальне підтвердження кваліфікації уповноваженої особи, відповідальної за фармаконагляд (пункт 4.3 цього додатка), гарантійний лист заявника.
2.5. Виробники
2.5.1. Виробник(-и) лікарського засобу і дільниця(-і) виробництва:
назва (найменування);
місцезнаходження;
країна;
телефон/факс;
e-mail.
Короткий опис технологічного процесу, здійснюваного виробником лікарського засобу
![]() Додайте схему виробництва із зазначенням дільниць виробництва, послідовно задіяних у виробничому процесі лікарського засобу (уключаючи дільниці для відбору зразків і контролю серій лікарського засобу) у пункті 4.5 цього додатка.
Додайте схему виробництва із зазначенням дільниць виробництва, послідовно задіяних у виробничому процесі лікарського засобу (уключаючи дільниці для відбору зразків і контролю серій лікарського засобу) у пункті 4.5 цього додатка.
![]() Додайте копію сертифіката з Належної виробничої практики (Good Manufacturing Practic — далі GMP) (крім вітчизняного виробника).
Додайте копію сертифіката з Належної виробничої практики (Good Manufacturing Practic — далі GMP) (крім вітчизняного виробника).
![]() Копію ліцензії на виробництво або документа, виданого уповноваженим органом країни виробника, який свідчить про наявність у виробника відповідної ліцензії на виробництво (пункт 4.4 цього додатка).
Копію ліцензії на виробництво або документа, виданого уповноваженим органом країни виробника, який свідчить про наявність у виробника відповідної ліцензії на виробництво (пункт 4.4 цього додатка).
Чи проводилася перевірка дільниці на дотримання вимог GMP уповноваженим органом України або органами країн, де діють процедури взаємного визнання![]()
![]() Ні
Ні ![]() Так
Так
Якщо так, додайте для кожної дільниці виробництва висновок компетентного органу, що проводив перевірку (пункт 4.6 цього додатка), включаючи:
дату останньої перевірки GMP;
назву вповноваженого органу, що проводив перевірку;
вид перевірки (перед-/післяреєстраційна/спеціальна/повторна);
категорію лікарських засобів, що перевіряються, і діючих речовин;
висновок:
відповідає GMP: ![]() Ні
Ні ![]() Так
Так
2.5.2. Виробник(-и) ДР
речовина;
назва (найменування) виробника;
місцезнаходження;
країна;
телефон/факс;
e-mail.
• Чи виданий сертифікат відповідності Європейській Фармакопеї для ДР
![]() Ні
Ні ![]() Так
Так
Якщо так:
речовина;
назва (найменування) виробника;
номер документа;
дата останнього перегляду (рік, місяць, число).
Надати копію (пункт 4.4 цього додатка)
• Чи використовуватиметься майстер-файл на виробництво ДР або посилання на нього
![]() Ні
Ні ![]() Так
Так
Якщо так:
діюча речовина;
назва (найменування) виробника;
реєстраційний номер;
дата надання (рік, місяць, число);
дата останнього перегляду (рік, місяць, число).
Додати лист(-и), що дає(-ють) змогу доступу до майстер-файла на виробництво діючої(-их) речовини(-н) уповноваженим органам України.
Додати копію письмового зобов’язання від виробника ДР інформувати заявника про зміни у виробничому процесі або специфікаціях відповідно до додатка 4 до Порядку (пункт 4.7 цього додатка)
2.5.3. Контрактні компанії, що залучалися для встановлення біодоступності або біоеквівалентності
Для кожної контрактної компанії вкажіть, де проводилися аналітичні перевірки і де зібрані клінічні дані:
назва (найменування);
місцезнаходження;
країна;
телефон/ факс;
e-mail.
Обов’язки відповідно до контракту
2.6. Якісний та кількісний склад лікарського засобу
2.6.1. Якісний та кількісний склад лікарського засобу (ДР та допоміжні речовини):
Необхідно вказати, на яку кількість розрахований склад (наприклад, 1 капсула)
Перерахуйте діючі речовини окремо від допоміжних речовин:
Назва ДР*
1.
2.
3.
Тощо
Кількість
Одиниця
Посилання/монографія
Назва допоміжної(-их) речовини(-н)
1.
2.
3.
Тощо
Кількість
Одиниця
Посилання/монографія
Інформацію про надлишкову кількість не слід указувати в колонках щодо складу, а необхідно викласти нижче:
діюча(-і) речовина(-и)
допоміжна(-і) речовина(-и)
* Для діючих речовин повинна бути зазначена їхня рекомендована непатентована назва із зазначенням їх сольової або гідратної форми.
2.6.2. Перелік матеріалів тваринного і людського походження, що містяться або використовуються у процесі виробництва лікарського засобу
НЕМАЄ ![]()
Назва
Функція
Тваринного походження, через які може передаватися ГЕ***
Інші тваринного походження
Людського походження
Сертифікат відповідності
ДР
Доп.Р*
Р**
1. ![]()
![]()
2. ![]()
![]()
3. ![]()
![]()
![]() Якщо є сертифікат про відповідність Європейській Фармакопеї щодо ГЕ або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків ГЕ, він повинен бути доданий у пункті 4.8 цього додатка
Якщо є сертифікат про відповідність Європейській Фармакопеї щодо ГЕ або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків ГЕ, він повинен бути доданий у пункті 4.8 цього додатка
2.6.3. Чи містить або складається лікарський засіб з генетично модифікованих організмів (далі — ГМО)![]()
![]() Ні
Ні ![]() Так
Так
Якщо так, то чи відповідає лікарський засіб установленим вимогам![]()
Зробіть необхідне посилання
![]() Ні
Ні ![]() Так
Так
![]() Укладіть копію документа, виданого уповноваженим органом країни виробника, про те, що виробник має право використовувати ГМО з дослідницькою метою (пункт 4.9 цього додатка)
Укладіть копію документа, виданого уповноваженим органом країни виробника, про те, що виробник має право використовувати ГМО з дослідницькою метою (пункт 4.9 цього додатка)
* Доп.Р — допоміжна речовина (уключаючи вихідні матеріали, які використовуються у виробництві діючої речовини/допоміжної речовини).
3. ІНШІ ВІДОМОСТІ
3.1. Чи захищений лікарський засіб патентами на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну![]()
![]() Ні
Ні ![]() Так
Так
Якщо так, то наведіть таку інформацію:
Номер патенту
Дата видачі
Діє до
Власник патенту
![]() Укладіть копії патентів згідно з пунктом 4.10 цього додатка
Укладіть копії патентів згідно з пунктом 4.10 цього додатка
![]() Для державної реєстрації лікарських засобів, що базуються або мають відношення до об’єктів інтелектуальної власності, на які відповідно до законів України видано патент, заявник подає копію патенту або ліцензії, якою дозволяються виробництво та продаж зареєстрованого лікарського засобу. Заявники подають лист, у якому вказується, що права третьої сторони, захищені патентом, не порушуються у зв’язку з реєстрацією лікарського засобу
Для державної реєстрації лікарських засобів, що базуються або мають відношення до об’єктів інтелектуальної власності, на які відповідно до законів України видано патент, заявник подає копію патенту або ліцензії, якою дозволяються виробництво та продаж зареєстрованого лікарського засобу. Заявники подають лист, у якому вказується, що права третьої сторони, захищені патентом, не порушуються у зв’язку з реєстрацією лікарського засобу
3.2. Чи захищена торгова марка в Україні![]()
![]() Ні
Ні ![]() Так
Так
Якщо так, то наведіть таку інформацію:
Номер документа
Дата видачі
Діє до
Власник документа
![]() Укладіть копії документів, що передбачені в пункті 4.11 цього додатка
Укладіть копії документів, що передбачені в пункті 4.11 цього додатка
3.3. Відмітити наявність дозвільного документу, виданого уповноваженим органом країни заявника або виробника, або країни зі строгою регуляторною системою, який видано для України
![]() Ні
Ні ![]() Так
Так
Якщо так, то вкладіть документи відповідно до пункту 4.12 цього додатка із зазначенням назви документу та назви компетентного органу, що його видав, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу)
![]() Перелік країн, де лікарський засіб зареєстрований/перереєстрований
Перелік країн, де лікарський засіб зареєстрований/перереєстрований
4. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхідне)
4.1. Доручення, оформлене для ведення переговорів/підписання документів від імені заявника.
4.2. Письмова згода власника реєстраційного посвідчення на зареєстрований лікарський засіб.
4.3. Документальне підтвердження кваліфікації уповноваженої особи, відповідальної за фармаконагляд.
4.4. Копія ліцензії на виробництво або документа, виданого уповноваженим органом країни виробника, який свідчить про наявність у виробника відповідної ліцензії на виробництво.
4.5. Схема виробництва із зазначенням дільниць виробництва, послідовно задіяних у виробничому процесі лікарського засобу (уключаючи дільниці для відбору зразків і контролю серій лікарського засобу).
4.6. Висновки компетентного органу, який здійснював перевірку кожної дільниці виробництва.
4.7. Копія письмового зобов’язання виробника діючої речовини інформувати заявника про зміни у виробничому процесі або специфікаціях відповідно до додатка 4 до Порядку.
4.8. Сертифікат про відповідність Європейській Фармакопеї щодо ГЕ або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрації в країні (за результатами клінічного та лабораторного контролю) випадків ГЕ.
4.9. Копія документа, виданого вповноваженим органом країни виробника, про те, що виробник має право використовувати ГМО з дослідницькою метою.
4.10. Копії патентів на винахід, корисну модель або промисловий зразок, дія яких розповсюджується на Україну.
4.11. Копії документів щодо захисту торговельної марки в Україні.
4.12. Копія реєстраційного посвідчення або іншого документу (сертифікат на фармацевтичний продукт, сертифікат на вільний продаж тощо), виданого уповноваженим органом країни виробника або заявника, або країни зі строгою регуляторної системою, який видано для України (достатньо ксерокопії сторінок із зазначенням номера, дати видачі і сторінки, підписаної уповноваженою особою компетентного органу), та перелік країн, де даний лікарський засіб зареєстрований/перереєстрований.
4.13. Перелік макетів графічного зображення, що надаються разом із заявою.
4.14. Гарантійний лист, у якому вказується, що права третьої сторони, захищені патентом, не порушуються у зв’язку з реєстрацією лікарського засобу.
4.15. Гарантійний лист заявника щодо забезпечення функціонування належної системи нагляду за безпекою лікарських засобів при їх медичному застосуванні в Україні.»
«Додаток 5
до пункту 4.1 Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
ВИМОГИ
до документів, що подаються для експертизи при внесенні змін типу
І до реєстраційних матеріалів протягом дії реєстраційного посвідчення
А. АДМІНІСТРАТИВНІ ЗМІНИ
| А.1. Зміна назви та/або адреси заявника (власника реєстраційного посвідчення) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2 | IА | |
| Умови | |||
| 1. Власником реєстраційного посвідчення повинна залишатися одна й та сама юридична особа. | |||
| Документація | |||
| 1. Документ відповідного компетентного уповноваженого органу, в якому зазначено нову назву та/або нову адресу виробника (власника реєстраційного посвідчення).2. Оновлені: коротка характеристика лікарського засобу, макет графічного зображення упаковки та інструкція для медичного застосування (за необхідності). | |||
| А.2. Зміна торгової назви лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2 | IБ | |
| Умови | |||
| 1. Запропонована назва не порушує права третіх сторін. | |||
| Документація | |||
| 1. Обґрунтування зміни торгової назви.2. Оновлені: коротка характеристика лікарського засобу, макет графічного зображення упаковки та інструкція для медичного застосування. | |||
| А.3. Зміна назви активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | 1, 2 | IА | |
| Умови | |||
| 1. Активна субстанція повинна залишатися тією ж самою. | |||
| Документація | |||
| 1. Підтвердження рекомендованих ВООЗ МНН або копія переліку МНН. Для рослинних лікарських засобів — заява про те, що назва відповідає чинним вимогам або Керівництву з якості рослинних лікарських засобів та керівним принципам для декларування рослинних субстанцій та рослинних препаратів, що входять до складу (традиційних) рослинних лікарських засобів (чинне видання).2. Оновлені: коротка характеристика лікарського засобу, макет графічного зображення упаковки та інструкція для медичного застосування. | |||
| А.4. Зміна назви та/або адреси виробника (включаючи, за необхідності місце проведення контролю якості) або постачальника активної субстанції/вихідного матеріалу/реагенту/проміжного продукту, що застосовуються у виробництві активної субстанції (за відсутності сертифікату відповідності Європейській Фармакопеї у затвердженому досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2, 3 | IА | |
| Умови | |||
| 1. Виробнича дільниця та усі виробничі операції залишаються незмінними. | |||
| Документація | |||
| 1. Документ відповідного уповноваженого органу, у якому зазначено нову назву та/або адресу.2. Зміни до відповідних розділів реєстраційного досьє.3. У разі зміни в назві власника мастер-файла на активну субстанцію — поновлений дозвіл на користування даними, що містяться у мастер-файлі. | |||
| А.5. Зміна назви та/або адреси виробника готового лікарського засобу, включаючи місце проведення контролю якості | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) виробнича дільниця випуску серій | 1 | 1, 2 | IА |
| б) усі інші дільниці | 1 | 1, 2 | IА |
| Умови | |||
| 1. Виробнича дільниця та всі виробничі операції залишаються незмінними. | |||
| Документація | |||
| 1. Копія оновленої ліцензії на виробництво (за наявності) або документ відповідного уповноваженого органу, у якому зазначено нову назва та/або адресу.2. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлені коротку характеристику лікарського засобу, макет графічного зображення упаковки та інструкцію для медичного застосування (за необхідності). | |||
| А.6. Зміна коду АТХ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1, 2 | IА | |
| Умови | |||
| 1. Присвоєння нового або зміна ВООЗ коду АТХ. | |||
| Документація | |||
| 1. Підтвердження присвоєння ВООЗ коду АТХ або копія переліку кодів АТХ.2. Оновлені: коротка характеристика лікарського засобу та інструкція для медичного застосування. | |||
| А.7. Вилучення виробничої дільниці (включаючи дільниці для активної субстанції, проміжного продукту або готового лікарського засобу, дільниці для проведення пакування, виробника відповідального за випуск серій, місце проведення контролю серії) або постачальника вихідного матеріалу, реагенту або допоміжної речовини (якщо зазначено у досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | 1, 2 | IА | |
| Умови | |||
| 1. Необхідно залишити принаймні одну затверджену дільницю/виробника, що виконують таку ж саму функцію, що й вилучені. | |||
| 2. Вилучення не обумовлено непередбаченими обставинами у виробничому процесі. | |||
| Документація | |||
| 1. Заява про внесення змін має чітко визначати затвердженого та запропонованого виробників, як зазначено в розділі 2.5 форми заяви для державної реєстрації лікарського засобу.2. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлені: коротку характеристику лікарського засобу, макет графічного зображення упаковки та інструкцію для медичного застосування (за необхідності). | |||
| Б. ЗМІНИ З ЯКОСТІ | |||
| Б. I. Активна субстанція | |||
| Б. I. а) Виробництво | |||
| Б. I. а.1. Зміна виробника вихідного/проміжного продукту/реагенту, що використовуються у виробничому процесі активної субстанції, або зміна виробника (включаючи де необхідно місце проведення контролю якості) активної субстанції (за відсутності сертифікату відповідності Європейській Фармакопеї у затвердженому досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) запропонований виробник належить до тієї ж виробничої групи підприємств, що й затверджений | 1, 2, 3 | 1, 2, 3, 4, 5, 6, 7 | IА |
| б) введення нового виробника активної субстанції з наданням мастер-файла на активну субстанцію | II | ||
| в) запропонований виробник використовує спосіб синтезу, що суттєво відрізняється від попереднього, або умови виробництва, які потенційно можуть змінити важливі характеристики якості активної субстанції, такі як якісний та/або кількісний профіль домішок, що потребує кваліфікації, або фізико-хімічні властивості активної субстанції, що впливають на біодоступність | II | ||
| г) новий виробник вихідного продукту, для якого вимагається попередня оцінка вірусної безпеки та/або ризику передачі збудників ГЕ | II | ||
| ґ) зміна у субстанції біологічного походження або вихідному матеріалі/реагенті/проміжному продукті, що використовуються для виробництва лікарського засобу біологічного походження | II | ||
| д) зміни у методах контролю якості активної субстанції або додавання дільниці для проведення контролю серії/випробування | 2, 4 | 1, 5 | IА |
| Умови | |||
| 1. Для вихідних матеріалів та реагентів специфікації (включаючи контроль під час виробництва, методи аналізу всіх матеріалів) ідентичні затвердженим. Для проміжних продуктів та активних субстанцій специфікації (включаючи контроль під час виробництва, методи аналізу всіх матеріалів), спосіб виробництва (включаючи розмір серії) та детальний опис методу синтезу ідентичні затвердженим.2. Активна субстанція не є стерильною або речовиною біологічного/імунобіологічного походження.3. Якщо у процесі виробництва використовуються матеріали людського або тваринного походження, виробник не використовує будь-якого нового постачальника, для якого потрібна експертиза вірусної безпеки або відповідності виробництва Керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами (чинне видання).4. Перенесення способу виробництва з попередньої дільниці до нової було успішно виконано. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє (за необхідності).2. Заява від власника реєстраційного посвідчення або від власника мастер-файла на активну субстанцію, за необхідності, що метод синтезу (або у разі лікарських засобів рослинного походження, за потреби, метод приготування, місце походження, виробництво рослинної субстанції та спосіб виробництва), методи контролю якості та специфікації на активну субстанцію та вихідний матеріал/реагент/проміжний продукт у виробництві активної субстанції не відрізняються від затверджених, за потреби.3. ГЕ Сертифікат відповідності Європейській Фармакопеї для нового вихідного продукту або, якщо необхідно, документальне підтвердження того, що вихідний продукт для отримання субстанції, який має ризик передачі збудників ГЕ, попередньо оцінений уповноваженим органом країни-виробника та продемонстрована його відповідність Керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами (чинне видання). Інформація повинна містити: назву виробника, види та тканини тварин, з яких одержано вихідний продукт, назву країни походження тваринної сировини, її використання та попередній дозвіл.4. Дані аналізів (у вигляді таблиць порівняння) принаймні для двох серій (мінімум дослідно-промислових) активної субстанції від затвердженого та запропонованого виробників/дільниць.5. Заява про внесення змін має чітко визначати «затвердженого» та «запропонованого» виробників, як зазначено в розділі 2.5 форми заяви для державної реєстрації лікарського засобу.6. Заява уповноваженої особи (УО) кожного з власників ліцензії на виробництво, перелічених у заяві, якщо активна субстанція використовується як вихідний матеріал, та заява УО кожного з власників ліцензії на виробництво, перелічених у заяві як відповідальних за випуск серії. У заявах необхідно зазначити, що виробник (-и) активної субстанції діє відповідно до Керівництву з належної виробничої практики щодо вихідних матеріалів (чинне видання). Узагальнена заява може бути прийнятною за певних умов — див. Примітку до п. Б. II. б.1.7. Зобов’язання виробника активної субстанції інформувати власника реєстраційного посвідчення про будь-які зміни у процесі виробництва, специфікаціях та методах аналізу активної субстанції (за необхідності). | |||
| Б. I. а.2. Зміни в процесі виробництва активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначна зміна у процесі виробництва активної субстанції | 1, 2, 3, 4, 5, 6, 7 | 1, 2, 3 | IА |
| б) значна зміна у процесі виробництва активної субстанції, що може мати істотний вплив на якість, безпеку або ефективність лікарського засобу | II | ||
| в) зміна стосується субстанції біологічного/імунологічного походження або використання хімічних субстанцій у виробництві лікарського засобу біологічного/імунологічного походження і не відноситься до протоколу | II | ||
| г) зміна у лікарському засобі рослинного походження та стосується однієї з таких характеристик: джерело походження сировини, спосіб виробництва або виготовлення | II | ||
| ґ) незначна зміна у закритій частині мастер-файла на активну субстанцію | 1, 2, 3, 4 | IБ | |
| Умови | |||
| 1. Не повинно бути жодних змін у якісному та кількісному складі домішок або змін фізико-хімічних властивостей субстанції.2. Шлях синтезу не змінюється, а саме проміжні продукти залишаються незмінними, і не використовуються нові реагенти, каталізатори або розчинники. Для рослинних лікарських засобів — джерело походження сировини, виробництво активної субстанції та спосіб виробництва залишаються незмінними.3. Специфікації на активну субстанцію або проміжні продукти залишаються незмінними.4. Зміна цілком описана у відкритій («для заявника») частині мастер-файла на активну субстанцію (за необхідності).5. Активна субстанція не є субстанцією біологічного/імунологічного походження.6. Зміна не стосується джерела походження та способу виробництва рослинного лікарського засобу.7. Зміна не стосується закритої частини мастер-файла на активну субстанцію. | |||
| Документація | |||
| 1 Зміни до відповідних розділів реєстраційного досьє та затвердженого мастер-файла на активну субстанцію (за можливості), включаючи результати порівняння узгодженого та запропонованого виробничого процесу.2. Дані аналізів (у вигляді таблиці порівняння) принаймні для двох серій (мінімум дослідно-промислових), виготовлених відповідно до узгодженого та запропонованого виробничого процесу.3. Копія затверджених специфікацій на активну субстанцію.4. Заява від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію, за необхідності, що будь-які зміни у якісному та кількісному профілі домішок або у фізико-хімічних властивостях відсутні, метод синтезу не змінюється, а також не змінюються специфікації на активну субстанцію або проміжні продукти. | |||
| Примітка до п. Б. I. а.2. б. Для хімічних активних субстанцій це стосується значних змін у методі синтезу або умовах виробництва, що потенційно можуть змінювати важливі характеристики активної субстанції, такі як якісний та/або кількісний профіль домішок, що потребує кваліфікації, або фізико-хімічні властивості активної субстанції, що впливають на біодоступність. | |||
| Б. I. а.3. Зміна розміру серії (включаючи діапазони) активної субстанції або проміжного продукту | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) збільшення до 10 разів порівняно із затвердженим розміром | 1, 2, 3, 4, 6, 7, 8 | 1, 2, 5 | IА |
| б) зменшення обсягу виробництва | 1, 2, 3, 4, 5 | 1, 2, 5 | IА |
| в) зміна, що потребує доведення подібності активної субстанції біологічного/імунологічного походження порівняно із затвердженою | II | ||
| г) збільшення у понад 10 разів порівняно із затвердженим розміром | 1, 2, 3, 4 | IБ | |
| ґ) розмір серії активної субстанції біологічного/імунологічного походження збільшився/зменшився без зміни параметрів процесу (наприклад, дублювання лінії) | 1, 2, 3, 4 | IБ | |
| Умови | |||
| 1. Зміни у процесі виробництва обумовлені тільки збільшенням або зменшенням об’єму виробництва, наприклад використанням обладнання іншої продуктивності.2. Наявність результатів аналізу принаймні двох серій запропонованого розміру відповідно до специфікацій.3. Активна субстанція не є речовиною біологічного/імунологічного походження.4. Зміна не повинна впливати на відтворюваність процесу виробництва.5. Зміна не повинна бути обумовлена непередбаченими обставинами в процесі виробництва або проблемами, пов’язаними зі стабільністю.6. Специфікації на активну субстанцію/проміжні продукти залишаються незмінними.7. Активна субстанція не є стерильною.8. Затверджений розмір серії не був схвалений при внесенні змін типу IА. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Номери серій запропонованого розміру, які проходять випробування.3. Дані аналізів (у вигляді таблиці порівняння) принаймні однієї промислової серії активної субстанції або проміжного продукту для затвердженого та запропонованого розмірів (за необхідності). Дані про наступні дві повні промислові серії мають бути надані на вимогу, а також заява про те, що у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією).4. Копія затверджених специфікацій на активну субстанцію (та проміжні продукти, за потреби).5. Заява від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію про те, що зміни у процесі виробництва обумовлені тільки збільшенням або зменшенням об’єму виробництва, наприклад, використанням обладнання іншої продуктивності, що зміна не впливає на відтворюваність процесу і не обумовлена непередбаченими обставинами в процесі виробництва або проблемами, пов’язаними зі стабільністю, і, що специфікації на активну субстанцію/проміжні продукти залишаються незмінними. | |||
| Б. I. а.4. Зміни випробувань або допустимих меж, що встановлені у специфікаціях, в процесі виробництва активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) додавання нового випробування та допустимих меж | 1, 2, 5, 6 | 1, 2, 3, 4, 6 | IА |
| в) вилучення несуттєвого випробування | 1, 2 | 1, 2, 5 | IА |
| г) розширення затверджених допустимих меж для показників, які можуть істотно вплинути на якість активної субстанції | II | ||
| ґ) вилучення випробування, що може мати істотний влив на загальну якість активної субстанції | II | ||
| д) додавання або заміна випробування за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 6 | IБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду показників, зазначених у специфікації (наприклад, проведеною під час подання заяви на реєстрацію або внесення змін типу II).2. Зміна не обумовлена непередбаченими обставинами в процесі виробництва, наприклад, появою нової некваліфікованої домішки; зміною меж загального вмісту домішок.3. Будь-які зміни не повинні виходити за допустимі межі затвердженої специфікації.4. Методи випробувань залишилися незмінними або такі зміни є незначними.5. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.6. Новий метод випробувань не належить до біологічних/імунобіологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу активної субстанції біологічного походження (за включенням стандартних фармакопейних мікробіологічних методів). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця щодо затверджених та запропонованих методів випробувань.3. Опис нового нефармакопейного методу випробування та дані з його валідації (за необхідності).4. Дані аналізів для двох промислових серій (трьох промислові серії для субстанцій біологічного походження, якщо не обґрунтовано інше) активної субстанції за всіма показниками, що зазначені у специфікації.5. Обґрунтування/оцінка ризику від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію (за необхідності), що підтверджує незначність зміненого параметру.6. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію (за необхідності) введення нового методу випробування або допустимих меж, визначених у специфікаціях. | |||
| Б.1. б) Контроль активної субстанції | |||
| Б. I. б.1. Зміна у параметрах специфікацій та/або допустимих меж, визначених у специфікаціях на активну субстанцію, або вихідний/проміжний продукт/реагент, що використовуються у процесі виробництва активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж, визначених у специфікації на лікарські засоби, що підлягають офіційному випуску серії | 1, 2, 3, 4 | 1, 2 | IA |
| б) звуження допустимих меж, визначених у специфікації | 1, 2, 3, 4 | 1, 2 | IA |
| в) доповнення специфікації новим показником якості та відповідним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 7 | IA |
| г) вилучення незначного показника якості, (наприклад, вилучення застарілого показника) | 1,2 | 1, 2, 6 | IА |
| ґ) вилучення параметра специфікації, який може мати суттєвий влив на якість активної субстанції та/або готового лікарського засобу | II | ||
| д) розширення допустимих меж, визначених у специфікації на активну субстанцію | II | ||
| е) розширення допустимих меж, визначених у специфікаціях на вихідні матеріали/проміжні продукти, які мають істотний вплив на якість активної субстанції та/або готового лікарського засобу | II | ||
| є) доповнення або заміна параметру специфікації за результатами досліджень з безпеки або якості (за виключенням субстанції біологічного або імунологічного походження) | 1, 2, 3, 4, 5, 7 | IБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду показників, зазначених у специфікації (наприклад, проведеною під час подання заяви на реєстрацію або внесення змін типу II).2. Зміна не обумовлена непередбаченими обставинами в процесі виробництва, наприклад, появою нової некваліфікованої домішки; зміною меж загального вмісту домішок.3. Будь-які зміни не повинні виходити за допустимі межі затвердженої специфікації.4. Методи випробувань залишилися незмінними або такі зміни є незначними.5. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.6. Новий метод випробувань не належить до біологічних/імунобіологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу активної субстанції біологічного походження (за включенням стандартних фармакопейних мікробіологічних методів).7. Зміна не стосується домішки, яка має генотоксичну дію. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця щодо затверджених та запропонованих специфікацій.3. Опис нового аналітичного методу та дані з його валідації (за необхідності).4. Результати аналізу для двох промислових серій (трьох промислові серії для субстанцій біологічного походження, якщо не обґрунтовано інше) активної субстанції за всіма показниками, що зазначені у специфікації.5. Порівняльні дані щодо профілю розчинення готового лікарського засобу для принаймні однієї дослідно-промисловій серії, що містить активну субстанцію за затвердженою та запропонованою специфікаціями (за необхідності) Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.6. Обґрунтування/оцінка ризику від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію (за необхідності), що підтверджує незначність зміненого параметру.7. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію (за необхідності) введення нового методу випробування або допустимих меж, визначених у специфікаціях. | |||
| Б. I. б.2. Зміна у методах випробування активної субстанції або вихідного/проміжного продукту/реагенту, що використовується у процесі виробництва активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробування | 1, 2, 3, 4, | 1, 2 | IА |
| б) видалення методу випробування для активної субстанції/реагенту/проміжного продукту, якщо альтернативний метод вже затверджений | 7 | 1 | IА |
| в) інші зміни в методах випробування (включаючи заміну або доповнення) для реагенту, що не спричиняє істотного впливу на якість активної субстанції | 1, 2, 3, 5, 6 | 1, 2 | IA |
| г) зміна (заміна) у біологічному/імунологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент для активної субстанції біологічного походження, наприклад, пептидні карти, глік-карти тощо | II | ||
| ґ) інші зміни у методах випробування (включаючи заміну або доповнення) активної субстанції або вихідного/проміжного продукту | 1, 2 | IБ | |
| Умови | |||
| 1. Дослідження з валідації, які були проведені відповідно до чинних фармакопейних вимог або Керівних принципів з валідації (чинне видання), підтверджують, що результати аналізу, отримані за затвердженою та запропонованою методиками, ідентичні.2. Не відбулося жодних змін меж загального вмісту домішок, не виявлено нових некваліфікованих домішок.3. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу але тип колонки або методика не змінні).4. Новий метод випробувань не належить до біологічних/імунобіологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу активної субстанції біологічного походження (за включенням стандартних фармакопейних мікробіологічних методів).5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.6. Активна субстанція не є речовиною біологічного/імунологічного походження.7. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні зміни типу ІА. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи опис методів контролю, звіт про дані з валідації, переглянуті допустимі межі для домішок (за необхідності).2. Порівняльні дані з валідації, або, якщо обґрунтовано, порівняльні результати аналізу, які підтверджують, що затверджена та запропонована методики випробування ідентичні. Ця вимога не стосується випадку додавання нової методики випробування. | |||
| Б. I. в) Система упаковка/укупорка | |||
| Б. I. в.1. Зміна у безпосередній упаковці активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) якісні та/або кількісні зміни складу | 1, 2, 3 | 1, 2, 3, 4, 6 | IА |
| б) якісні та/або кількісні зміни складу для стерильних та не заморожених активних субстанцій біологічного/імунологічного походження | II | ||
| в) рідких активних субстанцій (не стерильних) | 1, 2, 3, 5, 6 | IБ | |
| Умови | |||
| 1. Запропонований пакувальний матеріал має бути ідентичним затвердженому за відповідними показниками якості.2. Дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів ІСН щодо випробування стабільності (чинне видання) принаймні для двох дослідно-промислових або промислових серій активної субстанції і на момент випуску у розпорядженні заявника були задовільні дані по стабільності принаймні за три місяці. Однак, якщо запропонована упаковка більш стійкіша ніж затверджена, дані щодо стабільності за три місяці ще можуть бути не доступні. При цьому мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності невідповідності специфікаціям наприкінці терміну придатності/періоду повторного випробування (із запропонованими заходами).3. Не стосується стерильних рідких активних субстанцій та субстанцій біологічного/імунологічного походження. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Відповідні дані щодо нової упаковки (наприклад, порівняльні дані про проникність, наприклад, для О2, СО2, вологи), включаючи підтвердження того, що пакувальний матеріал відповідає чинним фармакопейним вимогам щодо пакувальних матеріалів або законодавству ЄС щодо безпеки взаємодії пластикових пакувальних матеріалів та упаковок з харчовими продуктами.3. Підтвердження відсутності будь-якої взаємодії між активною субстанцією та пакувальним матеріалом (наприклад, відсутнє перенесення компонентів запропонованого пакувального матеріалу до активної субстанції та немає жодних втрат компонентів упаковки), включаючи підтвердження того, що пакувальний матеріал відповідає чинним фармакопейним вимогам щодо пакувальних матеріалів або законодавству ЄС щодо безпеки взаємодії пластикових пакувальних матеріалів та упаковок з харчовими продуктами.4. Підтвердження від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію (за необхідності), що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів ІСН щодо випробування стабільності (чинне видання) (із зазначенням кількості та номерів серій активної субстанції) та підтвердження (за необхідності), що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності та наявні дані не вказують на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності невідповідності специфікаціям наприкінці терміну придатності/періоду повторного випробування (із запропонованими заходами).5. Результати дослідження стабільності, проведених згідно з Настановою або Керівними принципами ІСН щодо випробування стабільності (чинне видання) за відповідними параметрами принаймні для двох дослідно-промислових або промислових серій активної субстанції із задовільними показниками щодо стабільності принаймні за три місяці. При цьому мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності невідповідності специфікаціям наприкінці терміну придатності/періоду повторного випробування (із запропонованими заходами).6. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій (за необхідності). | |||
| Б. I. в.2. Зміни у параметрах специфікацій та/або допустимих меж, зазначених у специфікаціях, для первинної упаковки активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж, зазначених у специфікаціях | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфікації новим показником та відповідним методом випробування | 1, 2, 5 | 1, 2, 3, 4, 6 | IА |
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 5 | IА |
| г) доповнення або заміна показника за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 6 | IБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду показників, зазначених у специфікації (наприклад, проведеною під час подання заяви на реєстрацію або внесення змін типу II), якщо раніше вона була розглянута і затверджена як частина послідовних змін.2. Зміна не обумовлена непередбаченими обставинами, що виникли в процесі виробництва пакувального матеріалу або зберігання активної субстанції.3. Будь-які зміни не повинні виходити за допустимі межі затвердженої специфікації.4. Методи випробувань залишилися незмінними або такі зміни є незначними.5. Новий метод випробування не належить до нестандартного методу або стандартного методу, що використовується у новий спосіб. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій.3. Опис нового методу випробування та дані з його валідації (за необхідності).4. Результати аналізу для двох серій первинної упаковки за всіма показниками, зазначеними у специфікації.5. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію, що показник якості не є суттєвим (за необхідності).6. Обґрунтування від власника реєстраційного посвідчення або власника мастер-файла на активну субстанцію нових показників якості та допустимих меж, зазначених у специфікації. | |||
| Б. I. в.3. Зміна в методах випробування первинної упаковки активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробування | 1, 2, 3 | 1, 2 | IА |
| б) інші зміни в методах випробування (включаючи заміну або доповнення) | 1, 3, 4 | 1, 2 | IА |
| в) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | 5 | 1 | IА |
| Умови | |||
| 1. Відповідні дослідження з валідації, які були проведені відповідно до чинних фармакопейних вимог або Керівних принципів з валідації (чинне видання), та результати підтверджують, що затверджена та запропонована методики ідентичні.2. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу але тип колонки або методика не змінні).3. Новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.4. Активна субстанція не є речовиною біологічного/імунологічного походження.5. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні змін типу ІА. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, які включають опис методу випробування та звіт про дані з валідації.2. Порівняльні дані з валідації або, якщо обумовлено, порівняльні результати аналітичних випробувань, які підтверджують ідентичність результатів, отриманих за затвердженим та новим методами. Це не стосується випадків додавання нового методу випробування. | |||
| Б. I. г) Стабільність | |||
| Б. I. г.1. Зміна періоду повторних випробувань/терміну придатності або умов зберігання активної субстанції (за наявності у затвердженому досьє сертифікату відповідності Європейській фармакопеї, що включає період повторного випробування) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) період повторного випробування/термін придатності | |||
| 1. зменшення | 1 | 1, 2, 3 | ІА |
| 2. збільшення періоду повторного випробування на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності до Настанови або Керівних принципів ІСН щодо випробування стабільності (*) | ІІ | ||
| 3. збільшення терміну придатності активної субстанції біологічного/імунологічного походження на основі результатів досліджень, виконаних не у відповідності до затвердженого протоколу | ІІ | ||
| 4. збільшення або введення періоду повторного випробування/терміну придатності на основі результатів досліджень у реальному часі | 1, 2, 3 | ІБ | |
| б) умови зберігання | |||
| 1. обмеження умов зберігання | 1 | 1, 2, 3 | ІА |
| 2. зміна умов зберігання активної субстанції біологічного/імунологічного походження, якщо дослідження стабільності ще не проводились відповідно до затвердженого протоколу | II | ||
| 3. зміна умов зберігання активної субстанції | 1, 2, 3 | ІБ | |
| Умови | |||
| 1. Зміна не обумовлена непередбаченими обставинами у процесі виробництва або проблемами щодо стабільності. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє повинні містити результати досліджень стабільності, представлені у реальному часі, і проведені відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання), принаймні для двох (трьох — для лікарських засобів біологічного походження) дослідно-промислових або промислових серій активної субстанції у затвердженій упаковці з відповідним періодом повторного випробування у визначених умовах зберігання.2. Підтвердження, що дослідження стабільності виконані відповідно до затвердженого протоколу. Дослідження повинні підтвердити відповідність специфікацій.3. Копії затверджених специфікацій на активну субстанцію. | |||
| (*) Період повторного випробування не застосовується до активних субстанцій біологічного/імунологічного походження. | |||
| Б.1.ґ) Проектний простір | |||
| Б. I.ґ.1. Введення нового проектного простору або розширення затвердженого для активної субстанції щодо | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) одного елементу (блок, частина) виробничого процесу активної субстанції, включаючи контроль в процесі виробництва та/або методи випробування | 1, 2, 3 | II | |
| б) методів випробувань для вихідних матеріалів/реагентів/проміжних продуктів та/або активної субстанції | 1, 2, 3 | II | |
| Документація1. Проектний простір був розроблений відповідно до Європейських та міжнародних наукових керівництв. Результати досліджень розробки препарату, виробничого процесу або методів випробувань (наприклад, необхідно вивчити взаємодію різних параметрів, що формують проектний простір, включаючи дослідження оцінки ризику та багатомірні дослідження, якщо необхідно), які демонструють, що функціональна взаємодія властивостей матеріалу та параметрів процесу, які можуть впливати на критичні характеристики якості активної субстанції, була досягнута.2. Опис проектного простору у формі таблиці, включаючи змінні (властивості матеріалу та параметри процесу, якщо необхідно) та їх запропоновані діапазони.3. Зміни до відповідних розділів реєстраційного досьє. | |||
| Б. I.ґ.2. Внесення змін після затвердження протоколу для активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | II | ||
| Документація | |||
| 1. Детальний опис запропонованої зміни.2. Протокол управління змінами для активної субстанції. | |||
| Б. I.ґ.3. Вилучення затвердженого протоколу управління змінами для активної субстанції | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1 | 1 | IА | |
| Умови | |||
| 1. Вилучення не спричинене непередбаченими обставинами або результатами випробувань, що виходять за межі специфікацій під час внесення зміни, описаної у протоколі. | |||
| Документація | |||
| 1. Обґрунтування запропонованого вилучення. | |||
| Б. II. Готовий лікарський засіб | |||
| Б. II. а) Опис та склад | |||
| Б. II. а.1. Зміна або додавання штампів, потовщень або інших маркувань, уключаючи заміну або додавання фарб для маркування лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміни штампів, потовщень або інших маркувань | 1, 2, 3 | 1, 2 | IА |
| б) зміни риски, призначеної для розділу таблетки на рівні дози | 1, 2, 3 | IБ | |
| Умови | |||
| 1. Специфікації при випуску і наприкінці терміну придатності готового лікарського засобу не змінилися (крім зовнішнього вигляду).2. Будь-яка фарба повинна відповідати вимогам до фармацевтичної продукції.3. Риски не призначені для розділу на рівні дози. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи детальне зображення або опис затвердженого та запропонованого вигляду лікарського засобу, а також оновлену коротку характеристику лікарського засобу (за необхідності).2. Зразки готового лікарського засобу (за потреби).3. Результати фармакопейних випробувань, що підтверджують відповідність характеристик/правильного дозування лікарського засобу. | |||
| Б. II. а.2. Зміна форми або розмірів лікарської форми | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) таблетки з негайним вивільненням, капсули, супозиторії та песарії | 1, 2, 3, 4 | 1, 4 | IА |
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим або пролонгованим вивільненням та ділимі таблетки, призначені для розділу на рівні дози | 1, 2, 3, 4, 5 | IБ | |
| Умови | |||
| 1. Порівняння профілів розчинення лікарського засобу з новими та затвердженими формою або розмірами (за необхідності). Для рослинних лікарських засобів, коли випробування на розчинення не може бути проведене — дані щодо розпадання.2. Специфікації при випуску та наприкінці терміну придатності лікарського засобу не змінюються (крім розміру).3. Якісний та кількісний склад, середня маса лікарського засобу залишаються незмінними.4. Зміни не стосуються таблеток з рискою, призначеною для розподілу на рівні дози. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи детальне зображення затверджених та запропонованих форми або розміру, а також оновлену коротку характеристику лікарського засобу та інструкцію для медичного застосування.2. Порівняльні дані досліджень для принаймні однієї дослідно-промислової серії лікарського засобу, які підтверджують відсутність змін у профілі розчинення (див. Керівництво з дослідження біодоступності) для нового та затвердженого розміру або форми. Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.3. Обґрунтування відсутності необхідності проведення нового дослідження з біоеквівалентності у відповідності до Керівництва з дослідження біодоступності.4. Зразки готового лікарського засобу з новим розміром або формою (за потреби).5. Результати фармакопейних випробувань, що підтверджують відповідність характеристик/правильного дозування лікарського засобу. | |||
| Б. II. а.3. Зміна у складі (допоміжних речовинах) готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) смакові добавки або барвники | |||
| 1. додавання, вилучення або заміна | 1, 2, 3, 4, 5, 6, 7, 9 | 1, 2, 4, 5, 6 | IА |
| 2. збільшення або зменшення | 1, 2, 3, 4 | 1, 2, 4 | IА |
| б) інші допоміжні речовини | II | ||
| 1. будь-яка незначна зміна кількісного складу допоміжних речовин у готовому лікарському засобі | 1, 2, 4, 8, 9, 10 | 1, 2, 7 | IА |
| 2. якісні або кількісні зміни щодо однієї або декількох допоміжних речовин, які можуть значно вплинути на безпеку, якість або ефективність готового лікарського засобу | II | ||
| 3. зміна у лікарському засобі біологічного/імунологічного походження | II | ||
| 4. будь-яка нова допоміжна речовина, що включає використання матеріалів людського або тваринного походження, для яких вимагається оцінка даних з вірусної безпеки або ризику передачі збудників ГЕ | II | ||
| 5. зміна, яка підтверджується дослідженнями з біоеквівалентності | II | ||
| 6. заміна однієї допоміжної речовини на іншу з тими самими функціональними характеристиками та на тому ж рівні | 1, 3, 4, 5, 6, 7, 8, 9 | IБ | |
| Умови | |||
| 1. Не відбулося будь-яких змін функціональних характеристик лікарської форми, наприклад часу розпадання, профілю розчинення.2. Будь-яке незначне коригування складу для збереження загальної маси повинно проводитись у відношенні допоміжної речовини, що становить основну частину складу готового лікарського засобу.3. Специфікації готового лікарського засобу змінилися тільки у частині зовнішнього вигляду/запаху/смаку і, за потреби, вилучений показник якості щодо ідентифікації.4. Розпочаті дослідження стабільності відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) (із зазначенням номерів серій) для принаймні двох дослідно-промислових або промислових серій із задовільними результатами стабільності протягом трьох місяців (на час подання змін типу IA або IБ), які демонструють подібність профілю стабільності до вже затвердженого. Заявник гарантує, що дослідження будуть завершені, і одразу по завершенні досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або за наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (з відповідною пропозицією). Крім того, за потреби, мають бути проведені дослідження фотостабільності готового лікарського засобу.5. Будь-які нові компоненти повинні відповідати встановленим вимогам.6. Будь-який новий компонент не повинен передбачати використання матеріалів людського або тваринного походження, для яких необхідна оцінка вірусної безпеки або ризику передачі збудників ГЕ.7. Зміна не впливає на відмінності між дозуваннями та не спричиняє негативного впливу на смакову прийнятність педіатричних форм (за потреби).8. Профіль розчинення принаймні двох дослідно-промислових серій запропонованого складу та у затвердженим складом лікарського засобу демонструє відсутність будь-яких відмінностей (див. Керівництво з дослідження біодоступності) Для рослинних лікарських засобів, коли випробування на розчинення не може бути проведене — дані щодо розпадання.9. Зміна не обумовлена проблемами зі стабільністю та/або не повинна спричиняти можливі проблеми, пов’язані з безпекою, тобто розходження між дозуваннями.10. Лікарський засіб не відноситься до препаратів біологічного/імунологічного походження. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє (включаючи метод ідентифікації будь-якого нового барвника, за потреби), а також оновлені коротка характеристика лікарського засобу, інструкція для медичного застосування та зразки упаковок (за необхідності).2. Підтвердження, що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) (із зазначенням номерів серій) та підтвердження, за необхідності, що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності, а також наявні дані не вказували на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами).3. Результати дослідження стабільності, розпочатими до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання), за відповідними параметрами принаймні для двох дослідно-промислових або промислових серій активної субстанції із задовільними показниками стабільності принаймні за три місяці. При цьому мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами).4. Зразок готового лікарського засобу з новим складом (за потреби).5. ГЕ Сертифікат відповідності Європейській Фармакопеї на будь-який новий вихідний матеріал тваринного походження або, якщо необхідно, документальне підтвердження того, що вихідне джерело одержання допоміжної речовини, яке має ризик передачі збудників ГЕ, попередньо оцінено вповноваженими органами країни-виробника та відповідає Керівництву з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами (чинне видання). Інформація повинна містити: назву виробника, види та тканини тварин, з яких було одержано вихідний матеріал, назву країни-постачальника тваринної сировини, її використання.6. Дані, які підтверджують, що нова допоміжна речовина не впливає на методи контролю готового лікарського засобу, визначені у специфікації (за потреби).7. Обґрунтування зміни/вибору допоміжних речовин тощо, що має бути представлено відповідно до фармацевтичної розробки (включаючи стабільність та антимікробні консерванти, за необхідності).8. Для твердих лікарських форм порівняльні дані щодо профілю розчинення принаймні для двох дослідно-промислових серій лікарського засобу з новим та затвердженим складом. Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.9. Обґрунтування відсутності необхідності проведення нового дослідження біоеквівалентності відповідно до Настанови або Керівництва з дослідження біодоступності та біоеквівалентності (чинне видання). | |||
| Б. II. a.4. Зміна маси покриття лікарських форм для перорального застосування або зміна маси оболонки капсул | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) тверді лікарські форми для перорального застосування | 1, 2, 3, 4 | 1, 2 | IА |
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим вивільненням або пролонгованої дії, для яких покриття є вирішальним чинником механізму вивільнення | II | ||
| Умови | |||
| 1. Відсутність змін у профілі розчинення принаймні двох дослідно-промислових серій лікарського засобу із запропонованим та затвердженим складом. Для рослинних лікарських засобів, коли випробування на розчинення не може бути проведене — дані щодо розпадання.2. Покриття не є вирішальним чинником механізму вивільнення.3. Специфікації готового лікарського засобу змінені тільки щодо маси і розміру (за необхідності).4. Дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання), для принаймні двох дослідно-промислових або промислових серій із задовільними результатами стабільності протягом трьох місяців або більше. Заявник гарантує, що дослідження будуть завершені, і одразу по завершенні досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або за наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (з відповідною пропозицією). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Підтвердження, що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання), (із зазначенням номерів серій), та підтвердження, за необхідності, що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами). Крім того, у відповідних випадках, мають бути проведені дослідження фотостабільності готового лікарського засобу. | |||
| Б. II. a.5. Зміна концентрації в окремій дозі багатодозового лікарського засобу для парентерального застосування, коли кількість діючої речовини на одиницю дози (тобто сила дії) не змінюється | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| II | |||
| Б. II. a.6. Вилучення контейнера з розчинником з упаковки | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | IБ | ||
| Документація | |||
| 1. Обґрунтування вилучення, включаючи заяву щодо альтернативних шляхів отримання розчинника, необхідного для безпечного та ефективного застосування лікарського засобу.2. Оновлені коротка характеристика лікарського засобу, інструкція для медичного застосування та зразки упаковок. | |||
| Б. II. б) Виробництво | |||
| Б. II. б.1. Заміна або введення додаткової дільниці виробництва для частини або всього виробничого процесу готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) дільниця для вторинного пакування | 1, 2 | 1, 3, 8 | IА |
| б) дільниця для первинного пакування | 1, 2, 3, 4, 5 | 1, 2, 3, 4, 8, 9 | IА |
| в) дільниця, на якій проводяться будь-які виробничі стадії, за виключенням випуску серій, проведення контролю якості та вторинного пакування, для лікарських засобів біологічного/імунологічного походження | II | ||
| г) дільниця, яка вимагає проведення первинної перевірки виробництва або перевірки виробництва конкретного готового лікарського засобу | II | ||
| ґ) дільниця, на якій проводяться будь-які виробничі стадії, за виключенням випуску серій, контролю якості, первинного та вторинного пакування, для нестерильних лікарських засобів | 1, 2, 3, 4, 5, 6, 7, 8, 9 | IБ | |
| д) дільниця, на якій проводяться будь-які виробничі стадії, за виключенням випуску серії, контролю якості та вторинного пакування для стерильних лікарських засобів, що вироблені з використанням асептичного методу, за виключенням лікарських засобів біологічного/імунологічного походження | 1, 2, 3, 4, 5, 6, 7, 8 | IБ | |
| Умови | |||
| 1. Задовільні результати перевірки виробництва за останні три роки, що проведена компетентними уповноваженими органами держав, які входять до PIC/s, Інспекцією ВООЗ або Інспекцією України з GMP.2. Дільниця має дозвіл (ліцензію) на виробництво відповідних лікарських форм або лікарського засобу.3. Даний лікарський засіб є нестерильним.4. У разі необхідності, наприклад, для суспензій та емульсій проведена схема валідації процесу або успішно виконана валідація виробництва на новій дільниці, відповідно до затвердженого протоколу принаймні на трьох промислових серіях.5. Даний лікарський засіб не є лікарським засобом біологічного/імунологічного походження. | |||
| Документація | |||
| 1. Підтвердження, що запропонована дільниця виробництва має відповідний дозвіл на виробництво лікарської форми або лікарського засобу, а саме:— для виробничої дільниці в Україні — копія чинної ліцензії на виробництво;— для виробничої дільниці поза межами України, якщо існує взаємна угода про підтвердження GMP — копія чинної ліцензії на виробництво, копія сертифіката GMP або документа, еквівалентного сертифікату GMP, виданого уповноваженим органом країни-виробника протягом останніх трьох років;— для виробничої дільниці поза межами України, якщо відсутня взаємна угода про підтвердження GMP — документ, виданий Інспекцією України з GMP, або копія чинної ліцензії на виробництво, копія сертифіката GMP або документа, еквівалентного сертифікату GMP, виданого уповноваженим органом країни-виробника протягом останніх трьох років.2. Інформація щодо кількості.3. Заява про внесення змін має чітко визначати «затвердженого» та «запропонованого» виробників готового лікарського засобу, як зазначено в розділі 2.5 форми заяви про державну реєстрацію.4. Копії затверджених специфікацій при випуску та наприкінці терміну придатності (за потреби).5. Порівняльні дані аналізів однієї промислової та двох дослідно-промислових серій (або двох промислових серій), вироблених на запропонованій дільниці та трьох серій, вироблених на затвердженій дільниці. Дані про наступні дві промислові серії мають бути надані на вимогу або, у разі невідповідності специфікаціям, представлені в Центр (з відповідною пропозицією).6. Для м’яких та рідких лікарських форм, у яких активна субстанція присутня у нерозчиненій формі, мають бути надані необхідні дані з валідації, мікроскопічне зображення розподілу та морфологія часток.7. Якщо нова виробнича дільниця використовує активну субстанцію як вихідний матеріал — заява уповноваженої особи (УО), яка відповідає за випуск серії на дільниці, що активна субстанція виробляється згідно з Керівництвами з належної виробничої практики щодо вихідних матеріалів (чинне видання).8. Зміни до відповідних розділів реєстраційного досьє.9. Якщо виробництво готового лікарського засобу та його первинне пакування здійснюється на різних виробничих дільницях, слід визначити умови транспортування та зберігання готового лікарського засобу у формі in bulk та провести дослідження з їх валідації. | |||
| Примітки. За відсутності сертифіката GMP, визнаного в Україні, власникам реєстраційного посвідчення рекомендується проконсультуватися з відповідними вповноваженими органами до подання заяви та надати інформацію про будь-яку інспекцію виробництва за останні 2-3 роки та/або будь-які заплановані інспекції, включаючи дати інспекцій, категорію лікарського засобу, що інспектується, наглядовий орган та іншу відповідну інформацію. Це полегшить організацію інспекції України з GMP, якщо необхідно.Заяви УО щодо активних субстанційВиробники повинні використовувати у якості вихідних матеріалів тільки ті активні субстанції, що виробляються відповідно до GMP, таким чином, кожний виробник, що використовує активну субстанції у якості вихідного матеріалу, повинен надати необхідну заяву. Крім того, оскільки УО, відповідальна за сертифікацію серії, несе сукупну відповідальність за кожну серію, у разі, коли контроль серії відбувається на інший дільниці, необхідна додаткова заява від УО, відповідальної за контроль.У багатьох випадках виробником виступає одна особа, тож необхідно надавати тільки одну заяву. Однак, якщо виробників більше одного, то заявник може надати тільки одну заяву, підписану одною УО, за умови, що:— із заяви зрозуміло, що вона складена від імені всіх задіяних УО;— є у наявності письмовий контракт між заявником та виробником, у якому чітко визначені обов’язки кожної зі сторін (Глава 7 Керівництва з GMP; розділ 5.7 Настанови 42-01-2001. Належна виробнича практика), а УО, що надає заяву, зазначена у контракті як особа, що несе відповідальність за дотримання вимог GMP при виробництві активної субстанції.Примітка. Заходи підлягають інспектуванню компетентними органами.Заявником підтверджується, що він користується послугами, принаймні однієї УО, яка знаходиться в ЄЕЗ. Тому заяви від персоналу, який найнятий виробниками у третіх країнах, включаючи тих, що знаходяться у країнах-партнерах за угодою про взаємне визнання, не можуть бути прийняті.Виробництво лікарського засобу — діяльність, пов’язана з серійним випуском лікарських засобів, яка включає всі або хоча б одну зі стадій технологічного процесу, у тому числі процеси фасування, пакування і маркування, контроль якості в процесі виробництва, контроль якості готової продукції.Заява не вимагається для крові або компонентів крові, що повинні відповідати вимогам Директиви 2002/98/ЄС. | |||
| Б. II. б.2. Зміни, що стосуються випуску серії та контролю якості готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) заміна або додавання дільниці, на який здійснюється контроль/випробування серії | 2, 3, 4 | 1, 2, 5 | IА |
| б) заміна або додавання виробника, відповідального за випуск серії | |||
| 1. не включаючи контроль/випробування серії | 1, 2 | 1, 2, 3, 4, 5 | IА |
| 2. включаючи контроль/випробування серії | 1, 2, 3, 4 | 1, 2, 3, 4, 5 | IА |
| 3. включаючи контроль/випробування серії для лікарського засобу біологічного/імунологічного походження та один з методів аналізу, що застосовується на дільниці, є біологічним/імунологічним/імунохімічним методом | II | ||
| Умови | |||
| 1. Місцезнаходження виробника, відповідального за випуск серії, повинно бути на території ЄЕЗ.2. Дільниця повинна мати відповідний дозвіл.3. Лікарський засіб не є лікарським засобом біологічного/імунологічного походження.4. Перенесення з затвердженої до нової дільниці або лабораторії має бути успішно виконано. | |||
| Документація | |||
| 1. — для виробничої дільниці в Україні — копія чинної ліцензії на виробництво;— для виробничої дільниці поза межами України, якщо існує взаємна угода про підтвердження GMP — копія чинної ліцензії на виробництво, копія сертифіката GMP або документа, еквівалентного сертифікату GMP, виданого уповноваженим органом країни-виробника протягом останніх трьох років. Якщо відсутня взаємна угода про підтвердження GMP — документ, виданий Інспекцією України з GMP, або копія чинної ліцензії на виробництво, копія сертифіката GMP або документа, еквівалентного сертифікату GMP, виданого уповноваженим органом країни-виробника протягом останніх трьох років.2. Заява про внесення змін має чітко визначати «затвердженого» та «запропонованого» виробників готового лікарського засобу, як зазначено в розділі 2.5 форми заяви про державну реєстрацію.3. Заява уповноваженої особи (УО), яка відповідає за контроль якості серії, що активна субстанція виробляється згідно з Керівництвами з належної виробничої практики щодо вихідних матеріалів. Узагальнена заява може бути прийнятною за певних умов (див. примітку до п. Б. II. б.1.).4. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлені: коротку характеристику лікарського засобу, інструкцію для медичного застосування та зразки упаковки (за необхідності). | |||
| Б. II. б.3. Зміни у процесі виробництва готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначна зміна у процесі виробництва твердої лікарської форми для перорального застосування з негайним вивільненням або розчинів для перорального застосування | 1, 2, 3, 4, 5, 6, 7 | 1, 3, 4, 6, 7, 8 | ІА |
| б) зміна у процесі виробництва, яка може мати істотний влив на якість, безпеку та ефективність лікарського засобу | II | ||
| в) лікарський засіб є лікарським засобом біологічного/імунологічного походження та зміна вимагає проведення порівняльних досліджень | ІІ | ||
| г) ведення нестандартного методу кінцевої стерилізації | ІІ | ||
| ґ) введення або збільшення припустимого надлишку діючої речовини | ІІ | ||
| д) незначна зміна у процесі виробництва водної суспензії для перорального застосування | 1, 2, 4, 6, 7, 8 | IБ | |
| Умови | |||
| 1. Немає жодних змін у якісних або кількісних показниках профілю домішок або у фізико-хімічних властивостях.2. Даний лікарських засіб не є лікарським засобом біологічного/імунологічного або рослинного походження.3. Виробничий процес, який включає окремі стадії, залишається таким самим, наприклад обробка проміжних продуктів, і немає жодних змін у будь-якому розчиннику, що використовується у процесі виробництва.4. Затверджений виробничий процес має контролюватися відповідним методами і ці методи не потребують жодних змін (розширення або вилучення допустимих меж).5. Специфікації на готовий лікарський засіб або проміжні продукти залишаються незмінними.6. Новий виробничий процес повинен забезпечити виготовлення аналогічного попередньому лікарського засобу за усіма показниками якості, безпеки та ефективності.7. Розпочаті дослідження стабільності відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) для принаймні однієї дослідно-промислової або промислової серії із задовільними результатами стабільності протягом трьох місяців або більше. Заявник гарантує, що дослідження будуть завершені, і одразу по завершенні досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або за наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (з відповідною пропозицією). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи порівняння затвердженого та запропонованого виробничого процесу.2. Для м’яких та рідких лікарських форм, у яких діюча речовина міститься у нерозчинній формі — відповідні дані з валідації змін, включаючи мікроскопію часток для перевірки видимих змін у морфології; порівняльні дані гранулометричного складу за відповідним методом.3. Для твердих лікарських форм — дані, що підтверджують відсутність змін у профілі розчинення однієї промислової серії, виробленої за зміненою технологією порівняно з останніми трьома серіями, виробленими за узгодженою технологією. Дані про наступні дві повні промислові серії мають бути надані на вимогу, або у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією). Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.4. Обґрунтування відсутності необхідності проведення досліджень біоеквівалентності відповідно до Настанови або Керівництв з дослідження біодоступності (чинне видання).5. У разі зміни у процесі стерилізації, необхідно надати дані з валідації.6. Копія затверджених специфікацій при випуску та наприкінці терміну придатності.7. Дані аналізів (у вигляді порівняльної таблиці) принаймні однієї промислової серії, виробленої за узгодженою та запропонованою технологіями. Дані про наступні дві повні промислові серії мають бути надані на вимогу, або у разі невідповідності специфікаціям буде повідомлено Центр (з відповідною пропозицією). | |||
| 8. Підтвердження, що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) (із зазначенням номерів серій) для принаймні однієї дослідно-промислової або промислової серії із задовільними результатами стабільності протягом трьох місяців або більше та профілем стабільності, подібним до профілю стабільності лікарського засобу, виробленого за узгодженою технологією. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами). | |||
| Б. II. б.4. Зміна розміру серії (включаючи діапазон розміру серії) готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) збільшення до 10 разів порівняно із затвердженим розміром | 1, 2, 3, 4, 5, 7 | 1, 4 | IА |
| б) зменшення до 10 разів | 1, 2, 3, 4, 5, 6 | 1, 4 | IА |
| в) зміна вимагає оцінки порівнянності (проведення порівняльних досліджень) лікарського засобу біологічного/імунологічного походження | ІІ | ||
| г) зміна стосується всіх інших лікарських форм сукупного (комплексного) виробничого процесу | ІІ | ||
| ґ) збільшення більш ніж у 10 разів порівняно із затвердженим розміром для твердих лікарських форм з негайним вивільненням | 1, 2, 3, 4, 5, 6 | ІБ | |
| д) масштаб для лікарського засобу біологічного/імунологічного походження збільшився/зменшився без зміни виробничого процесу (наприклад, дублювання лінії) | 1, 2, 3, 4, 5, 6 | ІБ | |
| Умови | |||
| 1. Зміна не впливає на відтворюваність та/або постійність лікарського засобу.2. Зміна стосується тільки твердих лікарських форм з негайним вивільненням для перорального застосування або нестерильних рідких лікарських форм.3. Будь-які зміни методу виробництва та/або контролю у процесі виробництва спричинені зміною розміру серії, наприклад, використанням обладнання іншої продуктивності.4. Існує схема валідації або валідація виробництва була успішно проведена згідно із затвердженим протоколом з використанням принаймні трьох серій лікарського засобу нового розміру відповідно до Настанови або Керівництв з валідації виробництва (чинне видання).5. Даний лікарський засіб не є лікарським засобом біологічного/імунологічного походження.6. Зміна не обумовлена непередбаченими обставинами у процесі виробництва або пересторогами щодо стабільності.7. Попередній розмір серії не був затверджений під час внесення зміни типу ІА. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Дані аналізів (у вигляді порівняльної таблиці) принаймні однієї промислової серії затвердженого та запропонованого розмірів. Дані про наступні дві повні промислові серії мають бути надані на вимогу або у разі невідповідності специфікаціям, буде повідомлено Центр (з відповідною пропозицією).3. Копії затверджених специфікацій при випуску та наприкінці терміну придатності.4. Інформація щодо кількості.5. Дані з валідації.6. Підтвердження, що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) (із зазначенням кількості та номерів серій) для принаймні однієї дослідно-промислової або промислової серії із задовільними результатами стабільності протягом трьох місяців або більше. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами). Для лікарських засобів біологічного/імунологічного походження — підтвердження відсутності необхідності проведення порівняльних досліджень. | |||
| Б. II. б.5. Зміни випробувань або допустимих меж, встановлених у специфікаціях, під час виробництва готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення нового методу випробування та допустимих меж | 1, 2, 5, 6 | 1, 2, 3, 4, 5, 7 | IА |
| в) вилучення несуттєвого випробування | 1, 2 | 1, 2, 6 | IА |
| г) вилучення випробування у процесі виробництва, яке може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | ||
| ґ) розширення затверджених допустимих меж для показників, які можуть мати істотний вплив на загальну якість готового лікарського засобу | ІІ | ||
| д) доповнення або заміна випробування за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 5, 7 | ІБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду допустимих меж специфікацій (наприклад, проведеною під час подання заяви на реєстрацію або внесення змін типу II).2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва, наприклад утворенням нової некваліфікованої домішки, зміною допустимих меж загального вмісту домішок.3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.4. Метод випробування залишається незмінним або такі зміни є незначними.5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.6. Новий метод випробування не належить до біологічного/імунологічного/імунохімічного методу або методу, у якому використовується біологічний реактив для активної субстанції біологічного походження (за виключенням стандартних фармакопейних мікробіологічних методів). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця щодо затвердженого та запропонованого випробування та допустимих меж.3. Опис будь-якого нового методу випробування та дані з його валідації (за необхідності).4. Дані аналізів для двох промислових серій (трьох промислових серій для лікарських засобів біологічного походження, якщо не обґрунтовано інше) готового лікарського засобу за всіма показниками специфікацій.5. Порівняльні дані щодо профілю розчинення для готового лікарського засобу принаймні однієї дослідної серії, виготовленої за затверджених та запропонованих умов (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.6. Обґрунтування/оцінка ризику, що підтверджує незначність зміненого параметру.7. Обґрунтування введення нового випробування у процесі виробництва та допустимих меж. | |||
| Б. II. в) Контроль допоміжних речовин | |||
| Б. II. в.1. Зміна параметрів специфікацій та/або допустимих меж для допоміжної речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | ІА |
| б) доповнення специфікації новим показником з відповідним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 6, 8 | ІА |
| в) вилучення із специфікації незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 7 | ІА |
| г) зміна знаходиться поза затвердженими допустимими межами специфікацій | ІІ | ||
| ґ) вилучення із специфікації показника, який може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | ||
| д) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за виключенням лікарських засобів біологічного та імунологічного походження) | 1, 2, 3, 4, 5, 6, 8 | ІБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду допустимих меж специфікацій (наприклад, проведеною під час подання заяви на реєстрацію або внесення змін типу II).2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва, наприклад утворення нової некваліфікованої домішки; зміна допустимих меж загального вмісту домішок.3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.4. Метод випробування залишається незмінним або такі зміни є незначними.5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.6. Новий метод випробування не належить до біологічного/імунологічного/імунохімічного методу або методу, у якому використовується біологічний реактив для активної субстанції біологічного походження (за виключенням стандартних фармакопейних мікробіологічних методів).7. Зміни не стосуються домішки, яка має генотоксичну дію. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця щодо затвердженої та запропонованої специфікацій.3. Опис нового методу випробування та дані з його валідації (за необхідності).4. Дані аналізів для двох промислових серій (трьох промислових серій для лікарських засобів біологічного походження, якщо не обґрунтовано інше) готового лікарського засобу за всіма показниками специфікацій.5. Порівняльні дані профілю розчинення для готового лікарського засобу принаймні однієї дослідної серії з допоміжними речовинами, які відповідають затвердженій та запропонованій специфікаціям (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.6. Обґрунтування відсутності нових даних з біоеквіваленотності у відповідності до Настанови або Керівництв з дослідження біодоступності (чинне видання) (за необхідності).7. Обґрунтування/оцінка ризику, що підтверджує незначність зміненого параметру.8. Обґрунтування введення нового показника специфікації та допустимих меж. | |||
| Б. II. в.2. Зміна у методах випробування допоміжної речовини | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробувань | 1, 2, 3, 4 | 1, 2 | IА |
| б) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | 5 | 1 | ІА |
| в) заміна біологічного/імунологічного/імунохімічного методу випробування або методу, у якому використовується біологічний реагент | ІІ | ||
| г) інші зміни у методах випробування (включаючи заміну або додавання) | 1, 2 | IБ | |
| Умови | |||
| Б. II. в.3. Заміна джерела одержання допоміжної речовини або реактиву, що становить ризик передачі збудників ГЕ | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) матеріалу, що становить ризик передачі збудників ГЕ, на матеріал рослинного або синтетичного походження | |||
| 1. для допоміжних речовин або реактивів, які не використовуються у виробництві активної субстанції біологічного/імунологічного походження, або лікарського засобу біологічного/імунологічного походження | 1 | 1 | IА |
| 2. для допоміжних речовин або реактивів, які використовуються у виробництві активної субстанції біологічного/імунологічного походження, або лікарського засобу біологічного/імунологічного походження | 1, 2 | IБ | |
| б) заміна або додавання речовини, що становить ризик передачі збудників ГЕ, або заміна речовини, що становить ризик передачі збудників ГЕ, на іншу речовину, що становить ризик передачі збудників ГЕ, для якої немає ГЕ сертифіката відповідності Європейській фармакопеї | II | ||
| Умови | |||
| 1. Специфікації при випуску та наприкінці терміну придатності на допоміжну речовину та готовий лікарський засіб залишаються незмінними. | |||
| Документація | |||
| 1. Заява від виробника або заявника, яка підтверджує, що допоміжна речовина є речовиною виключно рослинного або синтетичного походження.2. Результати дослідження еквівалентності матеріалів, їх впливу на виробництво кінцевої речовини та впливу на характеристики (наприклад, параметри розчинення) готового лікарського засобу. | |||
| Б. II. в.4. Зміни в методі синтезу або регенерації нефармакопейної допоміжної речовини (за наявності в досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у методі синтезу або регенерації | 1, 2 | 1, 2, 3, 4 | IА |
| б) зміни у специфікації або зміна фізико-хімічних властивостей допоміжної речовини, що може мати вплив на якість готового лікарського засобу | ІІ | ||
| в) допоміжна речовина є речовиною біологічного/імунологічного походження | ІІ | ||
| Умови | |||
| 1. Метод синтезу та специфікації залишаються незмінними, відсутні зміни у якісних і кількісних показниках профілю домішок (за виключенням залишкових розчинників, за умови, що вони відповідають Керівним принципам ІСН) або фізико-хімічних властивостях.2. За виключенням ад’ювантів для вакцин. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Дані аналізів (у формі порівняльної таблиці) принаймні двох серій (мінімум дослідно-промислових) з використанням допоміжної речовини, виготовленої за узгодженим та новим методами виробництва.3. Порівняльні данні щодо профілю розчинення для готового лікарського засобу принаймні двох серій (мінімум дослідно-промислових) з використанням допоміжної речовини, виготовленої за узгодженим та новим методами виробництва (за необхідності). Для рослинних лікарських засобів можуть бути прийнятними дані щодо розпадання.4. Копії затверджених та нових (за необхідності) специфікацій на допоміжну речовину. | |||
| Б. II. г) Контроль готового лікарського засобу | |||
| Б. II. г.1. Зміна параметрів специфікацій та/або допустимих меж готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) звуження допустимих меж для лікарського засобу, що підлягає офіційному випуску серії | 1, 2, 3, 4 | 1, 2 | IА |
| в) доповнення специфікації новим показником з відповідним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 5, 7 | IА |
| г) вилучення незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 6 | IА |
| ґ) зміна знаходиться поза затвердженими допустимими межами | ІІ | ||
| д) вилучення показника, який може мати істотний вплив на якість готового лікарського засобу | ІІ | ||
| е) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за виключенням лікарських засобів біологічного/імунологічного походження) | 1, 2, 3, 4, 5, 7 | ІБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду допустимих меж специфікацій (наприклад, проведеною під час подання заяви на реєстрацію або внесення змін типу II).2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва, наприклад, утворенням нової некваліфікованої домішки; зміною допустимих меж загального вмісту домішок.3. Будь-яка зміна не повинна виходити за допустимі межі затвердженої специфікації.4. Метод випробування залишається незмінним або такі зміни є незначними.5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.6. Новий метод випробування не належить до біологічного/імунологічного/імунохімічного методу або методу, у якому використовується біологічний реактив для активної субстанції біологічного походження (за виключенням стандартних фармакопейних мікробіологічних методів).7. Зміни не стосуються домішки, яка має генотоксичну дію. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця щодо затверджених та запропонованих специфікацій.3. Опис нового методу випробування та дані з його валідації (за необхідності).4. Дані аналізів для двох промислових серій (трьох промислових серій для лікарських засобів біологічного походження, якщо не обґрунтовано інше) готового лікарського засобу за всіма показниками специфікацій.5. Порівняльні дані профілю розчинення для готового лікарського засобу принаймні однієї дослідної серії, які відповідають затвердженій та запропонованій специфікаціям (за необхідності). Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання.6. Обґрунтування/оцінка ризику, що підтверджує незначність зміненого параметру.7. Обґрунтування введення нового показника та допустимих меж. | |||
| Б. II. г.2. Зміна у методах випробування готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначна зміна у затверджених методах випробування | 1, 2, 3, 4 | 1, 2 | IА |
| б) вилучення методу випробування, якщо вже затверджений альтернативний метод | 4 | 1 | IА |
| в) зміна у біологічному/імунологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент | ІІ | ||
| г) інші зміни у методах випробувань (включаючи заміну або доповнення) | 1, 2 | IБ | |
| Умови | |||
| 1. Дослідження з валідації, які були проведені відповідно до чинних фармакопейних вимог або Керівних принципів з валідації (чинне видання), підтверджують, що нова методика ідентична затвердженій.2. Не було жодних змін меж загального вмісту домішок, не виявлено нових некваліфікованих домішок.3. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу але тип колонки або методика не змінні).4. Новий метод випробувань не належить до біологічних/імунобіологічних/імунохімічних методів або методу, при якому використовується біологічний реагент для аналізу активної субстанції біологічного походження (за включенням стандартних фармакопейних мікробіологічних методів). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи опис методу аналізу, звіт про дані з валідації, оновлені специфікації щодо домішок (якщо необхідно).2. Порівняльні дані валідації або, якщо обґрунтовано, порівняльні результати аналізу, які підтверджують, що затверджений та запропонований методи випробування є ідентичними. Ця вимога не стосується випадку додавання нового методу випробування. | |||
| Б. II. г.3. Зміни, які стосуються виробничого процесу у реальному часі або випуску за параметрами для готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| ІІ | |||
| Документація | |||
| Б. II.ґ) Система упаковка/укупорка | |||
| Б. II.ґ.1. Зміна у первинній упаковці готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| а) якісний та кількісний склад | |||
| 1. тверді лікарські форми | 1, 2, 3 | 1, 2, 3, 4, 6 | IА |
| 2. м’які та нестерильні рідкі лікарські форми | 1, 2, 3, 5, 6 | IБ | |
| 3. стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | ||
| 4. зміна стосується зниження ступеню захисту оновленої упаковки, якщо наявні відповідні зміни в умовах зберігання та/або скорочення терміну придатності | ІІ | ||
| б) тип контейнера | |||
| 1. тверді, м’які та нестерильні рідкі лікарські форми | 1, 2, 3, 5, 6, 7 | ІБ | |
| 2. стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | ||
| Умови | |||
| 1. Зміна стосується лише одного типу упаковки/контейнеру (наприклад, заміна блістера на блістер).2. Запропонований пакувальний матеріал повинен бути ідентичним затвердженому за відповідними властивостями.3. Розпочаті дослідження стабільності відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) для принаймні двох дослідно-промислових або промислових серій із задовільними результатами стабільності протягом трьох місяців або більше. Однак, якщо запропонований пакувальний матеріал більш стійкий, ніж затверджений (наприклад, матеріал блістера має більшу товщину) дані щодо стабільності за три місяці ще не доступні. Мають бути надані гарантії того, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлені коротку характеристику лікарського засобу, зразки упаковки та інструкцію для медичного застосування (за необхідності).2. Відповідні дані щодо нової упаковки (порівняльні дані про проникність, наприклад для O2, CO2, вологи).3. Підтвердження відсутності (за необхідності) будь-якої взаємодії між лікарським засобом та пакувальним матеріалом (наприклад, не було перенесення компонентів запропонованого пакувального матеріалу до лікарського засобу та навпаки), включаючи підтвердження того, що пакувальний матеріал відповідає чинним фармакопейним вимогам щодо пакувальних матеріалів або законодавству ЄС щодо безпеки взаємодії пластикових пакувальних матеріалів та упаковок з харчовими продуктами.4. Заява, що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) (із зазначенням номерів серій) та (за необхідності), що на момент внесення змін заявником було надано мінімум, який вимагається, даних щодо стабільності, а також наявні дані не вказували на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами).5. Результати дослідження стабільності, які були розпочаті згідно з Настановою або Керівними принципами щодо дослідження стабільності лікарських засобів (чинне видання), за відповідними параметрами, принаймні, для двох дослідно-промислових або промислових серій лікарського засобу із задовільними показниками щодо стабільності принаймні за три місяці. При цьому мають бути надані гарантії того, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами).6. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій (за необхідності).7. Зразки нової системи упаковка/укупорка (за необхідності). | |||
| Примітка до пункту Б. II.ґ.1. б. Будь-яка зміна, що призводить до нової лікарської форми, потребує подання заяви на реєстрацію. | |||
| Б. II.ґ.2. Зміна параметрів специфікацій та/або допустимих меж первинної упаковки готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) звуження допустимих меж, визначених у специфікації | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфікації новим показником з відповідним методом випробування | 1, 2, 5 | 1, 2, 3, 4, 6 | IА |
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | 1, 2 | 1, 2, 5 | ІА |
| г) додавання або заміна показника за результатами досліджень з безпеки або якості | 1, 2, 3, 4, 6 | ІБ | |
| Умови | |||
| 1. Зміна не обумовлена попередньою експертизою, що здійснювалась для перегляду параметрів специфікації (наприклад, проведеною під час подання заяви не реєстрацію або внесення змін типу II).2. Зміна не обумовлена непередбаченими обставинами у процесі виробництва.3. Будь-яка зміна на повинна виходити за допустимі межі затвердженої специфікації.4. Методи випробування залишаються незмінними або такі зміни є незначними.5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця вимог затвердженої та запропонованої специфікацій.3. Опис нового методу випробування та дані з його валідації (за необхідності).4. Результати аналізу для двох серій первинної упаковки за усіма показниками, зазначеними у специфікації.5. Обґрунтування того, що показник є незначним.6. Обґрунтування нового показника якості та допустимих меж. | |||
| Б. II.ґ.3. Зміна у методах випробування первинної упаковки готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) незначні зміни у затверджених методах випробувань | 1, 2, 3 | 1, 2 | IА |
| б) інші зміни у методах випробувань (включаючи заміну або додавання) | 1, 3, 4 | 1, 2 | IА |
| в) вилучення методу випробування, якщо вже затверджений альтернативний | 5 | 1 | ІА |
| Умови | |||
| 1. Дослідження з валідації, які були проведені відповідно до фармакопейних вимог або Керівництва з валідації, підтверджують, що нова методика ідентична затвердженій.2. Методи випробувань залишилися незмінними (наприклад, змінилась довжина колонки або температура проведення аналізу але тип колонки або методика не змінні).3. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосіб.4. Активна субстанція/готовий лікарський засіб не є речовиною/лікарським засобом біологічного/імунологічного походження.5. Існує затверджений метод випробування для показника специфікації і цей метод не затверджений при внесенні зміни типу ІА. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи опис методу випробування та звіт з його валідації.2. Порівняльні дані валідації або, якщо обумовлено, порівняльні результати аналізу, які підтверджують ідентичність результатів, отриманих за затвердженим та новим методами випробувань. Це не стосується випадків додавання нового методу випробування. | |||
| Б. II.ґ.4. Зміна форми або розміру контейнера чи закупорювального засобу (первинної упаковки) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) нестерильні лікарські засоби | 1, 2, 3 | 1, 2, 4 | IА |
| б) зміна форми або розміру основної частини пакувального матеріалу (вторинної упаковки), що може мати значний вплив на доставку, застосування, безпеку та стабільність готового лікарського засобу | IІ | ||
| в) стерильні лікарські засоби | 1, 2, 3, 4 | ІБ | |
| Умови | |||
| 1. Відсутні якісні або кількісні зміни складу пакувального матеріалу.2. Зміна не стосується основної частини пакувального матеріалу, яка впливає на доставку, застосування, безпеку та стабільність готового лікарського засобу.3. У разі зміни вільного простору над лікарським засобом або зміни у співвідношенні поверхня/об’єм, дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) принаймні для двох дослідно-промислових (трьох для лікарських засобів біологічного/імунологічного походження) або промислових серій лікарського засобу із задовільними показниками щодо стабільності принаймні за три місяці (шість місяців для лікарських засобів біологічного/імунологічного походження). При цьому мають бути надані гарантії того, що дослідження будуть завершені, і одразу по завершені досліджень отримані дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи опис, детальне зображення та склад контейнеру або закупорювального матеріалу та (за необхідності), оновлені: коротку характеристику лікарського засобу та інструкцію для медичного застосування.2. Зразок нового пакування (за необхідності).3. Дослідження з ревалідації проводилися для стерильних лікарських засобів, які пройшли кінцеву стерилізацію. Слід зазначити номери серій лікарського засобу, що використовувались в дослідженнях (за необхідності).4. У разі зміни вільного простору над лікарським засобом або зміни співвідношення поверхня/об’єм має бути надане підтвердження того, що дослідження стабільності розпочаті відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) (із зазначенням номерів серій лікарського засобу) та (за необхідності), що на момент подання заяви про внесення змін типу ІА або ІБ у розпорядженні заявника було мінімум, який вимагається, даних щодо стабільності, а також наявні дані не вказували на будь-яку проблему, пов’язану зі стабільністю. Мають бути надані гарантії, що дослідження будуть завершені, і одразу по завершені досліджень дані будуть представлені в Центр у разі невідповідності специфікаціям або наявної ймовірності відхилень від специфікацій наприкінці терміну придатності (із запропонованими заходами). | |||
| Б. II.ґ.5. Зміна розміру упаковки готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна кількості одиниць (наприклад, таблеток, ампул та ін.) в упаковці: | |||
| 1. зміна у діапазоні затверджених розмірів упаковки | 1, 2 | 1, 3 | IА |
| 2. зміна поза діапазоном затверджених розмірів упаковки | 1, 2, 3 | IБ | |
| б) вилучення упаковки певного розміру | 3 | 1, 2 | ІА |
| в) зміна маси/об’єму вмісту контейнера багатодозового стерильного лікарського засобу (або однодозового часткового використання) та багатодозового лікарського засобу біологічного/імунологічного походження для парентерального застосування | IІ | ||
| г) зміна маси/об’єму вмісту контейнера багатодозового лікарського засобу для непарентеральних застосування (або однодозового часткового використання) | 1, 2, 3 | ІБ | |
| Умови | |||
| 1. Новий розмір упаковки повинен відповідати дозуванню і тривалості лікування відповідно до затвердженої короткої характеристики лікарського засобу.2. Первинний пакувальний матеріал не змінився.3. Незмінені форми випуску лікарського засобу повинні відповідати інструкції з дозування та тривалості лікування, як вказано в короткій характеристиці лікарського засобу. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлені: коротку характеристику лікарського засобу (за необхідності).2. Обґрунтування нового/незміненого розміру упаковки та підтвердження, що новий/незмінений розмір упаковки відповідає схемі дозування та тривалості лікування, затвердженими у короткій характеристиці лікарського засобу.3. Заява, що дослідження стабільності будуть проводитись відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання), якщо показники стабільності будуть змінені. Дані мають бути представлені в Центр лише у разі невідповідності специфікаціям (з відповідною пропозицією). | |||
| Примітка до пункту Б. II.ґ.5. підпунктів в) та г). Будь-які зміни у силі дії лікарського засобу потребують подання заяви на реєстрацію. | |||
| Б. II.ґ.6. Зміна будь-якої частини матеріалу первинної упаковки, що не контактує з готовим лікарським засобом (наприклад, колір кришечок з контролем першого відкриття, колір кодових кілець на ампулах, контейнера для голок (різні види пластмаси) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна, яка впливає на коротку характеристику лікарського засобу | 1 | 1 | IА |
| б) зміна, яка не впливає на коротку характеристику лікарського засобу | 1 | 1 | IА |
| Умови | |||
| 1. Зміна не стосується тієї частини пакувального матеріалу, яка б могла вплинути на доставку, застосування, безпеку та стабільність готового лікарського засобу. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи оновлену коротку характеристику лікарського засобу (за необхідності). | |||
| Б. II.ґ.7. Зміна постачальника пакувальних матеріалів або комплектуючих (якщо зазначено в досьє) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) вилучення постачальника | 1 | 1 | IА |
| б) заміна або додавання постачальника | 1, 2, 3, 4 | 1, 2, 3 | IА |
| в) будь-яка зміна постачальника спейсерів для дозованих інгаляторів | ІІ | ||
| Умови | |||
| 1. Жодних вилучень у компонентах упаковки або комплектуючих не відбулося.2. Кількісний та якісний склад пакувального матеріалу/комплектуючих та проектні специфікації не змінилися.3. Специфікації та методи контролю якості принаймні ідентичні.4. Метод та умови стерилізації залишаються незмінними (за необхідності). | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Для комплектуючих для лікарських засобів — підтвердження CE-маркування та висновок, виданий МОЗ України, про безпеку комплектуючих.3. Порівняльна таблиця затвердженої та запропонованої специфікацій, якщо необхідно. | |||
| Примітка. СЕ-маркування (CE-marking) — обов’язкове маркування окремих груп продуктів для підтвердження відповідності вимогам Європейських директив щодо їх безпеки для здоров’я людини. Маркування «СЕ» — скорочення від Conformit Europeane (Європейська відповідність). До того як СЕ-маркування може бути застосоване до препарату, мають бути виконані встановлені Європейськими директивами вимоги. Крім того, відповідність препарату має бути підтверджена під час реєстрації та/або проведення випробувань. | |||
| Б. II. д) Стабільність | |||
| Б. II. д.1. Зміна у термінах придатності або умовах зберігання готового лікарського засобу: | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зменшення терміну придатності готового лікарського засобу: | |||
| 1. для торгової упаковки | 1 | 1, 2, 3 | IА |
| 2. після першого розкриття | 1 | 1, 2, 3 | IА |
| 3. після розчинення або відновлення | 1 | 1, 2, 3 | IА |
| б) збільшення терміну придатності готового лікарського засобу: | |||
| 1. для торгової упаковки (підтверджується даними реального часу) | 1, 2, 3 | IБ | |
| 2. після першого розкриття (підтверджується даними реального часу) | 1, 2, 3 | IБ | |
| 3. після розчинення або відновлення (підтверджується даними реального часу) | 1, 2, 3 | IБ | |
| 4. збільшення терміну придатності на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності з вимогами Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (*) | ІІ | ||
| 5. збільшення терміну придатності лікарського засобу біологічного/імунологічного походження на основі результатів досліджень стабільності, проведеними відповідно до затвердженого протоколу | 1, 2, 3 | IБ | |
| в) зміна в умовах зберігання лікарського засобу біологічного походження, якщо дослідження стабільності були проведені не у відповідності до затвердженого протоколу | ІІ | ||
| г) зміна в умовах зберігання готового лікарського засобу або після розчинення/відновлення | 1, 2, 3 | IБ | |
| Умови | |||
| 1. Зміна не обумовлена непередбаченими обставинами у процесі виробництва або пересторогами щодо стабільності. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє повинні містити результати досліджень стабільності у реальному часі (що включають повний термін придатності), проведених відповідно до Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (чинне видання) принаймні на двох дослідно-промислових серіях1 готового лікарського засобу у затвердженій упаковці та/або після першого розкриття, або відновлення (за необхідності). За потреби мають бути включені результати мікробіологічних досліджень.2. Оновлені: коротка характеристика лікарського засобу, інструкція для медичного застосування та зразки упаковки.3. Копія затвердженої специфікації наприкінці терміну придатності готового лікарського засобу та, якщо необхідно, специфікації на лікарський засіб після розчинення/відновлення або першого розкриття.____________________________________1дослідно-промислова серія може бути прийнята із зобов’язанням підтвердити термін придатності на промисловій серії. | |||
| (*) Екстраполяція не застосовується до лікарських засобів біологічного/імунологічного походження. | |||
| Б. II. е) Проектний простір | |||
| Б. II. е.1. Введення нового проектного простору або розширення затвердженого для готового лікарського засобу (за виключенням лікарських засобів біологічного походження) щодо | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) одного або більше елементів (блок, частина) виробничого процесу готового лікарського засобу, включаючи контроль в процесі виробництва та/або методи випробувань | 1, 2, 3 | II | |
| б) метоів випробувань для допоміжних речовин/проміжних продуктів та/або готового лікарського засобу | 1, 2, 3 | II | |
| Документація | |||
| 1. Результати досліджень розробки лікарського засобу або виробничого процесу (включаючи дослідження оцінки ризику та багатомірні дослідження, якщо необхідно), які демонструють, що функціональна взаємодія властивостей матеріалу та параметрів процесу, які можуть впливати на критичні характеристики якості готового лікарського засобу, була досягнута.2. Опис проектного простору у формі таблиці, включаючи змінні (властивості матеріалу та параметри процесу, якщо необхідно) та їх запропоновані діапазони.3. Зміни до відповідних розділів реєстраційного досьє. | |||
| Б. II. е.2. Внесення змін після затвердження протоколу для готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| 1, 2 | II | ||
| Документація1. Детальний опис запропонованої зміни.2. Протокол управління змінами для готового лікарського засобу. | |||
| Б. II. е.3. Вилучення затвердженого протоколу управління змінами для готового лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип змін |
| 1 | 1 | IА | |
| Умови | |||
| 1. Вилучення не спричинено непередбаченими обставинами або результатами випробувань, що виходять за межі специфікацій під час внесення зміни, описаної у протоколі. | |||
| Документація | |||
| 1. Обґрунтування запропонованого вилучення. | |||
| Б.ІІІ. Сертифікат відповідності/ГЕ-сертифікат відповідності Європейській Фармакопеї | |||
| Б.ІІІ.1. Подання нового або оновленого сертифіката відповідності Європейській Фармакопеї | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| Для активної субстанціїДля вихідного матеріалу/реагенту/проміжного продукту, що використовуються у виробництві активної субстанціїДля допоміжної речовини | |||
| а) сертифікат відповідності Європейській Фармакопеї | |||
| 1. новий сертифікат від вже затвердженого виробника | 1, 2, 3, 4, 5, 8 | 1, 2, 3, 4, 5 | IА |
| 2. оновлений сертифікат від вже затвердженого виробника | 1, 2, 3, 4, 8 | 1, 2, 3, 4, 5 | IА |
| 3. новий сертифікат від нового виробника (заміна або доповнення) | 1, 2, 3, 4, 5, 8 | 1, 2, 3, 4, 5 | IА |
| б) ГЕ Сертифіката відповідності Європейській Фармакопеї для активної субстанції/вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини | |||
| 1. новий сертифікат для активної субстанції від нового або вже затвердженого виробника | 3, 6 | 1, 2, 3, 4, 5 | IА |
| 2. новий сертифікат для вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини від нового або вже затвердженого виробника | 3, 6 | 1, 2, 3, 4, 5 | IА |
| 3. оновлений сертифікат від вже затвердженого виробника | 1, 2, 3, 4, 5 | IА | |
| Умови | |||
| 1. Специфікації при випуску та наприкінці терміну придатності готового лікарського засобу залишаються незмінними.2. Додаткові (до Європейської фармакопеї) специфікації щодо домішок (за виключенням залишкових розчинників, за умови, що вони відповідають Керівним принципам ІСН) та відповідні вимоги до лікарського засобу (наприклад, профілі розміру часток, поліморфна форма) залишаються незмінними, за виключенням звуження (за необхідності).3. Виробничий процес активної субстанції, вихідного матеріалу/реагенту/проміжного продукту не включає використання матеріалу людського або тваринного походження, для якого проводиться оцінка даних щодо вірусної безпеки.4. Тільки для активної субстанції необхідно проводити дослідження безпосередньо перед використанням, якщо період повторного випробування не включений до сертифікату відповідності Європейській Фармакопеї, або якщо дані щодо періодичності повторного випробування ще не представлені в досьє.5. Активна субстанція/вихідний матеріал/реагент/проміжний продукт/допоміжна речовина нестерильні.6. Для рослинних активних субстанцій: шлях виробництва, фізична форма, екстрагент та співвідношення лікарський засіб/екстрагент повинні залишатися незмінними. | |||
| Документація | |||
| 1. Копія затвердженого (оновленого) сертифікату відповідності Європейській Фармакопеї.2. У випадку додавання виробничої дільниці у заяві мають бути чітко визначені «затверджений» та «запропонований» виробники, як зазначено в розділі 2.5 форми заяви для державної реєстрації лікарського засобу.3. Зміни до відповідних розділів реєстраційного досьє.4. Якщо необхідно, документ в якому представлена інформація про будь-які матеріали, що підлягають оцінці вірусної безпеки відповідно до Керівництва з мінімізації ризику передачі збудників губчатої енцефалопатії тварин з лікарськими засобами, включаючи ті, що використовуються при виробництві активної субстанції/допоміжної речовини. Інформація повинна містити таке: назва виробника, види та тканини тварин, з яких було отримано матеріал, країна-постачальник тваринної сировини, її використання та попередній дозвіл.5. Для активної субстанції — заява від уповноваженої особи (УО) кожного з власників ліцензії на виробництво, які перелічені у заяві на внесення змін, якщо активна субстанція використовується як вихідний матеріал, та заява УО кожного з власників ліцензії на виробництво, які перелічені у заяві як відповідальні за випуск серії. Ці заяви повинні підтверджувати, що виробник (-и) активної субстанції, зазначений (-і) у заяві, діє відповідно до Керівництва з належної виробничої практики щодо вихідних матеріалів. Узагальнена заява може бути прийнятною за певних умов (див. примітку до п. Б. II. б.1.). Виробництво проміжних продуктів також вимагає заяви УО, так як будь-які зміни до сертифікатів на активну субстанцію та проміжні продукти пов’язані, заява УО надається лише у тому випадку, якщо порівняно з діючим сертифікатом є зміна у затвердженому переліку виробничих дільниць. | |||
| Б. III.2. Зміни, пов’язані з необхідністю приведення у відповідність до монографії ДФУ або Європейської Фармакопеї | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна у специфікації нефармакопейної субстанції для приведення у відповідність до вимог ДФУ або Європейської Фармакопеї | |||
| 1. активна субстанція | 1, 2, 3, 4, 5 | 1, 2, 3, 4, 5 | IА |
| 2. допоміжна речовина/вихідний матеріал для виробництва активної субстанції | 1, 2, 4 | 1, 2, 3, 4, 5 | IА |
| б) зміна у специфікаціях, пов’язана зі змінами в ДФУ або Європейській Фармакопеї | 1, 2, 4, 5 | 1, 2, 3, 4 | IА |
| в) зміна у специфікаціях, пов’язана із заміною вимог монографії ДФУ на вимогам монографії Європейської Фармакопеї | 1, 4, 5 | 1, 2, 3, 4 | IА |
| Умови | |||
| 1. Зміна вноситься виключно для приведення у відповідність до вимог Фармакопеї.2. Додаткові специфікації до Фармакопеї залишаються незмінними (наприклад, профіль розміру часток, поліморфна форма або, наприклад, біопроби, агреганти).3. Жодних значних змін у якісних та кількісних показниках профілю домішок не відбулося, якщо специфікації не звужені.4. Додаткова валідація нового або незміненого фармакопейного методу не потрібна.5. Для рослинних активних субстанцій: метод виробництва, фізична форма, екстрагент та співвідношення лікарський засіб/екстрагент повинні залишатися незмінними. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє.2. Порівняльна таблиця вимог затвердженої та оновленої специфікацій.3. Дані аналізу двох промислових серій активної субстанції за всіма показниками оновленої специфікації.4. Дані, які підтверджують придатність монографії для контролю субстанції, наприклад порівняння потенційних домішок із зазначеними у примітці до монографії.5. Дані аналізів (у формі таблиць порівняння) двох промислових серій готового лікарського засобу, що містять субстанцію, яка відповідає затвердженій та оновленій специфікаціям, а також порівняльні дані профілю розчинення принаймні однієї дослідно-промислової серії готового лікарського засобу. Для рослинних лікарських засобів можуть бути прийнятні дані щодо розпадання. | |||
| Примітка. Немає необхідності вносити зміни, пов’язані зі змінами у ДФУ або Європейській фармакопеї, у випадку, якщо відповідність вимогам оновленої монографії виконується протягом шести місяців з моменту її публікації та зроблено посилання на поточне видання Фармакопеї у досьє на затверджений лікарський засіб. | |||
| Б.ІV. Медичні пристрої | |||
| Б.ІV.1. Зміна пристроїв для вимірювання дози або введення лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) додавання або заміна пристрою, який не є невід’ємною частиною первинної упаковки | |||
| 1. пристрій, який має СЕ-маркування | 1, 2, 3 | 1, 2, 4 | IА |
| 2. спейсер для дозованих інгаляторів | ІІ | ||
| б) вилучення пристрою | 4, 5 | 1, 5 | IА |
| в) додавання або заміна пристрою, який є невід’ємною частиною первинної упаковки | IІ | ||
| Умови | |||
| 1. Запропонований пристрій для вимірювання дози повинен точно видавати необхідну дозу препарату згідно з затвердженим дозуванням. Необхідно надати результати відповідних досліджень.2. Новий пристрій повинен бути сумісним з лікарським засобом.3. Зміни не повинні призвести до суттєвих виправлень у коткій характеристиці лікарського засобу, інструкції для медичного застосування та маркуванні.4. Лікарський засіб, як і раніше, демонструє відтворюваність дози. | |||
| Документація | |||
| 1. Зміни до відповідних розділів реєстраційного досьє, включаючи опис, детальне зображення та склад матеріалу пристрою, та постачальника (за необхідності), оновлені коротку характеристику лікарського засобу, інструкцію для медичного застосування та зразки упаковок (за необхідності).2. Підтвердження СЕ-маркування або відповідний висновок МОЗ України про безпеку пристрою.3. Дані, які підтверджують безпеку, точність дозування та сумісність матеріалу пристрою та лікарського засобу.4. Зразки нового пристрою (за потреби).5. Обґрунтування виключення пристрою. | |||
| Примітка до п. B. IV.1. в. Будь-яка зміна, що призводить до нової лікарської форми, потребує подання заяви на реєстрацію. | |||
| Б. V. Зміни до реєстраційного посвідчення внаслідок інших регуляторних процедурБ. V. а) ПМФ/ВАМФ (мастер-файл на плазму/мастер-файл на антиген для приготування вакцини) | |||
| Б. V. а.1 Включення нового, оновленого або зміненого мастер-файла на плазму у реєстраційне досьє на лікарський засіб (процедура 2-го етапу для мастер-файла на плазму) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) первинне включення нового мастер-файла на плазму, що впливає на властивості готового лікарського засобу | II | ||
| б) первинне включення нового мастер-файла на плазму, що не впливає на властивості готового лікарського засобу | 1, 2, 3, 4 | IБ | |
| в) включення оновленого/зміненого мастер-файла на плазму, якщо зміни впливають на властивості готового лікарського засобу | 1, 2, 3, 4 | IБ | |
| г) включення оновленого/зміненого мастер-файла на плазму, якщо зміни не впливають на властивості готового лікарського засобу | 1 | 1, 2, 3, 4 | IA |
| Умови | |||
| 1. Оновлений або змінений мастер-файл на плазму (ПМФ) отримав сертифікат відповідності до законодавства Європейського Союзу згідно з Додатком 1 до Директиви 2001/83/ЄС. | |||
| Документація | |||
| 1. Заява, що Сертифікат відповідності на ПМФ та Звіт про оцінку цілком придатні для реєстрації лікарського засобу, власник ПМФ надав Сертифікат на ПМФ, Звіт про оцінку та Досьє на ПМФ заявнику (коли заявник не являється власником ПМФ), Сертифікат на ПМФ та Звіт про оцінку заміщають документацію на попередній ПМФ для даного реєстраційного посвідчення.2. Сертифікат на ПМФ та Звіт про оцінку.3. Експертний висновок, що дає стислий опис усіх змін, які внесені до сертифікованого ПМФ, та оцінює їх потенціальний вплив на готовий лікарський засіб, включаючи оцінки специфічного ризику.4. Форма заяви на внесення змін повинна чітко визначати «затверджений» та «запропонований» сертифікат ЕМЕА (Європейського агентства з лікарських засобів) на ПМФ (номер коду) у реєстраційному досьє на готовий лікарський засіб. Форма заяви на внесення зміни має чітко перераховувати також всі інші ПМФ, на які посилається заявник, навіть, якщо вони не є предметом заяви (за необхідності). | |||
| Б. V. а.2. Включення нового, оновленого або зміненого мастер-файла на антиген для виготовлення вакцини у реєстраційне досьє на готовий лікарський засіб (процедура 2-го етапу для ВАМФ) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) первинне включення нового мастер-файла на антиген для виготовлення вакцини | II | ||
| б) включення оновленого або зміненого мастер-файла на антиген для виготовлення вакцини, коли зміни впливають на властивості кінцевого продукту | 1, 2, 3, 4 | IБ | |
| в) включення оновленого або зміненого мастер-файла на антиген для виготовлення вакцини, коли зміни не впливають на властивості кінцевого продукту | 1 | 1, 2, 3, 4 | IA |
| Умови | |||
| 1. Оновлений або змінений мастер-файл на антиген для виготовлення вакцини отримав сертифікат відповідності до законодавства Європейського Союзу згідно з Додатком 1 до Директиви 2001/83/ЄС. | |||
| Документація | |||
| 1. Заява, що Сертифікат та Звіт про оцінку ВАМФ цілком придатні для реєстрації препарату, власник ВАМФ надав Сертифікат на ВАМФ, Звіт про оцінку та Досьє на ВАМФ заявнику (коли заявник не є власником ВАМФ), Сертифікат та Звіт про оцінку ВАМФ заміщають документацію попереднього ВАМФ для даного реєстраційного посвідчення.2. Сертифікат та Звіт про оцінку ВАМФ.3. Експертний висновок, що дає стислий опис усіх змін, внесених до сертифікованого ВАМФ, та оцінює їх потенціальний вплив на кінцеві продукти, включаючи оцінку специфічного ризику для продукту.4. Форма заяви на внесення зміни має чітко визначити «затверджений» та «»запропонований» сертифікат ЕМЕА (Європейського агентства з лікарських засобів) на ВАМФ (номер коду) у реєстраційному досьє. Форма заяви на внесення зміни має чітко перераховувати також всі інші АВМФ, на які посилається заявник, навіть, якщо вони не є предметом заяви (за необхідності). | |||
| Б. V. б) Процедура посилання | |||
| Б. V. б.1 Перегляд досьє з якості відповідно до Рішення Комісії за процедурою, що викладена у статті 30 та 31 Директиви 2001/83/ЄС (процедура посилання) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) зміна впроваджує результат процедури посилання (*) | 1 | IА | |
| б) Гармонізацію досьє з якості не включено до процедури посилання, перегляд призначений гармонізувати його. | II | ||
| Документація | |||
| 1. До супровідного листа до заяви про зміни: Посилання на відповідне Рішення Комісії. | |||
| (*) Застосовується у разі, коли власник торгової ліцензії має вжити заходів для надання державам ЄС можливості діяти відповідно до рішення Комісії протягом 30 днів після його повідомлення відповідно до Статті 34(3) Директиви 2001/83/ЄС | |||
| Б. V. в) Протокол управління змінами | |||
| Б. V. в.1. Перегляд досьє з якості для внесення змін на вимогу компетентного органу після оцінки протоколу управління змінами | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип зміни |
| а) внесення зміни не вимагає будь-яких додаткових супровідних даних | 1 | 1, 2, 4 | IА |
| б) внесення зміни вимагає додаткових супровідних даних | 1, 2, 3, 4 | IB | |
| в) внесення зміни для лікарського засобу біологічного/імунологічного походження | 1, 2, 3, 4, 5 | IB | |
| Умови | |||
| 1. Запропонована зміна введена цілком відповідно до затвердженого протоколу управління змінами, який вимагає негайного повідомлення після затвердження. | |||
| Документація | |||
| 1. Посилання на затверджений протокол управління змінами.2. Заява про те, що зміна відповідає затвердженому протоколу управління змінами, результати дослідження відповідають критеріям прийнятності, зазначеним в протоколі. Додатково заява про те, що оцінка порівнянності не вимагається для лікарських засобів біологічного/імунологічного походження.3. Результати досліджень, що проводилися згідно із затвердженим протоколом управління змінами.4. Зміни до відповідних розділів реєстраційного досьє.5. Копія затверджених специфікацій на активну субстанцію або готовий лікарський засіб. | |||
| В. ЗМІНИ ЩОДО БЕЗПЕКИ, ЕФЕКТИВНОСТІ, ФАРМАКОНАГЛЯДУ | |||
| В. I ЛІКАРСЬКІ ЗАСОБИ ДЛЯ ЛЮДИНИ | |||
| В. I.1 Зміна до короткої характеристики лікарського засобу, інструкції для медичного застосування або маркування | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип процедури |
| а) потребує негайного внесення, але не пізніше 30 днів | 1, 2, 3 | ІАнп | |
| б) потребує внесення відповідно до інформації референтного/оригінального лікарського засобу без надання жодних нових додаткових даних заявником | 1, 2, 3 | ІБ | |
| в) потребує внесення відповідно до посилань та нові додаткові дані надані заявником | 1, 3 | ІІ | |
| Документація | |||
| 1. До супровідного листа заяви на внесення зміни додати: обґрунтування внесення змін з доданням оновлених короткої характеристики лікарського засобу, інструкції для медичного застосування маркування2. Заява, що запропоновані Коротка характеристика лікарського засобу, інструкція для медичного застосування, маркування за даними розділами ідентичні щодо інформації на референтний/оригінальний лікарських засіб.3. Оновлена інформація про лікарський засіб | |||
| В. I.2 Зміна у Короткій характеристиці лікарського засобу, інструкції для медичного застосування, маркуванні генеричних/комбінованих/біоподібних лікарських засобів після оцінки тієї ж зміни щодо референтного/оригінального засобу. | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип процедури |
| а) Впровадження змін (и), щодо яких заявник не надає жодних нових додаткових даних | 1, 2 | IБ | |
| б) Впровадження змін (и), яку необхідно в подальшому обґрунтувати новими додатковими даними, які має надати заявник (наприклад, порівнянність) | II | ||
| Документація | |||
| 1. До супровідного листа заяви на внесення зміни додати: рішення МОЗ України, якщо необхідно.2. Обгрунтування внесення змін.3. Оновлена інформація про лікарській засіб (коротка характеристика та/або інструкцію для медичного застосування). | |||
| В. I.3 Внесення змін (и) за вимогою МОЗ України, згідно рекомендацій національних компетентних органів, міжнародних організацій, регуляторних агенцій інших країн після оцінки негайного обмеження щодо безпеки, маркування, характерного для певної фармакологічної та/або фармакотерапевтичної групи, регулярно оновлюваного звіту з безпеки, плану управління ризиком, контрольних заходів/спеціальних зобов’язань, змін для відображення короткої характеристики лікарського засобу та інструкції для медичного застосування | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип процедури |
| a) Впровадження узгоджених змін у формулюванні, щодо яких заявник не надає жодних нових додаткових даних | 1, 2 | IБ | |
| б) Впровадження змін, які вимагають подальшого обґрунтування новими додатковими даними, що надаються заявником | ІІ | ||
| Документація | |||
| 1. До супровідного листа заяви на внесення зміни додати: рішення МОЗ України з доданням відповідного звіту з оцінки, якщо необхідно.2. Обґрунтування внесення змін.3. Оновлена інформація про лікарській засіб (коротка характеристика та/або інструкцію для медичного застосування). | |||
| Примітка: Заявникам слід нагадати, що як тільки вони отримають нову інформацію, яка може внести зміну до реєстраційного посвідчення, її слід надати до компетентних органів у якості зміни, і не чекати оцінки цих даних за допомогою однією з процедур, що вказані вище. | |||
| В. I.4 Зміни, що пов’язані із значними змінами у Короткій характеристиці лікарського засобу, інструкції для медичного застосування згідно нових даних з якості, доклінічних, клінічних даних та даних фармаконагляду | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип процедури |
| ІІ | |||
| В. I.5 Зміна у правовому статусі лікарського засобу | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип процедури |
| а) Для генеричних/комбінованих/біоподібних лікарських засобів після зміни затвердженого правового статусу референтного/оригінального лікарського засобу | 1, 2 | ІB | |
| б) Усі інші зміни правового статусу | ІІ | ||
| Документація | |||
| 1. До супровідного листа заяви на внесення зміни додати: обґрунтування внесення змін з наданням документації, яка підтверджує зміну правового статусу лікарського засобу2. Оновлена інформація про лікарській засіб (коротка характеристика та/або інструкцію для медичного застосування)http://www.apteka.ua | |||
| В. I.6. Зміна (и) до терапевтичних показань (ня) | Умови, які мають бути виконані | Документи, які мають бути представлені | Тип процедури |
| а) додання нового терапевтичного показання або зміни затвердженого показання | ІІ | ||
| б) видалення терапевтичного показання | ІБ | ||
| 1. Обгрунтування внесення змін.2. Оновлена інформація про лікарській засіб (коротка характеристика та/або інструкцію для медичного застосування).Примітка: якщо додавання або зміна терапевтичного показання має місце в контексті застосування результату процедури посилання або змін до інформації на лікарський засіб генеричного/комбінованого/біоподібного продукту після оцінки тієї ж зміни для референтного/оригінального лікарського засобу, зміни В.1.1 та В.1.2 застосовуються відповідно. | |||
| В. I.7. Видалення | Умови, які необхідно виконати | Документи, які необхідно надати | Тип процедури |
| а) лікарська форма | 1, 2 | ІБ | |
| б) сила дії | 1, 2 | ІБ | |
| Документація | |||
| 1. Заява, що незмінені форми випуску лікарського засобу відповідають інструкцій з дозування та тривалості лікування, як вказано в короткій характеристиці лікарського засобу. | |||
| 2. Оновлена інформація про лікарській засіб (коротка характеристика та/або інструкцію для медичного застосування). | |||
| Примітка: У випадках, якщо на дану лікарську форму або силу дії отримано реєстраційне посвідчення, яке відрізняється від реєстраційного посвідчення на інші лікарські форми або сили дії, видалення попередньої не буде зміною, а буде відкликанням реєстраційного посвідчення. | |||
| В. I.8 Введення нової системи фармаконагляду | Умови, які необхідно виконати | Документи, які необхідно надати | Тип процедури |
| а) яка не пройшла оцінку відповідного національного компетентного органу/ЕМЕА для іншого продукту того ж заявника | ІІ | ||
| б) яка пройшла оцінку відповідного національного компетентного органу/інших регуляторних агенцій для іншого лікарського препарату того ж заявника (*) | 1 | IБ | |
| Документація | |||
| 1. Новий детальний опис системи фармаконагляду. | |||
| (*) Примітка: Ця зміна стосується випадків, коли прийнятність вже оціненої системи фармаконагляду буде оцінюватися для нового реєстраційного посвідчення (наприклад, під час перенесення реєстраційного посвідчення). | |||
| В. I.9 Зміни до існуючої системи фармаконагляду, як зазначено у описі системи фармаконагляду | Умови, які необхідно виконати | Документи, які необхідно надати | Тип процедури |
| а) зміна уповноваженої особи, відповідальної за фармаконагляд | 1 | 1 | IАнп |
| б) зміна в контактних даних уповноваженої особи, відповідальної за фармаконагляд | 1 | 2 | ІАнп |
| в) зміна до процедури підтримки уповноваженої особи, відповідальної за фармаконагляд | 1 | 2 | ІАнп |
| г) зміна до бази даних з безпеки (наприклад, введення нової бази даних з безпеки, включаючи перенесення зібраних даних з безпеки та/або аналіз та повідомлення до нової системи) | 1, 2, 3 | 2 | ІАнп |
| г’) Зміни в контрактних домовленостях з іншими особами або організаціями, що залучені до виконання вимог з фармаконагляду та описаними в системі фармаконагляду а саме, якщо складається договір субпідряду про подання електронних повідомлень про індивідуальний випадок, пов’язаний з безпекою лікарського засобу, про ведення головних баз даних, про відстеження сигналу або про підготовку регулярно оновлюваного звіту з безпеки | 1 | 2 | ІАнп |
| д) Виключення тем, що увійшли у письмову процедуру (и), яка описує діяльність з фармаконагляду | 1 | 2 | ІАнп |
| е) Зміна дільниці, що проводить діяльність з фармаконагляду | 1 | 2 | ІАнп |
| є) інші зміни до системи фармаконагляду, які не впливають на функціонування системи фармаконагляду (наприклад, зміни розташування основного сховища/архіву, адміністративні зміни, оновлення скорочень, зміни щодо назви функцій/процедур). | 1 | 2 | ІАнп |
| Умови | |||
| 1. Система фармаконагляду залишається незмінною | |||
| 2. Система бази даних була валідована. | |||
| 3. Перенесення даних з інших систем баз даних було валідовано. | |||
| 4. Такі ж зміни до системи фармаконагляду введені для всіх лікарських засобів того ж заявника (така ж кінцева версія опису системи фармаконагляду). | |||
| Документація | |||
| 1. Остання версія опису системи фармаконагляду, включаючи а) резюме нової уповноваженої особи, відповідальної за фармаконагляд б) доказ реєстрації уповноваженої особи, відповідальної за фармаконагляд в базі ЕМЕА Eudravigilance, в разі наявності та в) нове положення про заявника та уповноваженої особи, відповідальної за фармаконагляд, відносно їх наявності та засоби повідомлення про побічні реакції, які підписані новою уповноваженою особою, відповідальною за фармаконагляд, та заявником, та відображення будь-які наступних змін, наприклад, до організаційної схеми. | |||
| 2. Остання версія опису системи фармаконагляду та/або остання версія додатку стосовно лікарського засобу, якщо необхідно. Для б) якщо контактні дані уповноваженої особи, відповідальної за фармаконагляд, спочатку не включені до опису системи фармаконагляду, подання переглянутої версії опису системи фармаконагляду не вимагається/повинна подаватися лише форма заяви/повідомлення.3. Посилання на заяву/процедуру та лікарський засіб, до яких було внесено зміни.Примітка для з): Оцінка поданого опису системи фармаконагляду як частини нової заяви на отримання реєстраційного посвідчення/розширення/зміну може викликати зміни на вимогу національного компетентного органу у цьому описі системи фармаконагляду. Якщо це відбудеться, ті ж зміни можуть бути внесені до опису системи фармаконагляду в інших реєстраційних посвідченнях того ж заявника шляхом подання (згрупованої) зміни типу ІАнп. | |||
Примітка: У разі внесення протягом дії реєстраційного посвідчення змін в інструкцію для медичного застосування, які не стосуються клінічної інформації (назва лікарського засобу, склад, лікарська форма, основні фізико-хімічні властивості, виробник, місцезнаходження, умови зберігання, термін придатності та ін.) у складі інших реєстраційних документів разом із заявою Заявник подає до Центру інформацію за такою формою:
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
___________ № ____________
Реєстраційне посвідчення
№ ____________________
*Зміни внесено
Наказ Міністерства охорони здоров’я України
___________ № ____________
Заявник, країна:
Виробник, країна:
Зміни до інструкції для медичного застосування лікарського засобу
назва лікарського засобу, лікарська форма,
дозування, упаковка
| Попередня редакція | Нова редакція |
| Розділ «…» | Розділ «…» |
Повноважний представник (заявника в Україні) печатка, підпис
У правому кутку інструкції для медичного застосування, яка буде супроводжувати лікарський засіб, текст «Зміни внесено» друкувати не потрібно.»
«Додаток 6
до пункту 4.1 (з) Порядку проведення експертизи матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Зміни, які потребують нової реєстрації лікарського засобу
До змін, які потребують нової реєстрації лікарського засобу, належать:
1. Зміни активних речовин:
а) заміна активної речовини на іншу сіль, ефір, похідну тощо за умови, що характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих або
— використання інших ізомерів, або зміна ізомерного складу, або
— незначні зміни молекулярної структури речовин біологічного походження за виключенням зміни у активній речовині протигрипозних вакцин;
б) зміна вектора, який використовується для виробництва антигена або вихідного матеріалу, включаючи новий головний банк клітин від іншого джерела, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
в) новий ліганд або механізм з’єднання для радіофармацевтичного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих;
г) застосування інших екстрагентів або співвідношення рослинна лікарська сировина/рослинний препарат для рослинного лікарського засобу, якщо характеристики, які визначають співвідношення користь/ризик, суттєво не відрізняються від зареєстрованих.
2. Зміни сили дії, лікарської форми та способу застосування:
а) зміна біодоступності;
б) зміна фармакокінетики;
в) зміна або введення додаткового дозування лікарського засобу (додаткова доза);
г) зміна або додання нової лікарської форми;
ґ) зміна або додання нового шляху введення (для парентеральних форм у зв’язку з відмінностями в ефективності та безпечності лікарського засобу при внутрішньоартеріальному, внутрішньовенному, внутрішньом’язовому та інших шляхах уведення).
3. Одночасна зміна заявника та виробника.
При таких змінах заявник разом з обґрунтуванням потреби внесення змін подає до Центру відповідні розділи реєстраційних матеріалів, які обґрунтовують указані зміни і є достатніми для експертизи лікарського засобу.
«Додаток 7
до пункту 4.3 Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
ЗАЯВА
про внесення змін до реєстраційних матеріалів на лікарський засіб
Дата надходження заяви
___ ____________ 20_ року№ ______________
Цим я заявляю, що:
![]() немає інших змін, крім тих, що вказані у даній заяві (виняток: інші зміни, що подаються паралельно та вказані в розділі «інші заяви»);
немає інших змін, крім тих, що вказані у даній заяві (виняток: інші зміни, що подаються паралельно та вказані в розділі «інші заяви»);
![]() зміна(-и) не спричинить(-ять) негативного впливу на якість, ефективність та безпечність
зміна(-и) не спричинить(-ять) негативного впливу на якість, ефективність та безпечність
лікарського засобу;
![]() усі умови (згідно з додатком 5 цього Порядку), які стосуються змін(-и), виконані;
усі умови (згідно з додатком 5 цього Порядку), які стосуються змін(-и), виконані;
![]() необхідні документи, що стосуються змін(-и), подані;
необхідні документи, що стосуються змін(-и), подані;
![]() Заявник бере на себе відповідальність за ефективність, безпечність і якість лікарського засобу, гарантує достовірність інформації, що міститься в реєстраційних матеріалах, а також підтверджує, що всі наявні дані стосовно якості, безпечності та ефективності лікарського засобу представлені в реєстраційному дось
Заявник бере на себе відповідальність за ефективність, безпечність і якість лікарського засобу, гарантує достовірність інформації, що міститься в реєстраційних матеріалах, а також підтверджує, що всі наявні дані стосовно якості, безпечності та ефективності лікарського засобу представлені в реєстраційному дось
![]() усі внески сплачено/буде сплачено відповідно до вимог чинного законодавства,
усі внески сплачено/буде сплачено відповідно до вимог чинного законодавства,
Зміни будуть введені:
![]() з наступного виробничого циклу/наступного випуску
з наступного виробничого циклу/наступного випуску
![]() дата ________________________________
дата ________________________________
Основний підпис _____________________
Посада _______________________________
Прізвище ____________________________
Дата _________________________________
Другий підпис _______________________
(якщо необхідно)
Посада ______________________________
Прізвище ____________________________
Заява дійсна протягом 3- місяців з дати офіційного подання заяви. У разі ненадання матеріалів реєстраційного досьє протягом зазначеного терміну або листа з обґрунтуванням терміну, необхідного для їх підготовки, то заява знімається з розгляду. Надалі така заява може бути подана до Центру в установленому порядку.
Тип змін
![]() Тип ІА
Тип ІА
![]() Тип ІБ
Тип ІБ
![]() Тип ІІ
Тип ІІ
![]() безпечність
безпечність
![]() термінові обмеження, пов’язані із безпечністю
термінові обмеження, пов’язані із безпечністю
![]() якість
якість
![]() інші
інші
Торговельна назва
лікарського засобу
Назва, місцезнаходження заявника
Діюча(-і) речовина(-и)
Прізвище, ім’я та місцезнаходження уповноваженої особи
Лікарська форма та сила дії
Телефон
Факс
е-mail
Номер реєстраційного
посвідчення
Посилання заявника
Зміни типу ІА та ІБ (нижче позначте необхідне)
Примітки:
У разі змін типу II із заяви вилучають перелік змін типу I, наведений нижче.
У разі змін типу I вилучають ті зміни типу I, на які не поширюється заява.
У разі змін, які потребують нової реєстрації, вилучають ті зміни типу I та II, на які не розповсюджується заява.
| А. АДМІНІСТРАТИВНІ ЗМІНИ | ||||||
| Назва | Тип зміни | |||||
| А.1. Зміна назви та/або адреси заявника (власника реєстраційного посвідчення) | IА | IБ* | ||||
| А.2. Зміна торгової назви лікарського засобу | IБ | |||||
| А.3. Зміна назви активної субстанції | IА | IБ* | ||||
| А.4. Зміна назви та/або адреси виробника (включаючи, за необхідності місце проведення контролю якості) або постачальника активної субстанції/вихідного матеріалу/реагенту/проміжного продукту, що застосовуються у виробництві активної субстанції (за відсутності сертифікату відповідності Європейській Фармакопеї у затвердженому досьє) | IА | IБ* | ||||
| А.5. Зміна назви та/або адреси виробника готового лікарського засобу, включаючи місце проведення контролю якості | ||||||
| а) виробнича дільниця випуску серій | IА | IБ* | ||||
| б) усі інші дільниці | IА | IБ* | ||||
| А.6. Зміна коду АТХ | IА | IБ* | ||||
| А.7. Вилучення виробничої дільниці (включаючи дільниці для активної субстанції, проміжного продукту або готового лікарського засобу, дільниці для проведення пакування, виробника відповідального за випуск серій, місце проведення контролю серії) або постачальника вихідного матеріалу, реагенту або допоміжної речовини (якщо зазначено у досьє) | IА | IБ* | ||||
| Б. ЗМІНИ З ЯКОСТІ | ||||||
| Б. I. Активна субстанція | ||||||
| Б. I. а) Виробництво | ||||||
| Б. I. а.1. Зміна виробника вихідного/проміжного продукту/реагенту, що використовуються у виробничому процесі активної субстанції, або зміна виробника (включаючи де необхідно місце проведення контролю якості) активної субстанції (за відсутності сертифікату відповідності Європейській Фармакопеї у затвердженому досьє) | ||||||
| а) запропонований виробник належить до тієї ж виробничої групи підприємств, що й затверджений | IА | IБ* | ||||
| б) введення нового виробника активної субстанції з наданням мастер-файла на активну субстанцію | II | |||||
| в) запропонований виробник використовує спосіб синтезу, що суттєво відрізняється від попереднього, або умови виробництва, які потенційно можуть змінити важливі характеристики якості активної субстанції, такі як якісний та/або кількісний профіль домішок, що потребує кваліфікації, або фізико-хімічні властивості активної субстанції, що впливають на біодоступність | II | |||||
| г) новий виробник вихідного продукту, для якого вимагається попередня оцінка вірусної безпеки та/або ризику передачі збудників ГЕ | II | |||||
| ґ) зміна у субстанції біологічного походження або вихідному матеріалі/реагенті/проміжному продукті, що використовуються для виробництва лікарського засобу біологічного походження | II | |||||
| д) зміни у методах контролю якості активної субстанції або додавання дільниці для проведення контролю серії/випробування | IА | IБ* | ||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. а.2. Зміни в процесі виробництва активної субстанції | ||||||
| а) незначна зміна у процесі виробництва активної субстанції | IА | IБ* | ||||
| б) значна зміна у процесі виробництва активної субстанції, що може мати істотний вплив на якість, безпеку або ефективність лікарського засобу | II | |||||
| в) зміна стосується субстанції біологічного/імунологічного походження або використання хімічних субстанцій у виробництві лікарського засобу біологічного/імунологічного походження і не відноситься до протоколу | II | |||||
| г) зміна у лікарському засобі рослинного походження та стосується однієї з таких характеристик: джерело походження сировини, спосіб виробництва або виготовлення | II | |||||
| ґ) незначна зміна у закритій частині мастер-файла на активну субстанцію | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. а.3. Зміна розміру серії (включаючи діапазони) активної субстанції або проміжного продукту | ||||||
| а) збільшення до 10 разів порівняно із затвердженим розміром | IА | IБ* | ||||
| б) зменшення обсягу виробництва | IА | IБ* | ||||
| в) зміна, що потребує доведення подібності активної субстанції біологічного/імунологічного походження порівняно із затвердженою | II | |||||
| г) збільшення у понад 10 разів порівняно із затвердженим розміром | IБ | |||||
| ґ) розмір серії активної субстанції біологічного/імунологічного походження збільшився/зменшився без зміни параметрів процесу (наприклад, дублювання лінії) | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. а.4. Зміни випробувань або допустимих меж, що встановлені у специфікаціях, в процесі виробництва активної субстанції | ||||||
| а) звуження допустимих меж | IА | IБ* | ||||
| б) додавання нового випробування та допустимих меж | IА | IБ* | ||||
| в) вилучення несуттєвого випробування | IА | IБ* | ||||
| г) розширення затверджених допустимих меж для показників, які можуть істотно вплинути на якість активної субстанції | II | |||||
| ґ) вилучення випробування, що може мати істотний влив на загальну якість активної субстанції | II | |||||
| д) додавання або заміна випробування за результатами досліджень з безпеки або якості | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б.1. б) Контроль активної субстанції | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. б.1. Зміна у параметрах специфікацій та/або допустимих меж, визначених у специфікаціях на активну субстанцію, або вихідний/проміжний продукт/реагент, що використовуються у процесі виробництва активної субстанції | ||||||
| а) звуження допустимих меж, визначених у специфікації на лікарські засоби, що підлягають офіційному випуску серії | IA | ІБ* | ||||
| б) звуження допустимих меж, визначених у специфікації | IA | ІБ* | ||||
| в) доповнення специфікації новим показником якості та відповідним методом випробування | IA | ІБ* | ||||
| г) вилучення незначного показника якості, (наприклад, вилучення застарілого показника) | IА | ІБ* | ||||
| ґ) вилучення параметра специфікації, який може мати суттєвий влив на якість активної субстанції та/або готового лікарського засобу | II | |||||
| д) розширення допустимих меж, визначених у специфікації на активну субстанцію | II | |||||
| е) розширення допустимих меж, визначених у специфікаціях на вихідні матеріали/проміжні продукти, які мають істотний вплив на якість активної субстанції та/або готового лікарського засобу | II | |||||
| є) доповнення або заміна параметру специфікації за результатами досліджень з безпеки або якості (за виключенням субстанції біологічного або імунологічного походження) | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. б.2. Зміна у методах випробування активної субстанції або вихідного/проміжного продукту/реагенту, що використовується у процесі виробництва активної субстанції | ||||||
| а) незначні зміни у затверджених методах випробування | IА | ІБ* | ||||
| б) видалення методу випробування для активної субстанції/реагенту/проміжного продукту, якщо альтернативний метод вже затверджений | IА | ІБ* | ||||
| в) інші зміни в методах випробування (включаючи заміну або доповнення) для реагенту, що не спричиняє істотного впливу на якість активної субстанції | IA | ІБ* | ||||
| г) зміна (заміна) у біологічному/імунологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент для активної субстанції біологічного походження, наприклад, пептидні карти, глік-карти тощо | II | |||||
| ґ) інші зміни у методах випробування (включаючи заміну або доповнення) активної субстанції або вихідного/проміжного продукту | IБ | |||||
| Б. I. в) Система упаковка/укупорка | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. в.1. Зміна у безпосередній упаковці активної субстанції | ||||||
| а) якісні та/або кількісні зміни складу | IА | ІБ* | ||||
| б) якісні та/або кількісні зміни складу для стерильних та не заморожених активних субстанцій біологічного/імунологічного походження | II | |||||
| в) рідких активних субстанцій (не стерильних) | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. в.2. Зміни у параметрах специфікацій та/або допустимих меж, зазначених у специфікаціях, для первинної упаковки активної субстанції | ||||||
| а) звуження допустимих меж, зазначених у специфікаціях | IА | ІБ* | ||||
| б) доповнення специфікації новим показником та відповідним методом випробування | IА | ІБ* | ||||
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | IА | ІБ* | ||||
| г) доповнення або заміна показника за результатами досліджень з безпеки або якості | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. I. в.3. Зміна в методах випробування первинної упаковки активної субстанції | ||||||
| а) незначні зміни у затверджених методах випробування | IА | ІБ* | ||||
| б) інші зміни в методах випробування (включаючи заміну або доповнення) | IА | ІБ* | ||||
| в) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | IА | ІБ* | ||||
| Б. I. г) Стабільність | ||||||
| Б. I. г.1. Зміна періоду повторних випробувань/терміну придатності або умов зберігання активної субстанції (за наявності у затвердженому досьє сертифікату відповідності Європейській фармакопеї, що включає період повторного випробування) | ||||||
| а) період повторного випробування/термін придатності | ||||||
| 1. зменшення | ІА | ІБ* | ||||
| 2. збільшення періоду повторного випробування на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності до Настанови або Керівних принципів ІСН щодо випробування стабільності (*) | ІІ | |||||
| 3. збільшення терміну придатності активної субстанції біологічного/імунологічного походження на основі результатів досліджень, виконаних не у відповідності до затвердженого протоколу | ІІ | |||||
| 4. збільшення або введення періоду повторного випробування/терміну придатності на основі результатів досліджень у реальному часі | ІБ | |||||
| б) умови зберігання | ||||||
| 1. обмеження умов зберігання | ІА | ІБ* | ||||
| 2. зміна умов зберігання активної субстанції біологічного/імунологічного походження, якщо дослідження стабільності ще не проводились відповідно до затвердженого протоколу | II | |||||
| 3. зміна умов зберігання активної субстанції | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б.1.ґ) Проектний простір | ||||||
| а) одного елементу (блок, частина) виробничого процесу активної субстанції, включаючи контроль в процесі виробництва та/або методи випробування | II | |||||
| б) методів випробувань для вихідних матеріалів/реагентів/проміжних продуктів та/або активної субстанції | II | |||||
| Б. I.ґ.2. Внесення змін після затвердження протоколу для активної субстанції | ||||||
| II | ||||||
| Б. I.ґ.3. Вилучення затвердженого протоколу управління змінами для активної субстанції | ||||||
| IА | ІБ* | |||||
| Б. II. Готовий лікарський засіб | ||||||
| Б. II. а) Опис та склад | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. а.1. Зміна або додавання штампів, потовщень або інших маркувань, уключаючи заміну або додавання фарб для маркування лікарського засобу | ||||||
| а) зміни штампів, потовщень або інших маркувань | IА | ІБ* | ||||
| б) зміни риски, призначеної для розділу таблетки на рівні дози | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. а.2. Зміна форми або розмірів лікарської форми | ||||||
| а) таблетки з негайним вивільненням, капсули, супозиторії та пес арії | IА | ІБ* | ||||
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим або пролонгованим вивільненням та ділимі таблетки, призначені для розділу на рівні дози | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. а.3. Зміна у складі (допоміжних речовинах) готового лікарського засобу | ||||||
| а) смакові добавки або барвники | ||||||
| 1. додавання, вилучення або заміна | IА | ІБ* | ||||
| 2. збільшення або зменшення | IА | ІБ* | ||||
| б) інші допоміжні речовини | II | |||||
| 1. будь-яка незначна зміна кількісного складу допоміжних речовин у готовому лікарському засобі | IА | ІБ* | ||||
| 2. якісні або кількісні зміни щодо однієї або декількох допоміжних речовин, які можуть значно вплинути на безпеку, якість або ефективність готового лікарського засобу | II | |||||
| 3. зміна у лікарському засобі біологічного/імунологічного походження | II | |||||
| 4. будь-яка нова допоміжна речовина, що включає використання матеріалів людського або тваринного походження, для яких вимагається оцінка даних з вірусної безпеки або ризику передачі збудників ГЕ | II | |||||
| 5. зміна, яка підтверджується дослідженнями з біоеквівалентності | II | |||||
| 6. заміна однієї допоміжної речовини на іншу з тими самими функціональними характеристиками та на тому ж рівні | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. a.4. Зміна маси покриття лікарських форм для перорального застосування або зміна маси оболонки капсул | ||||||
| а) тверді лікарські форми для перорального застосування | IА | ІБ* | ||||
| б) лікарські форми, стійкі до дії шлункового соку, з модифікованим вивільненням або пролонгованої дії, для яких покриття є вирішальним чинником механізму вивільнення | II | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. a.5. Зміна концентрації в окремій дозі багатодозового лікарського засобу для парентерального застосування, коли кількість діючої речовини на одиницю дози (тобто сила дії) не змінюється | ||||||
| II | ||||||
| Б. II. a.6. Вилучення контейнера з розчинником з упаковки | ||||||
| IБ | ||||||
| Б. II. б) Виробництво | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. б.1. Заміна або введення додаткової дільниці виробництва для частини або всього виробничого процесу готового лікарського засобу | ||||||
| а) дільниця для вторинного пакування | IА | ІБ* | ||||
| б) дільниця для первинного пакування | IА | ІБ* | ||||
| в) дільниця, на якій проводяться будь-які виробничі стадії, за виключенням випуску серій, проведення контролю якості та вторинного пакування, для лікарських засобів біологічного/імунологічного походження | II | |||||
| г) дільниця, яка вимагає проведення первинної перевірки виробництва або перевірки виробництва конкретного готового лікарського засобу | II | |||||
| ґ) дільниця, на якій проводяться будь-які виробничі стадії, за виключенням випуску серій, контролю якості, первинного та вторинного пакування, для нестерильних лікарських засобів | IБ | |||||
| д) дільниця, на якій проводяться будь-які виробничі стадії, за виключенням випуску серії, контролю якості та вторинного пакування для стерильних лікарських засобів, що вироблені з використанням асептичного методу, за виключенням лікарських засобів біологічного/імунологічного походження | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. б.2. Зміни, що стосуються випуску серії та контролю якості готового лікарського засобу | ||||||
| а) заміна або додавання дільниці, на який здійснюється контроль/випробування серії | IА | ІБ* | ||||
| б) заміна або додавання виробника, відповідального за випуск серії | ||||||
| 1. не включаючи контроль/випробування серії | IА | ІБ* | ||||
| 2. включаючи контроль/випробування серії | IА | ІБ* | ||||
| 3. включаючи контроль/випробування серії для лікарського засобу біологічного/імунологічного походження та один з методів аналізу, що застосовується на дільниці, є біологічним/імунологічним/імунохімічним методом | II | |||||
| Б. II. б.3. Зміни у процесі виробництва готового лікарського засобу | ||||||
| а) незначна зміна у процесі виробництва твердої лікарської форми для перорального застосування з негайним вивільненням або розчинів для перорального застосування | ІА | ІБ* | ||||
| б) зміна у процесі виробництва, яка може мати істотний влив на якість, безпеку та ефективність лікарського засобу | II | |||||
| в) лікарський засіб є лікарським засобом біологічного/імунологічного походження та зміна вимагає проведення порівняльних досліджень | ІІ | |||||
| г) ведення нестандартного методу кінцевої стерилізації | ІІ | |||||
| ґ) введення або збільшення припустимого надлишку діючої речовини | ІІ | |||||
| д) незначна зміна у процесі виробництва водної суспензії для перорального застосування | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. б.4. Зміна розміру серії (включаючи діапазон розміру серії) готового лікарського засобу | ||||||
| а) збільшення до 10 разів порівняно із затвердженим розміром | IА | ІБ* | ||||
| б) зменшення до 10 разів | IА | ІБ* | ||||
| в) зміна вимагає оцінки порівнянності (проведення порівняльних досліджень) лікарського засобу біологічного/імунологічного походження | ІІ | |||||
| г) зміна стосується всіх інших лікарських форм сукупного (комплексного) виробничого процесу | ІІ | |||||
| ґ) збільшення більш ніж у 10 разів порівняно із затвердженим розміром для твердих лікарських форм з негайним вивільненням | ІБ | |||||
| д) масштаб для лікарського засобу біологічного/імунологічного походження збільшився/зменшився без зміни виробничого процесу (наприклад, дублювання лінії) | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. б.5. Зміни випробувань або допустимих меж, встановлених у специфікаціях, під час виробництва готового лікарського засобу | ||||||
| а) звуження допустимих меж | IА | ІБ* | ||||
| б) доповнення нового методу випробування та допустимих меж | IА | ІБ* | ||||
| в) вилучення несуттєвого випробування | IА | ІБ* | ||||
| г) вилучення випробування у процесі виробництва, яке може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | |||||
| ґ) розширення затверджених допустимих меж для показників, які можуть мати істотний вплив на загальну якість готового лікарського засобу | ІІ | |||||
| д) доповнення або заміна випробування за результатами досліджень з безпеки або якості | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. в) Контроль допоміжних речовин | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. в.1. Зміна параметрів специфікацій та/або допустимих меж для допоміжної речовини | ||||||
| а) звуження допустимих меж | ІА | ІБ* | ||||
| б) доповнення специфікації новим показником з відповідним методом випробування | ІА | ІБ* | ||||
| в) вилучення із специфікації незначного показника (наприклад, вилучення застарілого показника) | ІА | ІБ* | ||||
| г) зміна знаходиться поза затвердженими допустимими межами специфікацій | ІІ | |||||
| ґ) вилучення із специфікації показника, який може мати істотний вплив на загальну якість готового лікарського засобу | ІІ | |||||
| д) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за виключенням лікарських засобів біологічного та імунологічного походження) | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. в.2. Зміна у методах випробування допоміжної речовини | ||||||
| а) незначні зміни у затверджених методах випробувань | IА | ІБ* | ||||
| б) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | ІА | ІБ* | ||||
| в) заміна біологічного/імунологічного/імунохімічного методу випробування або методу, у якому використовується біологічний реагент | ІІ | |||||
| г) інші зміни у методах випробування (включаючи заміну або додавання) | IБ | |||||
| Б. II. в.3. Заміна джерела одержання допоміжної речовини або реактиву, що становить ризик передачі збудників ГЕ | ||||||
| а) матеріалу, що становить ризик передачі збудників ГЕ, на матеріал рослинного або синтетичного походження | ||||||
| 1. для допоміжних речовин або реактивів, які не використовуються у виробництві активної субстанції біологічного/імунологічного походження, або лікарського засобу біологічного/імунологічного походження | IА | ІБ* | ||||
| 2. для допоміжних речовин або реактивів, які використовуються у виробництві активної субстанції біологічного/імунологічного походження, або лікарського засобу біологічного/імунологічного походження | IБ | |||||
| б) заміна або додавання речовини, що становить ризик передачі збудників ГЕ, або заміна речовини, що становить ризик передачі збудників ГЕ, на іншу речовину, що становить ризик передачі збудників ГЕ, для якої немає ГЕ сертифіката відповідності Європейській фармакопеї | II | |||||
| Б. II. в.4. Зміни в методі синтезу або регенерації нефармакопейної допоміжної речовини (за наявності в досьє) | ||||||
| а) незначні зміни у методі синтезу або регенерації | IА | ІБ* | ||||
| б) зміни у специфікації або зміна фізико-хімічних властивостей допоміжної речовини, що може мати вплив на якість готового лікарського засобу | ІІ | |||||
| в) допоміжна речовина є речовиною біологічного/імунологічного походження | ІІ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. г) Контроль готового лікарського засобу | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. г.1. Зміна параметрів специфікацій та/або допустимих меж готового лікарського засобу | ||||||
| а) звуження допустимих меж | IА | ІБ* | ||||
| б) звуження допустимих меж для лікарського засобу, що підлягає офіційному випуску серії | IА | ІБ* | ||||
| в) доповнення специфікації новим показником з відповідним методом випробування | IА | ІБ* | ||||
| г) вилучення незначного показника (наприклад, вилучення застарілого показника) | IА | ІБ* | ||||
| ґ) зміна знаходиться поза затвердженими допустимими межами | ІІ | |||||
| д) вилучення показника, який може мати істотний вплив на якість готового лікарського засобу | ІІ | |||||
| е) доповнення або заміна показника специфікації за результатами досліджень з безпеки або якості (за виключенням лікарських засобів біологічного/імунологічного походження) | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. г.2. Зміна у методах випробування готового лікарського засобу | ||||||
| а) незначна зміна у затверджених методах випробування | IА | ІБ* | ||||
| б) вилучення методу випробування, якщо вже затверджений альтернативний метод | IА | ІБ* | ||||
| в) зміна у біологічному/імунологічному/імунохімічному методі випробування або методі, у якому використовується біологічний реагент | ІІ | |||||
| г) інші зміни у методах випробувань (включаючи заміну або доповнення) | IБ | |||||
| Б. II. г.3. Зміни, які стосуються виробничого процесу у реальному часі або випуску за параметрами для готового лікарського засобу | ||||||
| ІІ | ||||||
| Б. II.ґ) Система упаковка/укупорка | ||||||
| з) інші зміни | IА | IБ | II | |||
| Б. II.ґ.1. Зміна у первинній упаковці готового лікарського засобу | ||||||
| а) якісний та кількісний склад | ||||||
| 1. тверді лікарські форми | IА | ІБ* | ||||
| 2. м’які та нестерильні рідкі лікарські форми | IБ | |||||
| 3. стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | |||||
| 4. зміна стосується зниження ступеню захисту оновленої упаковки, якщо наявні відповідні зміни в умовах зберігання та/або скорочення терміну придатності | ІІ | |||||
| б) тип контейнера | ||||||
| 1. тверді, м’які та нестерильні рідкі лікарські форми | ІБ | |||||
| 2. стерильні лікарські засоби та лікарські засоби біологічного/імунологічного походження | ІІ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II.ґ.2. Зміна параметрів специфікацій та/або допустимих меж первинної упаковки готового лікарського засобу | ||||||
| а) звуження допустимих меж, визначених у специфікації | IА | ІБ* | ||||
| б) доповнення специфікації новим показником з відповідним методом випробування | IА | ІБ* | ||||
| в) вилучення незначного показника (наприклад, вилучення застарілого показника) | ІА | ІБ* | ||||
| г) додавання або заміна показника за результатами досліджень з безпеки або якості | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II.ґ.3. Зміна у методах випробування первинної упаковки готового лікарського засобу | ||||||
| а) незначні зміни у затверджених методах випробувань | IА | ІБ* | ||||
| б) інші зміни у методах випробувань (включаючи заміну або додавання) | IА | ІБ* | ||||
| в) вилучення методу випробування, якщо вже затверджений альтернативний | ІА | ІБ* | ||||
| Б. II.ґ.4. Зміна форми або розміру контейнера чи закупорювального засобу (первинної упаковки) | ||||||
| а) нестерильні лікарські засоби | IА | ІБ* | ||||
| б) зміна форми або розміру основної частини пакувального матеріалу (вторинної упаковки), що може мати значний вплив на доставку, застосування, безпеку та стабільність готового лікарського засобу | IІ | |||||
| в) стерильні лікарські засоби | ІБ | |||||
| Б. II.ґ.5. Зміна розміру упаковки готового лікарського засобу | ||||||
| а) зміна кількості одиниць (наприклад, таблеток, ампул та ін.) в упаковці: | ||||||
| 1. зміна у діапазоні затверджених розмірів упаковки | IА | ІБ* | ||||
| 2. зміна поза діапазоном затверджених розмірів упаковки | IБ | |||||
| б) вилучення упаковки певного розміру | ІА | ІБ* | ||||
| в) зміна маси/об’єму вмісту контейнера багатодозового стерильного лікарського засобу (або однодозового часткового використання) та багатодозового лікарського засобу біологічного/імунологічного походження для парентерального застосування | IІ | |||||
| г) зміна маси/об’єму вмісту контейнера багатодозового лікарського засобу для непарентеральних застосування (або однодозового часткового використання) | ІБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II.ґ.6. Зміна будь-якої частини матеріалу первинної упаковки, що не контактує з готовим лікарським засобом (наприклад, колір кришечок з контролем першого відкриття, колір кодових кілець на ампулах, контейнера для голок (різні види пластмаси) | ||||||
| а) зміна, яка впливає на коротку характеристику лікарського засобу | IА | ІБ* | ||||
| б) зміна, яка не впливає на коротку характеристику лікарського засобу | IА | ІБ* | ||||
| Б. II.ґ.7. Зміна постачальника пакувальних матеріалів або комплектуючих (якщо зазначено в досьє) | ||||||
| а) вилучення постачальника | IА | ІБ* | ||||
| б) заміна або додавання постачальника | IА | ІБ* | ||||
| в) будь-яка зміна постачальника спейсерів для дозованих інгаляторів | ІІ | |||||
| Б. II. д) Стабільність | ||||||
| Б. II. д.1. Зміна у термінах придатності або умовах зберігання готового лікарського засобу: | ||||||
| а) зменшення терміну придатності готового лікарського засобу: | ||||||
| 1. для торгової упаковки | IА | ІБ* | ||||
| 2. після першого розкриття | IА | ІБ* | ||||
| 3. після розчинення або відновлення | IА | ІБ* | ||||
| б) збільшення терміну придатності готового лікарського засобу: | ||||||
| 1. для торгової упаковки (підтверджується даними реального часу) | IБ | |||||
| 2. після першого розкриття (підтверджується даними реального часу) | IБ | |||||
| 3. після розчинення або відновлення (підтверджується даними реального часу) | IБ | |||||
| 4. збільшення терміну придатності на основі екстраполяції результатів дослідження стабільності, проведених не у відповідності з вимогами Настанови або Керівних принципів щодо дослідження стабільності лікарських засобів (*) | ІІ | |||||
| 5. збільшення терміну придатності лікарського засобу біологічного/імунологічного походження на основі результатів досліджень стабільності, проведеними відповідно до затвердженого протоколу | IБ | |||||
| в) зміна в умовах зберігання лікарського засобу біологічного походження, якщо дослідження стабільності були проведені не у відповідності до затвердженого протоколу | ІІ | |||||
| г) зміна в умовах зберігання готового лікарського засобу або після розчинення/відновлення | IБ | |||||
| з) інші зміни | IА | IБ | II | |||
| Б. II. е) Проектний простір | ||||||
| Б. II. е.1. Введення нового проектного простору або розширення затвердженого для готового лікарського засобу (за виключенням лікарських засобів біологічного походження) щодо | ||||||
| а) одного або більше елементів (блок, частина) виробничого процесу готового лікарського засобу, включаючи контроль в процесі виробництва та/або методи випробувань | II | |||||
| б) метоів випробувань для допоміжних речовин/проміжних продуктів та/або готового лікарського засобу | II | |||||
| Б. II. е.2. Внесення змін після затвердження протоколу для готового лікарського засобу | ||||||
| II | ||||||
| Б. II. е.3. Вилучення затвердженого протоколу управління змінами для готового лікарського засобу | ||||||
| IА | ІБ* | |||||
| Б.ІІІ.1. Подання нового або оновленого сертифіката відповідності Європейській Фармакопеї | ||||||
| Для активної субстанціїДля вихідного матеріалу/реагенту/проміжного продукту, що використовуються у виробництві активної субстанціїДля допоміжної речовини | ||||||
| а) сертифікат відповідності Європейській Фармакопеї | ||||||
| 1. новий сертифікат від вже затвердженого виробника | IА | ІБ* | ||||
| 2. оновлений сертифікат від вже затвердженого виробника | IА | ІБ* | ||||
| 3. новий сертифікат від нового виробника (заміна або доповнення) | IА | ІБ* | ||||
| б) ГЕ Сертифіката відповідності Європейській Фармакопеї для активної субстанції/вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини | ||||||
| 1. новий сертифікат для активної субстанції від нового або вже затвердженого виробника | IА | ІБ* | ||||
| 2. новий сертифікат для вихідного матеріалу/реагенту/проміжного продукту або допоміжної речовини від нового або вже затвердженого виробника | IА | ІБ* | ||||
| 3. оновлений сертифікат від вже затвердженого виробника | IА | ІБ* | ||||
| Б. III.2. Зміни, пов’язані з необхідністю приведення у відповідність до монографії ДФУ або Європейської Фармакопеї | ||||||
| а) зміна у специфікації нефармакопейної субстанції для приведення у відповідність до вимог ДФУ або Європейської Фармакопеї | ||||||
| 1. активна субстанція | IА | ІБ* | ||||
| 2. допоміжна речовина/вихідний матеріал для виробництва активної субстанції | IА | ІБ* | ||||
| б) зміна у специфікаціях, пов’язана зі змінами в ДФУ або Європейській Фармакопеї | IА | ІБ* | ||||
| в) зміна у специфікаціях, пов’язана із заміною вимог монографії ДФУ на вимогам монографії Європейської Фармакопеї | IА | ІБ* | ||||
| В.ІV. Медичні пристрої | ||||||
| з) інші зміни | IА | IБ | II | |||
| В.ІV.1. Зміна пристроїв для вимірювання дози або введення лікарського засобу | ||||||
| а) додавання або заміна пристрою, який не є невід’ємною частиною первинної упаковки | ||||||
| 1. пристрій, який має СЕ-маркування | IА | ІБ* | ||||
| 2. спейсер для дозованих інгаляторів | ІІ | |||||
| б) вилучення пристрою | IА | ІБ* | ||||
| в) додавання або заміна пристрою, який є невід’ємною частиною первинної упаковки | IІ | |||||
| Б. V. Зміни до реєстраційного посвідчення внаслідок інших регуляторних процедур | ||||||
| Б. V. а) ПМФ/ВАМФ (мастер-файл на плазму/мастер-файл на антиген для приготування вакцини) | ||||||
| Б. V. а.1 Включення нового, оновленого або зміненого мастер-файла на плазму у реєстраційне досьє на лікарський засіб (процедура 2-го етапу для мастер-файла на плазму) | ||||||
| а) первинне включення нового мастер-файла на плазму, що впливає на властивості готового лікарського засобу | II | |||||
| б) первинне включення нового мастер-файла на плазму, що не впливає на властивості готового лікарського засобу | IБ | |||||
| в) включення оновленого/зміненого мастер-файла на плазму, якщо зміни впливають на властивості готового лікарського засобу | IБ | |||||
| г) включення оновленого/зміненого мастер-файла на плазму, якщо зміни не впливають на властивості готового лікарського засобу | IA | ІБ* | ||||
| Б. V. а.2. Включення нового, оновленого або зміненого мастер-файла на антиген для виготовлення вакцини у реєстраційне досьє на готовий лікарський засіб (процедура 2-го етапу для ВАМФ) | ||||||
| а) первинне включення нового мастер-файла на антиген для виготовлення вакцини | II | |||||
| б) включення оновленого або зміненого мастер-файла на антиген для виготовлення вакцини, коли зміни впливають на властивості кінцевого продукту | IБ | |||||
| в) включення оновленого або зміненого мастер-файла на антиген для виготовлення вакцини, коли зміни не впливають на властивості кінцевого продукту | IA | ІБ* | ||||
| Б. V. б) Процедура посилання | ||||||
| Б. V. б.1 Перегляд досьє з якості відповідно до Рішення Комісії за процедурою, що викладена у статті 30 та 31 Директиви 2001/83/ЄС (процедура посилання) | ||||||
| а) зміна впроваджує результат процедури посилання (*) | IА | |||||
| б) Гармонізацію досьє з якості не включено до процедури посилання, перегляд призначений гармонізувати його. | II | |||||
| Б. V. в) Протокол управління змінами | ||||||
| Б. V. в.1. Перегляд досьє з якості для внесення змін на вимогу компетентного органу після оцінки протоколу управління змінами | ||||||
| а) внесення зміни не вимагає будь-яких додаткових супровідних даних | IА | ІБ* | ||||
| б) внесення зміни вимагає додаткових супровідних даних | IB | |||||
| в) внесення зміни для лікарського засобу біологічного/імунологічного походження | IB | |||||
| В.I.1 Зміна до короткої характеристики лікарського засобу, інструкції для медичного застосування або маркування | ||||||
| а) потребує негайного внесення, але не пізніше 30 днів | ІАнп | ІБ* | ||||
| б) потребує внесення відповідно до інформації референтного/оригінального лікарського засобу без надання жодних нових додаткових даних заявником | ІБ | |||||
| в) потребує внесення відповідно до посилань та нові додаткові дані надані заявником | ІІ | |||||
| В.I.2 Зміна у Короткій характеристиці лікарського засобу, інструкції для медичного застосування, маркуванні генеричних/комбінованих/біоподібних лікарських засобів після оцінки тієї ж зміни щодо референтного/оригінального засобу. | ||||||
| а) Впровадження змін(и), щодо яких заявник не надає жодних нових додаткових даних | IБ | |||||
| б) Впровадження змін(и), яку необхідно в подальшому обґрунтувати новими додатковими даними, які має надати заявник (наприклад, порівнянність) | II | |||||
| В.I.3 Внесення змін(и) за вимогою МОЗ України, згідно рекомендацій національних компетентних органів, міжнародних організацій, регуляторних агенцій інших країн після оцінки негайного обмеження щодо безпеки, маркування, характерного для певної фармакологічної та/або фармакотерапевтичної групи, регулярно оновлюваного звіту з безпеки, плану управління ризиком, контрольних заходів/спеціальних зобов’язань, змін для відображення короткої характеристики лікарського засобу та інструкції для медичного застосування | ||||||
| a) Впровадження узгоджених змін у формулюванні, щодо яких заявник не надає жодних нових додаткових даних | IБ | |||||
| б) Впровадження змін, які вимагають подальшого обґрунтування новими додатковими даними, що надаються заявником | ІІ | |||||
| В.I.4 Зміни, що пов’язані із значними змінами у Короткій характеристиці лікарського засобу, інструкції для медичного застосування згідно нових даних з якості, доклінічних, клінічних даних та даних фармаконагляду | ||||||
| ІІ | ||||||
| В.I.5 Зміна у правовому статусі лікарського засобу | ||||||
| а) Для генеричних/комбінованих/біоподібних лікарських засобів після зміни затвердженого правового статусу референтного/оригінального лікарського засобу | ІB | |||||
| б) Усі інші зміни правового статусу | ІІ | |||||
| В.I.6. Зміна(и) до терапевтичних показань(ня) | ||||||
| а) додання нового терапевтичного показання або зміни затвердженого показання | ІІ | |||||
| б) видалення терапевтичного показання | ІБ | |||||
| В.I.7. Видалення | ||||||
| а) лікарська форма | ІБ | |||||
| б) сила дії | ІБ | |||||
| В.I.8 Введення нової системи фармаконагляду | ||||||
| а) яка не пройшла оцінку відповідного національного компетентного органу/ЕМЕА для іншого продукту того ж заявника | ІІ | |||||
| б) яка пройшла оцінку відповідного національного компетентного органу/інших регуляторних агенцій для іншого лікарського препарату того ж заявника (*) | IБ | |||||
| В.I.9 Зміни до існуючої системи фармаконагляду, як зазначено у описі системи фармаконагляду | ||||||
| а) зміна уповноваженої особи, відповідальної за фармаконагляд | IАнп | ІБ* | ||||
| б) зміна в контактних даних уповноваженої особи, відповідальної за фармаконагляд | ІАнп | ІБ* | ||||
| в) зміна до процедури підтримки уповноваженої особи, відповідальної за фармаконагляд | ІАнп | ІБ* | ||||
| г) зміна до бази даних з безпеки (наприклад, введення нової бази даних з безпеки, включаючи перенесення зібраних даних з безпеки та/або аналіз та повідомлення до нової системи) | ІАнп | ІБ* | ||||
| г’) Зміни в контрактних домовленостях з іншими особами або організаціями, що залучені до виконання вимог з фармаконагляду та описаними в системі фармаконагляду а саме, якщо складається договір субпідряду про подання електронних повідомлень про індивідуальний випадок, пов’язаний з безпекою лікарського засобу, про ведення головних баз даних, про відстеження сигналу або про підготовку регулярно оновлюваного звіту з безпеки | ІАнп | ІБ* | ||||
| д) Виключення тем, що увійшли у письмову процедуру(и), яка описує діяльність з фармаконагляду | ІАнп | ІБ* | ||||
| е) Зміна дільниці, що проводить діяльність з фармаконагляду | ІАнп | ІБ* | ||||
| є) інші зміни до системи фармаконагляду, які не впливають на функціонування системи фармаконагляду (наприклад, зміни розташування основного сховища/архіву, адміністративні зміни, оновлення скорочень, зміни щодо назви функцій/процедур). | ІАнп | ІБ* | ||||
| з) інші зміни | IА | IБ | II | |||
* Якщо одна з умов не виконується і дану зміну неможливо віднести до змін ІІ тип
Для документів, що подавалися на реєстрацію у форматі,
|
|||
| Зміни у частині I реєстраційного досьє | 0 | Звіт експерта
0 Оновлення 0 Доповнення 0 |
|
| Зміни у частині II реєстраційного досьє | 0 | ||
| Зміни у частині III реєстраційного досьє | 0 | ||
| Зміни у частині IV реєстраційного досьє | 0 | ||
Для документів, що подавалися на реєстрацію у форматі ЗТД
|
|||
| Зміни у модулі I реєстраційного досьє | 0 | Короткий огляд
0 Резюме 0 Оновлення 0 Доповнення 0 |
|
| Зміни у модулі 2 реєстраційного досьє | 0 | ||
| Зміни у модулі 3 реєстраційного досьє | 0 | ||
| Зміни у модулі 4 реєстраційного досьє | 0 | ||
| Зміни у модулі 5 реєстраційного досьє | 0 | ||
| Інша(-і) заява(-и) (дайте коротке обґрунтування запропонованим змінам або іншим змінам, заявленим паралельно, або поновленню заяви, або розширенню заяви) |
|||
| Зміст (перерахуйте всі зміни в стислій формі) | |||
| Передумови для внесення змін та обґрунтування послідовних змін (якщо необхідно)(дайте коротке пояснення передумовам для пропонованих змін, а також обґрунтування у разі послідовних змін) | |||
| Діюча редакція* | Пропонована редакція* | ||
* Укажіть точну діючу та пропоновану редакцію або специфікацію; у разі змін, які стосуються етикетки та листка-вкладиша, підкресліть або виділіть змінені слова, указані в таблиці вище, або подайте окремим додатком.
Додайте (за необхідності) пропоновані тексти інформації про лікарський засіб (коротка характеристика лікарського засобу, інструкція для медичного застосування, етикетки) та інші матеріали, які обґрунтовують внесення змін.»
«Додаток 8
до пункту 6.1 (з) Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Коротка характеристика лікарського засобу
Коротка характеристика лікарського засобу (Summary of Product Characteristics — SPC) затверджується на бажання заявника та повинна містити таку інформацію:
1. Назва лікарського засобу до складу якої за необхідності може входити дозування та лікарська форма.
2. Якісний і кількісний склад із зазначенням діючих та тих допоміжних речовин, інформація про які є важливою для належного застосування лікарського засобу. Необхідно використовувати загальноприйняті або хімічні назви.
3. Лікарська форма.
4. Клінічна інформація:
4.1. Терапевтичні показання.
4.2. Нозологія і спосіб застосування для дорослих і, якщо необхідно, для дітей.
4.3. Протипоказання.
4.4. Особливі застереження та запобіжні заходи щодо застосування і, у випадку імунологічного лікарського засобу, будь-які особливі запобіжні заходи, що повинні бути вжиті лікарями, які працюють з цими засобами та вводять їх пацієнтам, а також усі запобіжні заходи, яких повинен дотримуватися пацієнт.
4.5. Взаємодія з іншими лікарськими засобами та інші форми взаємодії.
4.6. Застосування під час вагітності та годування груддю.
4.7. Вплив на здатність керувати транспортними засобами або працювати з іншими автоматизованими системами.
4.8. Побічні реакції.
4.9. Передозування (ознаки, термінові заходи лікування, протиотрути).
5. Фармакологічні властивості:
5.1. Фармакодинамічні властивості.
5.2. Фармакокінетичні властивості.
5.3. Доклінічні дані з безпеки.
6. Фармацевтична інформація:
6.1. Перелік допоміжних речовин.
6.2. Основні випадки несумісності.
6.3. Термін придатності, у разі необхідності вказується термін придатності після розведення лікарського засобу або після першого розкриття первинної упаковки.
6.4. Особливі запобіжні заходи при зберіганні.
6.5. Тип та вміст первинної упаковки.
6.6. Спеціальні заходи безпеки при поводженні з невикористаним лікарським засобом або відходами лікарського засобу (у разі необхідності).
7. Власник реєстраційного посвідчення.
8. Номер (-и) реєстраційного (-их) посвідчення (-нь).
9. Дата першої реєстрації або перереєстрації лікарського засобу.
10. Дата перегляду тексту короткої характеристики.
11. Для радіофармацевтичних лікарських засобів — повний докладний опис внутрішньої радіаційної дозиметрії; додаткові інструкції для приготування лікарських засобів для негайного застосування та контролю якості такого лікарського засобу і, за необхідності, максимальний термін зберігання, протягом якого будь-який проміжний препарат, наприклад елюат, або готовий до застосування лікарський засіб будуть відповідати своїм специфікаціям.»
«Додаток 9
до Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Структура короткої характеристики лікарського засобу
1. Назва лікарського засобу, дозування, лікарська форма.
2. Якісний і кількісний склад.
3. Лікарська форма.
4. Клінічна інформація.
4.1. Терапевтичні показання.
4.2. Дози та спосіб застосування.
4.2.1. Діти
4.3. Протипоказання.
4.4. Особливі застереження та запобіжні заходи щодо застосування.
4.5. Взаємодія з іншими лікарськими засобами та інші форми взаємодій.
4.6. Застосування під час вагітності та годуванні груддю.
4.7. Вплив на здатність керувати транспортними засобами або працювати з іншими автоматизованими системами.
4.8. Побічні реакції.
4.9. Передозування.
5. Фармакологічні властивості. Фармакотерапевтична група. Код АТХ.
5.1. Фармакодинамічні властивості.
5.2. Фармакокінетичні властивості.
5.3. Доклінічні дані з безпеки.
6. Фармацевтична інформація
6.1. Допоміжні речовини.
6.2. Основні випадки несумісності.
6.3. Термін придатності.
6.4. Особливі запобіжні заходи при зберіганні.
6.5. Тип та вміст первинної упаковки.
6.6. Спеціальні заходи безпеки при поводженні з невикористаним лікарським засобом або відходами лікарського засобу (у разі необхідності).
7. Власник реєстраційного посвідчення.
8. Номер реєстраційного посвідчення.
9. Дата першої реєстрації лікарського засобу.
10. Дата останнього перегляду.».
«Додаток 11
до Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Інструкція для медичного застосування, що супроводжує лікарський засіб
Інструкція для медичного застосування, що супроводжує лікарський засіб, повинна бути складена відповідно до короткої характеристики лікарського засобу.
1. Упаковку лікарського засобу повинна супроводжувати інструкція для медичного застосування, якщо викладена в ній інформація не наведена безпосередньо на вторинній або первинній упаковці.
2. Текст Інструкції для медичного застосування лікарського засобу повинен бути викладений чітко, виразно, українською або іншою мовою міжнаціонального спілкування, яка визначена законодавством України. Це не виключає можливості надання відомостей декількома мовами, одна з яких мова виробника, за умови, що в текстах усіма мовами буде наведена ідентична інформація.
3. Стосовно певних лікарських засобів може бути відмінена вимога щодо необхідності вказувати в Інструкції для медичного застосування деякі дані українською або іншою мовою міжнаціонального спілкування в Україні за умови, якщо лікарський засіб не призначений для самостійного застосування пацієнтом.
4. При друкуванні Інструкцій для медичного застосування повинен використовуватися шрифт не менше 9 пунктів Дідо з відстанню між рядками не менше 1 мм.
5. Інструкція для медичного застосування повинна містити таку інформацію:
5.1. Назва лікарського засобу;
5.2. Якісний та кількісний склад з вказанням активних субстанцій і тих допоміжних речовин, інформація про які важлива для правильного застосування лікарського засобу (використовується загальноприйнята назва або хімічний опис).
Однак, якщо лікарський засіб призначений для парентерального введення або є лікарським засобом для місцевого застосування, або для офтальмології, повинні бути вказані всі допоміжні речовини;
5.3. Лікарська (фармацевтична) форма;
5.4. Фармакотерапевтична група. Код АТХ.
5.5. Клінічні характеристики:
5.5.1. Терапевтичні показання;
5.6. Дози і спосіб уведення для дорослих
5.6.1. Діти;
5.7. Протипоказання;
5.8. Особливі заходи безпеки (за необхідності), яких треба дотримуватися особам, які працюють з лікарськими засобами та вводять їх пацієнтам, а також усі заходи безпеки, яких необхідно дотримуватися пацієнту;
5.9. Лікарські взаємодії та інші форми взаємодій;
5.10. Застосування у період вагітності або годування груддю;
5.11. Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами;
5.12. Побічні реакції (частота та вираженість за умови їх наявності);
5.13. Передозування (симптоми, невідкладні заходи лікування та антидоти);
*5.14. Фармакологічні властивості та, якщо така інформація корисна для лікування, фармакокінетичні характеристики;
5.15. Фармацевтичні характеристики:
основні фізико-хімічні властивості
5.16. Несумісність (основні види) за наявності інформації;
5.17. Спеціальні перестороги;
5.18. Термін придатності (за необхідності вказується термін зберігання після підготовки лікарського засобу для безпосереднього застосування, наприклад після розчинення або після першого відкриття первинної упаковки);
5.19. Особливі заходи безпеки при зберіганні;
5.20. Тип і місткість первинної упаковки;
5.21. Особливі заходи безпеки, якщо це необхідно, при поводженні з невикористаними лікарськими засобами або відходами лікарських засобів;
5.22. Назва (найменування) та місцезнаходження виробника, а за бажанням заявника й назва (найменування) і місцезнаходження заявника;
5.23. Категорія відпуску лікарського засобу, установлена Центром, відповідно до критеріїв, затверджених МОЗ України;
5.24. Дата останнього перегляду інструкції для медичного застосування;
У розділі «Дата останнього перегляду» в інструкції для медичного застосування зазначається дата наказу МОЗ України (при реєстрації)/дата його подовження (при перереєстрації)/дата перегляду тексту інструкції для медичного застосування (при внесенні змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення)
5.25. Для радіофармацевтичних лікарських засобів — повний докладний опис внутрішньої радіаційної дозиметрії, а також додаткові докладні інструкції щодо приготування для негайного застосування та контролю якості таких лікарських засобів, а за необхідності — максимальний термін зберігання, протягом якого будь-який проміжний лікарський засіб, такий як елюат, або готовий для застосування фармацевтичний лікарський засіб, буде відповідати власним специфікаціям.
Крім того, інструкція для медичного застосування радіофармацевтичного лікарського засобу повинна містити опис усіх заходів безпеки, яких необхідно дотримуватися медичним працівникам та пацієнтам під час приготування та використання лікарського засобу, а також спеціальні заходи безпеки з утилізації упаковки або її невикористаного вмісту.
7. В інструкції для медичного застосування можуть бути розміщені символи або піктограми, які дають змогу пояснити інформацію, надану в ній, а також інша інформація, яка відповідає короткій характеристиці лікарського засобу і є корисною для санітарної просвіти, за винятком будь-яких елементів, які сприяють просуванню лікарського засобу на ринку.
8. Інструкція для медичного застосування традиційного лікарського засобу рослинного походження повинна містити вказівки на те, що:
лікарський засіб є звичайним лікарським засобом рослинного походження для використання відповідно до показань, підтверджених тривалим застосуванням;
користувач повинен проконсультуватися у лікаря, якщо симптоми захворювання не зникли під час застосування лікарського засобу або спостерігаються побічні реакції, не вказані в інструкції для медичного застосування лікарського засобу.
9. Інструкція для медичного застосування гомеопатичних лікарських засобів, які відповідають умовам, викладеним у додатку 13 до цього Порядку, повинна містити:
формулювання «гомеопатичний лікарський засіб без затверджених терапевтичних показань для застосування»;
застереження для споживача про необхідність консультації з лікарем, якщо симптоми зберігаються при застосуванні лікарського засобу.
10. Власник реєстраційного посвідчення повинен гарантувати, що, у разі необхідності, зможе надати організаціям пацієнтів з обмеженим зором текст інструкції для медичного застосування у доступному для них форматі.
«Додаток 12
до пункту 6.6 Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Структура реєстраційного досьє для традиційних лікарських засобів рослинного походження та лікарських засобів, що виробляються згідно із затвердженим прописом
Модуль 1. Адміністративна інформація
1.1. Зміст.
1.2. Форма заяви.
1.3. Коротка характеристика лікарського засобу, маркування та
інструкція для медичного застосування:
1.3.1. Коротка характеристика лікарського засобу.
1.3.2. Маркування.
1.3.3. Інструкція для медичного застосування.
1.3.4. Макети та зразки.
1.3.5. Коротка характеристика лікарського засобу, уже
затверджена в країні виробника/заявника.
1.4. Інформація про незалежних експертів:
1.4.1. Інформація про експерта з якості.
1.4.2. Інформація про експерта з доклінічних даних.
1.4.3. Інформація про експерта з клінічних даних.
1.5. Спеціальні вимоги до різних типів заяв.
Додаток до Модуля 1. Оцінка небезпеки для довкілля.
Модуль 3. Якість.
Хімічна, фармацевтична та біологічна інформація про лікарські засоби, що містять хімічні та/або біологічні діючі речовини
3.1. Зміст.
3.2. Основні дані.
3.2.S. Діюча(і) речовина(и).
3.2.S.1. Загальна інформація:
3.2.S.1.1. Назва.
3.2.S.1.2. Структура.
3.2.S.1.3. Загальні властивості.
3.2.S.2. Процес виробництва діючої(их) речовини(н):
3.2.S.2.1. Виробник(и).
3.2.S.2.2. Опис виробничого процесу та його контролю.
3.2.S.2.3. Контроль матеріалів.
3.2.S.2.4. Контроль критичних стадій і проміжної продукції.
3.2.S.2.5. Валідація процесу та/або його оцінка.
3.2.S.2.6. Розробка виробничого процесу.
3.2.S.3. Опис характеристик діючої(их) речовини(н):
3.2.S.3.1. Доказ структури та інші характеристики.
3.2.S.3.2. Домішки.
3.2.S.4. Контроль діючої(их) речовини(н):
3.2.S.4.1. Специфікація.
3.2.S.4.2. Аналітичні методики.
3.2.S.4.3. Валідація аналітичних методик.
3.2.S.4.4. Аналізи серій.
3.2.S.4.5. Обґрунтування специфікації.
3.2.S.5. Стандартні зразки або лікарські засоби.
3.2.S.6. Система упаковка/укупорка.
3.2.S.7. Стабільність:
3.2.S.7.1. Резюме щодо стабільності та висновки.
3.2.S.7.2. Протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності.
3.2.S.7.3. Дані про стабільність.
3.2.Р. Готовий лікарський засіб:
3.2.Р.1. Опис і склад лікарського засобу.
3.2.Р.2. Фармацевтична розробка:
3.2.Р.2.1. Компоненти лікарського засобу.
3.2.Р.2.1.1. Діюча(і) речовина(и).
3.2.Р.2.1.2. Допоміжні речовини.
3.2.Р.2.2. Лікарський засіб.
3.2.Р.2.2.1. Розробка складу.
3.2.Р.2.2.2. Надлишки.
3.2.Р.2.2.3. Фізико-хімічні та біологічні властивості.
3.2.Р.2.3. Розробка виробничого процесу.
3.2.Р.2.4. Система упаковка/укупорка.
3.2.Р.2.5. Мікробіологічні характеристики.
3.2.Р.2.6. Сумісність.
3.2.Р.3. Процес виробництва лікарського засобу:
3.2.Р.3.1. Виробник(и).
3.2.Р.3.2. Склад на серію.
3.2.Р.3.3. Опис виробничого процесу та контролю процесу.
3.2.Р.3.4. Контроль критичних стадій і проміжної продукції.
3.2.Р.3.5. Валідація процесу та/або його оцінка.
3.2.Р.4. Контроль допоміжних речовин:
3.2.Р.4.1. Специфікації.
3.2.Р.4.2. Аналітичні методики.
3.2.Р.4.3. Валідація аналітичних методик.
3.2.Р.4.4. Обґрунтування специфікацій.
3.2.Р.4.5. Допоміжні речовини людського або тваринного походження.
3.2.Р.4.6. Нові допоміжні речовини.
3.2.Р.5. Контроль лікарського засобу:
3.2.Р.5.1. Специфікація(ї).
3.2.Р.5.2. Аналітичні методики.
3.2.Р.5.3. Валідація аналітичних методик.
3.2.Р.5.4. Аналізи серій.
3.2.Р.5.5. Характеристика домішок.
3.2.Р.5.6. Обґрунтування специфікації(й).
3.2.Р.6. Стандартні зразки та лікарські засоби.
3.2.Р.7. Система упаковка/укупорка.
3.2.Р.8. Стабільність:
3.2.Р.8.1. Резюме щодо стабільності та висновки.
3.2.Р.8.2. Протокол післяреєстраційного вивчення стабільності та зобов’язання щодо стабільності.
3.2.Р.8.3. Дані про стабільність.
3.2.А. Додаток:
3.2.А.1. Приміщення та обладнання.
3.2.А.2. Оцінка безпечності щодо сторонніх агентів.
3.2.А.3. Нові допоміжні речовини.
3.2.R. Додаткова інформація.
3.3. Посилання на джерела літератури.
Примітка. У разі проведення державної реєстрації лікарських засобів, що виробляються згідно із затвердженим прописом Заявник надає листа-підтвердження щодо того, що склад, виробництво та контроль лікарського засобу відповідають пропису, наведеному у Переліку лікарських засобів, що виробляються згідно із затвердженими прописами.
Для традиційних лікарських засобів рослинного походження повинні виконуватись наступні вимоги:
а) показання для їх застосування мають відповідати традиційним лікарським засобам рослинного походження, які завдяки своєму складу призначені для використання без контролю з боку лікаря;
б) вони повинні застосовуватися в певній концентрації та певному дозуванні;
в) мають бути лікарськими засобами для перорального, зовнішнього або інгаляційного застосування;
г) період традиційного застосування має становити понад 30 років у світі і понад 15 років в Україні;
ґ) є достатня кількість даних про традиційне використання лікарського засобу, зокрема його використання у звичайних умовах не завдає шкоди, фармакологічна ефективність лікарського засобу доведена тривалим використанням і досвідом. Наявність у складі лікарського засобу рослинного походження вітамінів та мінералів, безпечність яких представлена задокументованими доказами, не повинна бути перешкодою для реєстрації таких лікарських засобів, як традиційних лікарських
засобів рослинного походження за умови, що дія вітамінів або мінералів є допоміжною або здатна підсилювати дію активних інгредієнтів рослинного походження щодо заявлених показань.
У разі проведення державної реєстрації традиційних лікарських засобів, за вимогою Центру Заявник повинен надати додаткові матеріали, які підтверджують ефективність та безпечність цього лікарського засобу.»
«Додаток 13
до Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Інструкція
для медичного застосування лікарського засобу
Назва лікарського засобу (укр.)
Склад:
діюча (і) речовина (и): (міжнародна або в разі ії відсутності скорочена хімічна назва)
склад на 1 одиницю лікарської форми
допоміжні речовини:
Лікарська форма.
Фармакотерапевтична група. Код АТХ.
Клінічні характеристики.
Показання.
Спосіб застосування та дози.
Діти.
Протипоказання.
Особливі заходи безпеки. (при наявності)
Особливості застосування.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Застосування у період вагітності або годування груддю.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Побічні реакції.
Передозування.
Фармакологічні властивості.
Фармакодинаміка.Фармакокінетика.
Фармацевтичні характеристики.
Основні фізико-хімічні властивості
Несумісність. (за наявності)
Термін придатності.
Умови зберігання.
Упаковка.
Категорія відпуску.
Виробник/Заявник
Місцезнаходження.
Дата останнього перегляду.
«Додаток 16
до Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Перелік документів для проведення експертизи матеріалів для державної перереєстрації лікарського засобу
1. Супровідний лист.
2. Повний зміст.
3. Заява про проведення експертизи матеріалів на лікарські засоби, які подаються для державної перереєстрації (додатки 14, 17).
4. Інформація про уповноважених осіб (кваліфікований працівник заявника в Україні для здійснення фармаконагляду);
5. Висновки про відповідність належній виробничій практиці (GMP) (зроблені уповноваженим органом не пізніше як за три роки);*
6. Копія ліцензії на виробництво або документа, виданого уповноваженим органом країни виробника, який свідчить про наявність у виробника права на здійснення такого виробництва;
7. Перелік країн, де препарат представлений на ринку, якщо такі є;*
8. Перелік одержаних за останні 5 років в Україні рекламацій на препарат (у хронологічному порядку) з докладним аналізом причин, що слугували підставами для видачі рекламацій, та заходів, вжитих заявником для запобігання їх у майбутньому.
9. Перелік гарантій заявника та зобов’язань, наданих після реєстрації/перереєстрації (у хронологічному порядку) із зазначенням характеру, стану виконання, дати подання та дати, коли проблема була вирішена (рекомендації щодо післяреєстраційних досліджень, усунення будь-яких недоліків, що надавалися контролюючими та іншими органами тощо).
10. Переглянутий перелік усіх гарантій та зобов’язань, які залишилися, і підписаний лист зобов’язань (за необхідності).
11. Проекти оновлених короткої характеристики препарату*, упаковки (етикетки) та інструкції для медичного застосування (додатково надається на електронному носії). Діюча редакція короткої характеристики препарату українською чи російською та англійською мовами (з відповідним перекладом) або, якщо необхідно, пропонована редакція короткої характеристики препарату українською, російською чи англійською мовами (з відповідним перекладом) з усіма змінами, чітко виділеними*;
12. Затверджені при реєстрації та оновлені методи контролю якості готового лікарського засобу.
13. Відомості про технологію виробництва та інформація щодо відсутності/наявності змін у технологічному процесі.
14. Звіт за результатами післяреєстраційного нагляду (регулярно оновлюваний звіт з безпеки) та/або узагальнюючий звіт.*
15. Зведені дані заявника про стан безпеки медичного застосування лікарського засобу в Україні за період дії останнього реєстраційного посвідчення згідно з формою, передбаченою вимогами чинного законодавства.
Примітка. Якщо при поданні заяви для державної перереєстрації лікарського засобу декларуються зміни до реєстраційних матеріалів, які не були схвалені у встановленому порядку під час дії реєстраційного посвідчення, заявник разом з заявою для державної перереєстрації подає заяву про внесення змін до реєстраційних матеріалів та документи, які обґрунтовують внесення таких змін.»
«Додаток 20
до Порядку проведення експертизи матеріалів на лікарські засоби,
що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів
про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
1. Загальні положення
1.1. Концепція біоеквівалентності є основною. Мета встановлення біоеквівалентності полягає в демонстрації еквівалентності біофармацевтичної якості між генеричним і референтним лікарськими засобами.
1.2. Для оцінки еквівалентності застосовуються такі методи:
порівняльні фармакокінетичні дослідження, під час яких вимірюється концентрація діючої речовини та/або її метаболіту (-ів) у доступній біологічній рідині, а саме: крові, плазмі, сироватці або сечі для отримання фармакокінетичних показників типу АUC і Смакс, які відображають системну дію;
порівняльні фармакодинамічні дослідження за участю людини;
порівняльні клінічні дослідження;
дослідження еквівалентності in vitro.
1.3. Прийнятність для доведення еквівалентності кожного з зазначених у пункті 1.2 методів визначається на підставі комплексної експертизи факторів, які включають характеристики діючої речовини та форму лікарського засобу.
1.4. При виборі методів доведення еквівалентності, за можливості заявника, застосовують такі підходи:
— якщо можливо отримати вимірювані концентрації діючої речовини в наявній біологічній рідині, такій як плазма, проводяться порівняльні фармакокінетичні дослідження in vivo на людяї;
— якщо неможливо отримати вимірювані концентрації діючої речовини у відповідній біологічній рідині, проводяться порівняльні фармакодинамічні дослідження на людях;
— якщо неможливо визначити фармакокінетичний профіль, знайти прийнятні фармакодинамічні кінцеві точки, можуть застосовуватися порівняльні клінічні дослідження;
— для лікарських засобів орального застосування з негайним вивільненням може застосовуватися дослідження еквівалентності in vitro в разі виконання всіх відповідних вимог до процедури біовейвер.
2. Відсутність необхідності проведення дослідження еквівалентності
2.1. Генеричні лікарські засоби, наведені у цьому розділі, вважаються еквівалентними при виконанні зазначених умов і не потребують досліджень in vivo за умов дотримання вимог до виробництва та стандартів якості, встановлених чинним законодавством України та міжнародними нормами:
а) лікарські засоби, які застосовуються парентерально (наприклад, внутрішньовенно, підшкірно або внутрішньом’язово) у вигляді водних розчинів, котрі містять таку саму діючу речовину і в тій самій молярній концентрації, що і референтний препарат, і з такими ж самими або подібними допоміжними речовинами в порівнюваних з референтним препаратом концентраціях. Деякі допоміжні речовини (наприклад, буферні розчини, консерванти та антиоксиданти) можуть відрізнятися за умови доведення, що у даних концентраціях вони не впливають на безпечність та/або ефективність лікарського засобу;
б) лікарські засоби, які є фармацевтично еквівалентними і є розчинами для орального застосування (наприклад, сиропи, еліксири і настойки), котрі містять діючу речовину в тій же молярній концентрації, що і референтний препарат, і містять, в основному, такі ж допоміжні речовини в порівнюваних концентраціях. При експертизі звертається увага на концентрації та властивості допоміжних речовин, про які відомо, що вони впливають на проходження шлунково-кишковим трактом (ШКТ), проникність у ШКТ і таким чином абсорбцію або стабільність діючої речовини в ШКТ;
в) лікарські засоби, які є фармацевтично еквівалентними і знаходяться у формі порошків для приготування розчинів, і якщо розчин відповідає вищенаведеним вимогам «а» або «б»;
г) лікарські засоби, які є фармацевтично еквівалентними і є газами;
д) лікарські засоби, які є фармацевтично еквівалентними і є вушними або очними лікарськими засобами, виготовленими у вигляді водних розчинів, котрі містять таку ж саму діючу речовину (-и) в такій самій молярній концентрації (-ях) і, по суті, такі ж допоміжні речовини у порівнюваних концентраціях, що й референтний препарат. Деякі допоміжні речовини (наприклад, консерванти, буфери, речовини, які коригують густину, або згущувачі) можуть відрізнятися за умови доведення, що при їх використанні не передбачається вплив на безпечність та/або ефективність лікарського засобу;
е) лікарські засоби, які є фармацевтично еквівалентними і є препаратами місцевої дії, виготовленими у вигляді водних розчинів, котрі містять таку ж діючу речовину (-и) в такій самій молярній концентрації (-ях) і, по суті, такі ж самі допоміжні речовини у порівнюваних концентраціях, що і референтний препарат;
ж) лікарські засоби, які є фармацевтично еквівалентними і є водними розчинами у формі інгаляційно-розпилюючих небулайзером препаратів, або назальні спреї, які застосовуються за допомогою практично однакових пристроїв і котрі містять таку ж саму діючу речовину (-и) в такій самій молярній концентрації (-ях) і, по суті, такі ж допоміжні речовини у порівнюваних концентраціях, що і референтний препарат. Лікарський засіб може містити інші допоміжні речовини за умови доведення, що при їх використанні не передбачається вплив на безпечність та/або ефективність лікарського засобу.
з) лікарські засоби, які є фармацевтично еквівалентними і є водними розчинами системної дії для ректального або вагінального застосування, котрі містять діючу речовину в тій же молярній концентрації, що і референтний препарат, і містять, в основному, такі ж допоміжні речовини в порівнюваних концентраціях.
2.2. Для випадків «б», «в», «ґ», «д» «е», «ж» та «з» заявник повинен довести, що допоміжні речовини у фармацевтично еквівалентному препараті є, по суті, такі самі і в порівнюваних концентраціях, як і в референтному препараті або, за можливості (наприклад, у випадках «ґ» і «е»), якщо при їх використанні не передбачається вплив на безпечність та/або ефективність препарату. У випадку, коли заявник не може надати таку інформацію і не має доступу до відповідних даних, він повинен провести відповідні дослідження для доказу відсутності впливу різних допоміжних речовин або допоміжних пристроїв на безпечність та/або ефективність лікарського засобу.
3. Необхідність проведення дослідження еквівалентності in vivo
3.1. Дослідження еквівалентності in vivo проводять у випадку, коли існує ризик того, що можливі відмінності у біодоступності можуть призвести до терапевтичної нееквівалентності лікарського засобу. Наприклад, для:
а) Оральних препаратів системної дії з негайного вивільнення, якщо до них застосовані один або декілька таких критеріїв:
застосування лікарського засобу для невідкладної допомоги;
вузький спектр терапевтичної дії (межі ефективність/безпечність), крутий нахил кривої доза-відклик;
документально підтверджені проблеми щодо біодоступності або біонееквівалентності, які пов’язані з діючою речовиною або її формами (що не мають відношення до розчинення);
дані стосовно того, що на біоеквівалентність може впливати поліморфізм діючої речовини, допоміжні речовини та/або технологічні процеси.
б) Препаратів системної дії, що не призначені для орального або парентерального застосування (таких, як трансдермальні пластирі, супозиторії, нікотинові жувальні гумки, гелі тестостерону та трансдермальні контрацептиви).
в) Препаратів системної дії з модифікованим вивільненням.
г) Препаратів системної дії із фіксованою комбінацією діючих речовин, у яких, як мінімум, для однієї діючої речовини потрібне проведення дослідження in vivo.
д) Препаратів несистемної дії (наприклад, для орального, назального, офтальмологічного, дерматологічного, ректального або вагінального застосування), без системної абсорбції, що не є розчинами. У такому разі еквівалентність доводять шляхом проведення, наприклад, порівняльних клінічних або фармакодинамічних, дерматофармакокінетичних досліджень та/або досліджень in vitro.
е) Генеричний препарат містить інший ефір, складний ефір, ізомер, суміш ізомерів, комплекс або похідне речовини діючої речовини, на відміну від референтного продукту, оскільки ці відмінності можуть привести до різної біодоступності.
4. Особливі вимоги для різних лікарських форм:
4.1. Таблетки, що диспергуються в ротовій порожнині.
4.1.1. Якщо може бути продемонстровано, що діюча речовина, не абсорбується в ротовій порожнині, а проковтується та абсорбується через шлунково-кишковий тракт, у такому випадку лікарський препарат може розглядатися для процедури біовейвер на підставі БСК. Якщо не можливо продемонструвати, що діюча речовина, не абсорбується в ротовій порожнині, біоеквівалентність необхідно оцінювати в дослідженнях in vivo.
4.1.2. Якщо таблетки, що диспергуються в ротовій порожнині є розширенням до іншого перорального складу, рекомендується 3-фазове дослідження з або без супутнього прийняття рідини. Проте якщо біоеквівалентність між лікарським засобом, що приймається без води і референтним складом з водою демонструється в 2-фазовому дослідженні, біоеквівалентність лікарського препарату, що приймається з водою, може передбачатися.
4.1.3. Якщо таблетки, що диспергують в ротовій порожнині є генериком відносно вже затвердженого референтного препарату, наступні рекомендації щодо дизайну використовуються:
- якщо референтний лікарський засіб може прийматися з або без води, біоеквівалентність повинна демонструватися без води, оскільки ця умова має найбільша схожість з передбачуваним використанням складу. Це особливо важливо, якщо речовина може розчинятися і частково абсорбуватися в ротовій порожнині. Якщо біоеквівалентність демонструється, коли лікарський засіб приймається без води, може передбачатися біоеквівалентність, коли склад приймається з водою.
- якщо референтний лікарський засіб приймається лише одним способом (наприклад, лише з водою) біоеквівалентність має бути продемонстрована в цьому випадку (у звичайному двохфазовому перехресному дизайні).
- якщо референтний лікарський засіб приймається лише одним способом (наприклад, лише з водою) і продукт тестування призначений для додаткових умов введення (наприклад, без води), звичайний і новий метод необхідно порівняти з референтним при звичайному способі застосування (дизайн з 3 видами лікуваннями, 3 фазовий, 6 послідовний).
4.1.4. У дослідженнях, оцінюючих таблетки, що диспергуються в ротовій порожнині без води, рекомендується зволожувати ротову порожнину шляхом ковтання 20 мл води безпосередньо перед вживанням лікарського засобу. Не рекомендується приймати рідину раніше, ніж за годину після вживання.
4.1.5. Інші пероральні склади, такі як плівки, що диспергують в ротовій порожнині, пігулки або плівки для повільного розчинення в щоковій кишені, сублінгвальні пігулки і жувальні пігулки можуть використовуватися точно так як і таблетки, що диспергують в ротовій порожнині. Дослідження біоеквівалентності повинні проводитися згідно з рекомендованим використанням лікарського препарату.
4.2. Пероральні розчини
4.2.1. Якщо допоміжні речовини можуть вплинути на транзит в шлунково-кишковому тракті (наприклад, сорбітол, манітол, тощо), абсорбцію (наприклад, сурфактанти або допоміжні речовини, які можуть вплинути на транспортний білок), розчинність in vivo (наприклад, со-розчинники) або in vivo стабільність діючої речовини, дослідження біоеквівалентності повинне проводитися, якщо відмінності в кількості цих допоміжних речовин не можуть бути адекватно обґрунтовано шляхом посилання на інші дані.
4.2.2. У тих випадках, якщо генеричний лікарський препарат є пероральним розчином, який має бути біоеквівалентним до іншої лікарської форми з негайним вивільненням для перорального вживання, потрібні дослідження по біоеквівалентності.
4.3. Комбіновані препарати генерики
Залежить від типа комбінації:
4.3.1. Лікарський препарат є генериком до існуючого референтного препарату, потрібні дослідження по біоеквівалентності. Процедура біовейвер може бути застосована для твердих оральних лікарських форм з негайним вивільненням, коли для всіх діючих речовин комбінації можливо проведення процедури та генеричний лікарський препарат відповідає вимогам цієї процедури за найгіршим випадком.
4.3.2. Комбінація містить відомі діючі речовини, які використовувались раніше одночасно при тих же дозах як і в комбінації, але у вигляді окремих препаратів. У цих випадках слід продемонструвати біоеквівалентність між референтними препаратами індивідуальних монокомпонентів і генеричним препаратом (комбінацією).
4.4. Парентеральні розчини
4.4.1. Якщо будь-які допоміжні речовини взаємодіють з діючою речовиною (наприклад, утворюють комплекс) або інакше впливають на розподіл діючої речовини, потрібне дослідження біоеквівалентності.
4.4.2. Для парентеральних лікарських засобів, що вводяться внутрішньом’язово або підшкірно потрібне дослідження біоеквівалентності, якщо обидва препарати не містять ті ж допоміжні речовини в дуже схожій кількості та коли допоміжні речовини впливають на в’язкість.
4.4.3. Ліпосомні, міцелярні та емульсійні лікарські форми для внутрішньовенного введення.
4.4.3.1. Емульсії: емульсії звичайно потребують дослідження біоеквівалентності.
Проте можливі випадки, коли можливо не проводити дослідження біоеквівалентності:
(а) лікарський засіб не предназначений для контролю вивільнення чи розподілу.
(б) метод і швидкість введення являється такою ж, як і в затвердженого в даний час лікарського засобу.
В цих випадках склад повинен бути якісно та кількісно однаковим із затвердженою в даний час емульсією, і задовільні дані необхідно представляти для демонстрації дуже подібних фізико-хімічних характеристик, включаючи розмір розподілу дисперсної рідкої фази і підтверджувати іншим характеристиками емульсії, які вважаються значимими, наприклад, поверхневі характеристики, такі як дзета-потенціал та реологічні властивості.
4.4.3.2. Ліпіди для внутрішньовенного парентерального живлення можуть не потребувати дослідження біоеквівалентності, якщо представлені задовільні дані для демонстрації порівняльних фізико-хімічних характеристик. Відмінність в складі може бути обґрунтована, враховуючи основну властивість і терапевтичні цілі таких лікарських форм.
4.4.3.3. Міцелле формуючі склади: міцельні розчини для внутрішньовенного введення можуть вважатися «комплексними» розчинами і тому звичайно потребують дослідження біоеквівалентності.
Міцелле формуючі склади можуть не потребувати дослідження біоеквівалентності, якщо:
(а) відбувається швидкий розподіл міцел при розведенні, і лікарський засіб не предназначений для контролю вивільнення чи розподілу
(б) метод і швидкість введення є такою, як і у затвердженого в даний момент продукта
(в) допоміжні речовини не впливають на розподіл лікарського засобу.
В цих випадках склад міцел інфузії безпосередньо перед вживанням повинен бути якісно та кількісно однаковим із затвердженим на даний час, і необхідно представляти задовільні дані для демонстрації подібності фізико-хімічних характеристик. Наприклад, критичні міцелові концентрації, здатність складу до розчинення (такі як максимальна додаткова концентрація), вільна і зв’язана діюча речовина та розмір міцел.
Це також застосовується у випадку незначних змін в кількісному чи якісному складі, при умові, що не включаються будь-які зміни в кількості чи типі сурфактантів.
4.4.4. Лікарські форми з модифікованим вивільненням системної дії
4.4.4.1. Лікарські форми з модифікованим вивільненням системної дії і трансдермальні лікарські форми.
Потребують дослідження біоеквівалентності.
5. Умови проведення процедури біовейвер.
5.1. Введення
Використання процедури біовейвер обмежене швидко розчинними діючими речовинами, з відомою абсорбцією у людини, які не мають вузького терапевтичного індексу та критичного застосування.
Концепція застосовується до твердих лікарських засобів з негайним вивільненням для перорального вживання і системної дії, що мають одну і ту ж лікарську форму. Процедура біовейвер не застосовується для сублінгвальних, буккальних лікарських препаратів і препаратів з модифікованим вивільненням. Для лікарських препаратів, що диспергують в ротовій порожнині підхід біовейвера на підставі БСК може застосовуватися лише тоді, коли абсорбція в ротовій порожнині може бути виключена.
5.2. Загальні вимоги
Біовейвер на підставі БСК застосовується для лікарського засобу з негайним вивільненням, якщо:
було доведено, що діюча речовина, проявляє швидку розчинність і повну абсорбцію (клас I за БСК)
дуже швидкі (’85% в межах 15 хвилин) або швидкі (85% в межах 30 хвилин) характеристики розчинення in vitro генеричного і референтного препарату були продемонстровані
допоміжні речовини, які можуть вплинути на біодоступність, є якісно і кількісно подібними. В основному використання подібних допоміжних речовин в схожих кількостях є переважним.
Біовейвер на підставі БСК також застосовується для лікарського засобу з негайним вивільненням, якщо:
- було доведено, що діюча речовина, проявляє швидку розчинність і обмежену абсорбцію (клас III за БСК)
- дуже швидкі (’85% в межах 15 хвилин) характеристики розчинення in vitro генеричного і референтного препаратів були продемонстровані
- допоміжні речовини, які можуть вплинути на біодоступність є якісно і кількісно подібними і інші допоміжні речовини є якісно подібними і кількісно дуже схожими.
Біовейвер на підставі БСК також застосовується для лікарського засобу з негайним вивільненням, якщо:
було доведено, що діюча речовина, проявляє швидку розчинність при рН 6,8 та повну абсорбцію (клас II за БСК)
швидкі (’85% в межах 30 хвилин) характеристики розчинення in vitro генеричного і референтного препаратів були продемонстровані при рН 6,8
профілі розчинення генеричного і референтного препаратів при рН 1,2 та 4,5 показали подібність
допоміжні речовини, які можуть вплинути на біодоступність є якісно і кількісно подібними і інші допоміжні речовини є якісно подібними і кількісно дуже схожими.
В основному ризики невідповідного рішення щодо можливості проведення процедури біовейвер мають бути критичніше розглянуті для лікарських препаратів, діюча речовина яких відноситься до ІІ та ІІІ класів за БСК (наприклад, абсорбція специфічна для певної ділянки, ризик взаємодії транспортних протеїнів в ділянці абсорбції, склад допоміжних речовин і терапевтичні ризики). Для лікарських препаратів, діюча речовина яких відноситься до ІІ класу за БСК може запроваджуватися дослідження еквівалентності in vitro як альтернатива дослідженню біоеквівалентності in vivo у разі наявності монографії «Біовейвер» для твердої дозованої форми системної дії орального застосування Міжнародної фармацевтичної організації (FIP) та/або рекомендацій WHO.
5.3. Розчинність
Профіль рН-розчинності діючої речовини, повинен визначатися і розглядатися. Діюча речовина, вважається швидко розчинною, якщо найвища разова доза, яка застосовується повністю розчиняється в 250 мл буферного розчину в межах рН 1-6.8 при температурі 37±1оС. Ця демонстрація вимагає дослідження, принаймні, в трьох буферних розчинах в межах цього діапазону (переважно при рН 1.2, 4.5 і 6.8) і на додаток при рКа (негативний логарифм константи іонізації кислоти), якщо він в межах вказаного рН діапазону. Проведення дослідження, для кожного рН, необхідно для досягнення чіткої класифікації розчинності використовуємої діючої речовини. Розчин рН має бути перевірений до і після додавання діючої речовини, в буфер. Рекомендується вивчення розчинності діючої речовини проводити в трьох повторах при кожному із вибраних значень рН для кожного заявленого виробника діючої речовини. Використання ультразвуку неприємно.
5.4. Абсорбція
Демонстрація повної абсорбції у людини є переважною для проведення процедури біовейвер. З цією метою вважається, що повна абсорбція має бути встановлена, якщо вимірюваний рівень абсорбції складає більше 85%. Повна абсорбція в основному пов’язана з високою проникністю. Повна абсорбція препарату має бути обґрунтовано на підставі надійних досліджень у людини.
Дані досліджень абсолютної біодоступності або балансу маси можуть використовуватися для підтвердження цього твердження.
Якщо дані з досліджень балансу маси використовуються для підтвердження повної абсорбції, повинно гарантуватися, що метаболіти, які враховуються при визначенні абсорбованої фракції, формуються після абсорбції.
Дослідження проникності in vitro (in vivo або in situ кишкова перфузія з використанням моделей тварин; — in vitro проникнення крізь ізольовані тканини тварин або моношар культури епітеліальних клітин), можуть також вважатися підтверджуючими для даних in vivo.
5.5. Лікарський препарат
Вивчення кінетики вивільнення діючої речовини генеричного та референтного лікарських засобів проводять у порівнянні з референтним препаратом за таких умов:
прилади — прилад із кошиком, що обертається
(100 об/мін), або прилад із лопаттю (50 об/мін);
— об’єм середовища розчинення — 900 мл або менше, але не менше 500 мл;
— температура середовища розчинення — (37±0.5) °С;
— середовище розчинення:
— розчин кислоти хлористоводневої р Н 1.2;
—буферний розчин рН 4.5;
— фосфатний буферний розчин рН 6.8;
— кількість досліджуваних зразків — 12 для досліджуваного препарату і 12 для референтного препарату;
— точки контролю — не менше 3 точок (не враховуючи нульову), а саме: через 10 хв, 15хв, 20 хв, 30 хв та 45 хв, причому точка «15 хв» є визначальною;
— буфер: рН 1.0-1.2 (зазвичай 0.1 N HCL або штучний шлунковий сік без ферментів), рН 4.5 і рН 6.8 (або штучна кишкова рідина без ферментів); (рН повинен гарантуватися впродовж всього дослідження; рекомендуються буферні розчини Ph. Eur.)
— інші умови: відсутність сурфактанту; в разі желатинових капсул або таблеток з желатиновим покриттям використання ферментів може бути допустимим.
5.6. Оцінка результатів розчинення in vitro
Лікарські засоби вважаються «дуже швидко» розчинними, якщо більше 85% вказаної кількості розчиняється протягом 15 хвилин. У випадках, якщо це гарантується для генеричного і референтного лікарських засобів, схожість профілів розчинення може бути прийняте, що продемонстроване без будь-якого математичного обчислення.
Відсутність відповідних відмінностей (схожість) має бути продемонстрована у випадках, якщо потрібно більше 15 хвилин, але не більше 30 хвилин для досягнення майже повного розчинення (принаймні, 85% від вказаної кількості). Фактор подібності або інші відповідні тести повинні використовуватися для демонстрації профілю схожості генеричного та референтного продукту.
5.7. Допоміжні речовини
Для лікарських засобів І класу за БСК рекомендується використовувати схожу кількість тих же допоміжних речовин у складі генеричного лікарського засобу, що і у складі референтного лікарського засобу.
Якщо біовейвер застосовується до діючих речовин ІІ та III класу за БСК, допоміжні речовини мають бути якісно однаковими і кількісно дуже схожим з метою виключення різних ефектів на мембранні транспортери.
Допоміжні речовини, які можуть впливати на біодоступність (наприклад, сорбітол, манітол, лаурілсульфат натрію або інші сурфактанти) мають бути якісно і кількісно однаковими в генеричному та референтному лікарських засобах
6. Вимоги щодо надання матеріалів дослідження біоеквівалентності в разі проведення порівняльних фармакокінетичних досліджень.
Звіт дослідження по біоеквівалентності повинен включати повну документацію щодо його протоколу, проведення і оцінки.
Він має бути написаний відповідно до керівництва ICH E3 і підписаний дослідником.
Повинні вказуватися імена та приналежність до організації відповідального дослідника (ів), ділянка дослідження і період його використання. Сертифікати аудитів, якщо є, повинні включатися в звіт.
Звіт має містити обґрунтування вибору референтного препарату.
Він повинен включати назву референтного продукту, силу дії, лікарську форму, номер серії, виробника, закінчення терміну придатності і країну закупівлі.
Повинні надаватися назва і склад генеричного препарату лікарського засобу, використовуваного в дослідженні. Необхідно вказувати розмір серії, номер серії, дату виробництва і, якщо можливо, закінчення терміну придатності генеричного препарату. Крім того, повинна бути надана інформація, що використовуємий у досліджені препарат має той же кількісний склад і виробляється шляхом того ж процесу та є тим, який поданий для одержання реєстраційного посвідчення.
Сертифікати аналізу референтної і досліджуваної серій, використовуваних в дослідженні, повинні входити в додаток до звіту дослідження.
Дані концентрації і фармакокінетичні дані та дані статистичного аналізу мають бути представлені детально.
Всі дані окремих концентрацій і фармакокінетичні параметри мають бути перераховані згідно із складом разом із загальною статистикою, такою як середнє геометричне, медіана, середнє арифметичне, стандартне відхилення, коефіцієнт варіації, мінімум і максимум.
Мають бути наведені дані щодо кожного суб’єкта дослідження, а також індивідуальні криві залежності «концентрація — час», представлені в лінейно/лінійній та логаріфмічно/лінійній шкалі.
Необхідно вказувати використовуваний метод для отримання фармакокінетичних параметрів з вихідних даних.
Звіт має бути досить детальним для повторення фармакокінетичного і статистичного аналізу, наприклад, необхідно надати дані по дійсному часу відбору зразків крові після дози, концентрації препаратів, показникам фармакокінетичних параметрів для кожного суб’єкта в кожен період та схемі рандомізації.
Усі результати слід надавати у чіткій формі; мають бути включені дані щодо суб’єктів, які вибули з дослідження. Вибуття та виключення суб’єктів із дослідження необхідно повністю документувати і пояснювати. Має бути обґрунтовано вилучення даних. Дозволені причини для виключення мають бути заздалегідь вказані в протоколі.
Повторний аналіз досліджуваних зразків слід заздалегідь визначити в протоколі дослідження (і СОПах) до реального початку аналізу зразків. Як правило, повторний аналіз зразків, узятих в досліджуваного, неприйнятний з фармакокінетичної причини.
Для фармакокінетичних параметрів, які піддавалися статистичному аналізу, мають бути представлені дані оцінки і 90% довірчий інтервал для співвідношення досліджуваний і референтний препарат. Критерії оцінки 90 % довірчого інтервалу мають бути заздалегідь вказані в протоколі.
Біоаналітичний метод необхідно документувати у валідаційному звіті перед дослідженням.
Біоаналітичний звіт необхідно надавати. Біоаналітичний звіт повинен включати короткий опис використовуваного біоаналітичного методу і результати всіх калібрувальних стандартів і зразків контролю якості. Використовувані біоаналітичні методи необхідно належним чином охарактеризувати, провести валідацію і задокументувати з тим, щоб отримати надійні результати, які можна задовільно інтерпретувати. В рамках дослідження слід проводити валідацію з використанням зразків контролю якості в кожній серії аналітичних дослідів. Основні характеристики біоаналітичного методу, які необхідні для забезпечення прийнятності виконання і надійності аналітичних результатів, такі: специфічність, нижня межа кількісного визначення, функція відгуку (характеристика калібрувальної кривої), точність/правильність, точность/прецизіонность та стабільність. Необхідно представити репрезентативну кількість хроматограм або інші вихідні дані, включаючи весь діапазон концентрацій для всіх стандартних зразків і зразків контролю якості, а також аналізованих зразків. Дані повинні включати всі хроматограми, принаймні, по 20% суб’єктів із зразками контролю якості і калібраційними стандартами серії дослідів, включаючи цих суб’єктів.
Валідаційний звіт біоаналітичного методу необхідно надавати разом з репрезентативними хроматограмами.
Статистичний звіт має бути досить докладним, щоб забезпечити можливість повторення статистичного аналізу; він має містити, наприклад, схему рандомізації, демографічні дані, значення фармакокінетичних параметрів для кожного суб’єкта, описову статистику для кожного препарату та періоду. Слід включити також дисперсійний аналіз та/або непараметричний аналіз, точечні оцінки і відповідні довірчі інтервали, включаючи метод їх оцінювання.
Мають бути надані порівняльні профілі розчинення.
7.0 Вимоги до надання матеріалів дослідження біоеквівалентності в разі проведення процедури біовейвер.
У звіті має наводитись обґрунтування порівняльних досліджень in vitro як альтернативи оцінки біоеквівалентності in vivo.
Мають зазначатись дані, які б підтверджували співпадання якісного складу допоміжних речовин та співвідношення діючої речовини і допоміжних речовин генеричного лікарського засобу у порівнянні з референтним лікарським засобом.
У випадку внесення нових допоміжних речовин або нетипово великої кількості зазвичай використовуваної допоміжної речовини, необхідно наводити експериментальні дані, які підтверджують, що допоміжні речовини не впливають на кінетику розчинення лікарської форми і не взаємодіють між собою.
Має бути наданий детальний опис діючої речовини, тобто інформація щодо хімічної структури, молекулярної маси, природи (кислота, основа, амфотерна сполука) тощо, стислий опис референтного та генеричного лікарських засобів — номери серій, об’єми серій, терміни придатності тощо.
Крім того, необхідно викласти короткий опис виробництва, в якому вказати інформацію щодо задіяних виробничих процесів (вологе гранулювання, сухе пресування тощо), їх вплив на терапевтичну активність і стабільність лікарського засобу, призначення кожної допоміжної речовини на кожній стадії виробництва.
Має бути надано короткий опис референтного та генеричного лікарських засобів — номери серій, об’єми серій, терміни придатності та ін.
Має бути надано експериментальні дані розчинності (біофармацевтичної) найвищої разової дози діючої речовини в 250 мл трьох буферних розчинах у діапазоні рН 1.2-6.8 (рекомендовані значення рН 1.2, 4.5 та 6.8) при температурі (37 ± 1) о С.
По тесту «Абсорбція» мають наводитись літературні матеріали, а разі їх відсутності, дані, отримані при проведенні досліджень.
В результатах порівняльних досліджень кінетики вивільнення діючої речовини мають бути надані експериментальні докази подібності профілів розчинення досліджуваного і референтного препаратів. При цьому має наводитись повна інформація, яка містить:
обґрунтування вибору приладу для проведення дослідження (з кошиком, лопаттю, проточного чи іншого);
дані щодо приготування середовищ розчинення (характеристика вихідних речовин, методи дегазації) або посилання на фармакопею;
дані щодо внесення одиниць препаратів в середовище розчинення (для виключення систематичної похибки рекомендується використання рандомізованого методу);
дані щодо методики відбору проб (ручний чи автоматичний спосіб, місце відбору та об’єм проби) і їх підготовки (матеріали, використані для фільтрування, методика відбору фільтрату тощо).
Будь-які відхилення від рекомендованих умов необхідно аргументувати.
Має бути надано аналітичні методики контролю та їх валідація. В розділі має бути наведена повна інформація щодо:
кваліфікації приладів;
валідації методики кількісного визначення діючої речовини при різних значеннях рН, яка охоплює весь діапазон можливих концентрацій діючої речовини при різних значеннях рН;
методики визначення рН в присутності діючої речовини і продуктів її розкладу (при вивченні розчинності діючої речовини);
валідації методики визначення ступеня проникнення діючої речовини;
процедури відбору проб і пробопідготовки (при вивченні форми профілю розчинення) тощо.
Має бути надано експериментальні дані у вигляді графіків і таблиць, а також необхідні статистичні оцінки:
таблиці з даними щодо розчинності діючої речовини у вигляді рН — розчинність для визначення (підтвердження) класу розчинності згідно БСК (включаючи всі повтори);
таблиці з результатами вимірювань ступеню проникнення діючої речовини, а також модельної речовини, обчислені коефіцієнти проникнення діючої речовини та модельної речовини, статистична оцінка достовірності отриманих експериментальних даних;
таблиці з результатами вимірювань розчинення (значення для кожної з 12 одиниць досліджуваного препарату і кожної з 12 одиниць референтного препарату) для отриманих значень (площа піку, оптична густина, тощо) та результатів розрахунку,
криві розчинення (концентрація — час відбору) проби досліджуваного і референтного препаратів, які б доводили їх подібність.
Таблиці повинні містити результати паралельних повторів при всіх значеннях рН, статистична оцінка має надаватись у вигляді розрахунків середнього значення, відносного стандартного відхилення та фактора подібності f2.
В звіті має бути доведено, що діюча речовина відноситься до певного класу за БСК та генеричний препарат витримую всі вимоги для цього класу БСК.
7.0 Вимоги щодо доказу біоеквівалентності додаткових доз лікарського препарату
Якщо заявка подається на кілька дозувань досліджуваного продукту, достатньо встановити біоеквівалентність тільки при одній або двох дозах в залежності від пропорційності складу різних дозувань та інших питань, пов’язаних з продуктом і описаних нижче. Дозування для оцінки залежить від лінійності в фармакокінетиці діючої речовини.
7.1. У випадку нелінійної фармакокінетики (тобто не пропорційного збільшення в AUC при підвищеній дозі) може бути відмінність між різними дозами в чутливості для виявлення потенційних відмінностей між складами / формами.
Фармакокінетика вважається лінійною, якщо різниця в середніх AUCах, відкоригованих по відношенню до дози, становить не більше 25% при порівнянні досліджуваного дозування (або дозування в планованому дослідженні біоеквівалентності) і дозування (дозувань), щодо яких розглядається відмова від досліджень in vivo.
Для оцінки лінійності заявник повинен розглянути всі дані для публічного користування на предмет пропорційності дози та критично розглянути дані. Оцінка лінійності буде розглядати, чи відповідають відмінності в AUC, відкориговані по відношенню до дози, критерієм ± 25%.
Якщо біоеквівалентність було продемонстровано при дозуванні (-ях), які найбільш чутливі до виявлення потенційного відмінності між лікарськими засобами, на дослідження біоеквівалентності in vivo для іншого (-их) дозування (-нь) може бути прийнята відмова від досліджень in vivo.
7.2. Загальні критерії прийняття відмови від досліджень in vivo для додаткових доз лікарського препарату.
У разі розгляду можливості відмови від досліджень in vivo для додаткової дозування необхідно виконати наступні загальні вимоги:
а) лікарські засоби виробляються з використанням того ж самого виробничого процесу,
б) якісний склад різних дозувань однаковий,
в) склад дозувань кількісно пропорційний, тобто співвідношення між кількістю кожної допоміжної речовини до кількості діючої речовини однакове для всіх доз (що стосується засобів з негайним вивільненням, компоненти покриття, оболонка капсули, барвники та ароматизатори/смакові добавки не повинні виконувати це правило).
Якщо є деякі відхилення від кількісно пропорційного складу, умова в) як і раніше вважається виконаною, якщо умови i) і ii) або i) і iii) нижче застосовані до дозування, використовуваної в дослідженні біоеквівалентності, і дозування, для якої розглядається відмова від досліджень in vivo.
і) кількість діючої речовини становить менше 5% від ваги ядра таблетки, ваги вмісту капсули
iі) кількості різних допоміжних речовин ядра або вмісту капсули однакові для розглянутих дозувань, і змінюється тільки кількість діючої речовини
ііі) кількість наповнювача змінюється, щоб врахувати зміну кількості діючої речовини. Кількість інших допоміжних речовин ядра таблетки або вмісту капсули повинні бути однаковими для розглянутих дозувань
г) відповідні дані розчинення in vitro повинні підтвердити адекватність відмови від досліджень in vivo на додаткове дозування
Для лікарських засобів з лінійною фармакокінетикою, для яких виконані вище зазначені умови від а) до г), досить встановити біоеквівалентність тільки для одного дозування.
Як правило, дослідження біоеквівалентності слід проводити з максимальним дозуванням. Для засобів з лінійною фармакокінетикою, і коли діюча речовина має високу розчинність, також прийнятний відбір меншою дози, ніж максимальна доза. Відбір меншого дозування можливо також обґрунтувати, якщо максимальне дозування не можна вводити здоровим добровольцям з причин безпеки/переносимості. Крім того, якщо проблеми чутливості аналітичного методу перешкоджають досить точним вимірам концентрації в плазмі після одноразового введення максимального дозування, можна вибрати більш високу дозу (переважно з використанням таблеток для багаторазового введення з максимальним дозуванням). Обрана доза може бути вище максимальної терапевтичної дози, за умови, що ця одноразова доза добре переноситься у здорових добровольців, і що немає жодних обмежень з абсорбції або розчинності при цій дозі.
Для лікарських засобів з нелінійною фармакокінетикою, яка характеризується більш, ніж пропорційним збільшенням AUC зі зростанням дози в діапазоні терапевтичної дози, дослідження біоеквівалентності, як правило, слід проводити при максимальному дозуванні.
Для лікарських засобів з менш, ніж пропорційним збільшенням AUC зі зростанням дози в діапазоні терапевтичної дози біоеквівалентність, в більшості випадків, слід встановлювати як при максимальному дозуванні, так і при мінімальному дозуванні (або дозуванні в лінійному діапазоні), тобто в цій ситуації потрібно два дослідження біоеквівалентності. Якщо не лінійність не викликана обмеженою розчинністю, а, наприклад, насиченням транспортерами захоплення за умови виконання вищевказаних умов від а) до г), а генеричний і референтний лікарські засоби не містять будь-яких допоміжних речовин, які можуть вплинути на моторику шлунково-кишкового тракту або транспортні білки, досить продемонструвати біоеквівалентність при мінімальному дозуванні (або дозуванні в лінійному діапазоні).
Відбір інших дозувань можна обґрунтувати, якщо існують проблеми аналітичної чутливості, що перешкоджають дослідженню при мінімальному дозуванні або, якщо максимальне дозування не можна вводити здоровим добровольцям з причин безпеки / переносимості.
8.0 Загальні аспекти проведення тесту «Розчинення».
Тимчасових точок відбору зразків повинно бути достатньо для отримання значущих профілів розчинення і, принаймні, через кожні 15 хвилин. Рекомендується проводити більш частий відбір зразків під час періоду найбільшої зміни в профілі розчинення. Для швидкорозчиних продуктів, якщо повне розчинення відбувається протягом 30 хвилин, може бути необхідний відповідний профіль шляхом відбору зразків через кожні 5-10 хвилин.
Якщо більш ніж 85% лікарського засобу розчиняється протягом 15 хвилин, профілі розчинення можуть прийматися як подібні без подальшої математичної оцінки.
У разі якщо більш ніж 85% не розчиняється за 15 хвилин, а розчиняється протягом 30 хвилин, то потрібні, принаймні, три тимчасових точки: перша точка відбору до 15 хвилин, друга — в 15 хвилин і третя, коли вивільнення досягає 85 %.
Для лікарських засобів з модифікованим вивільненням, необхідно дотримуватися рекомендації:
профілі розчинення форм з відстроченим вивільненням мають бути подібними в точках контролю, що відповідають механізму їх розчинення, тобто і при рН 1.2 (протягом 2 год.), і при рН 6.8;
профілі розчинення форм з пролонгованим вивільненням мають бути подібними у трьох середовищах розчинення у точках контролю, які відповідають швидкості їх розчинення, наприклад, через 1 год, 2 год, 4 год, 6 год, 8 год і 16 год
Фактор подібності f2 чином обчислюють за формулою:
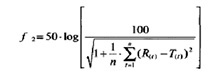
У цьому рівнянні
f2 — це фактор подібності,
n-це кількість тимчасових точок,
R (t) є середнім відсотком розчиненого референтного лікарського засобу за час t після початку дослідження;
T (t) є середнім відсотком розчиненого генеричного лікарського засобу за час t після початку дослідження .
Для обох референтного та генеричного лікарських засобів повинен визначатися відсоток розчинення.
Оцінка фактора подібності ґрунтується на наступних умовах:
— Мінімум три тимчасових точки (нуль виключається)
— Точки контролю повинні бути однаковими для двох складів
— Дванадцять окремих показників для кожної точки контролю для кожного складу
— Не більше ніж один середній показник’ 85% розчинення для будь-якого з складів
— Відносне відхилення від стандарту або коефіцієнт варіації будь-якого продукту повинен бути менше 20% для першої точки і менше 10% від другої до останньої тимчасової точки.
Показник f2 між 50 і 100 передбачає, що два профілі розчинення є подібними.
Слід надати чіткий опис та пояснення дій, розпочатих при застосуванні процедур, з відповідними зведеними таблицями.»
«Додаток 21
до Порядку проведення експертизи матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
Перелік документів для проведення експертизи матеріалів для державної перереєстрації лікарського засобу, що виробляється згідно з затвердженим прописом.
1. Заява про проведення експертизи матеріалів на лікарські засоби, які подаються для державної перереєстрації згідно із додатком 17 до Порядку експертизи матеріалів;
2. Інформація про уповноважену особу заявника для здійснення фармаконагляду;
3. Перелік одержаних за останні 5 років в Україні рекламацій на препарат (у хронологічному порядку) з докладним аналізом причин, що слугували підставами для видачі рекламацій, та заходів, вжитих заявником для запобігання їм у майбутньому;
4. Переглянутий перелік усіх гарантій та зобов’язань, які залишились, і підписаний лист зобов’язань (за необідності);
5. Копія ліцензії на виробництво або документа, виданого уповноваженим органом країни виробника, який свідчить про наявність у виробника права на здійснення такого виробництва;
6. Затверджені при реєстрації та оновлені методи контролю якості готового лікарського засобу
7. Лист-підтвердження щодо того, що склад, виробництво та контроль лікарського засобу відповідають пропису, наведеному у Переліку лікарських засобів, що виробляються згідно із затвердженими прописами.».
Коментарі