Звіт
щодо відповідності процедури державної реєстрації лікарських засобів в Україні законодавству та стандартам ЄС
Автори:
Томаш Яворскі, Марчин Томашик, Лешек Борковскі, Агнєшка Буксовіч (Польща) Борис Даневич, Юлія Гулима, Артем Грудінін, Сергій Хома, Антон Делікатний, Тала Стеценко (Україна), Мартінс Сітхс, Таліс Талентс, Айнарс Бідерс (Латвія)
Проект фінансується Європейським банком реконструкції та розвитку
Договір про надання консультаційних послуг № С33648/1753/9351 для України: Рекомендації щодо вдосконалення законодавства у фармацевтичномусекторі України
ЗМІСТ
| СПИСОК АБРЕВІАТУР ТА СКОРОЧЕНЬ |
8 |
| СПИСОК ВИКОРИСТАНИХ ЗАКОНОДАВЧИХ АКТІВ |
11 |
| I. РЕЗЮМЕ | 14 |
| І.1 РЕЗУЛЬТАТИ ПОРІВНЯННЯ ЗАКОНОДАВСТВА УКРАЇНИ ПРОРЕЄСТРАЦІЮ ЛІКАРСЬКИХ ЗАСОБІВ ЗІ СТАНДАРТАМИ ЄС | 14 |
| І.2 СПОСТЕРЕЖЕННЯ У СФЕРІ СТРАТЕГІЇ І ПЛАНУВАННЯ | 14 |
| І.3 СПОСТЕРЕЖЕННЯ У СФЕРІ ЗАКОНОДАВСТВА | 15 |
| І.4 СПОСТЕРЕЖЕННЯ У СФЕРІ ОРГАНІЗАЦІЇ ПРОЦЕСІВ | 16 |
| І.4.1 Щодо тривалості процедури | 16 |
| І.4.2 Щодо ефективності процесу | 17 |
| І.4.3 Щодо прийнятої моделі процедури | 17 |
| І.4.4 Щодо участі Держлікслужби | 17 |
| І.5 СПОСТЕРЕЖЕННЯ У СФЕРІ КОМУНІКАЦІЙ ТА ПРОЗОРОСТІ | 21 |
| І.6 СПОСТЕРЕЖЕННЯ У СФЕРІ ФІНАНСУВАННЯ | 22 |
| І.7 СПОСТЕРЕЖЕННЯ У СФЕРІ ЛЮДСЬКИХ РЕСУРСІВ | 23 |
| І.8 CПОСТЕРЕЖЕННЯ У СФЕРІ ВИКОРИСТАННЯ ЕЛЕКТРОННИХ ІНФОРМАЦІЙНИХ СИСТЕМ | 25 |
| II. ВСТУП | 28 |
| II.1. ЦІЛЬ | 28 |
| II.2. МЕТОДОЛОГІЯ | 28 |
| II.3. ПРИПУЩЕННЯ | 29 |
| II.4. ЗАГАЛЬНІ ПРИЦИПИ | 30 |
| ІІ.5. СТРУКТУРА ЗВІТУ | 30 |
| II.6. ПОДЯКА | 31 |
| III. АНАЛІЗ ВІДПОВІДНОСТІ УКРАЇНСЬКОГО ЗАКОНОДАВСТВАСТАНДАРТАМ ЄС | 32 |
| III.1 НОРМАТИВНО-ПРАВОВА БАЗА В УКРАЇНІ | 32 |
| III.2. ВИЗНАЧЕННЯ | 35 |
| III.2.1. Визначення поняття «лікарський засіб» | 35 |
| III.2.2. Посилання на не Європейські фармакопеї при визначенні «гомеопатичних лікарських засобів» | 37 |
| III.2.3. Визначення «конфіденційної реєстраційної інформації» (Стаття II.1.29 Наказу МОЗ № 426) | 37 |
| III.2.4. Визначення «незалежного експерта» (Стаття II.1.35 Наказу МОЗ № 426) | 38 |
| III.2.5. Визначення «референтний лікарський засіб » (Стаття II.1.53 Наказу МОЗ № 426) та «оригінальний (інноваційний) лікарський засіб» (Стаття ІІ.1.37 Наказу МОЗ № 426) | 38 |
| III.2.6. Визначення «неправомірне використання реєстраційної інформації про безпеку та ефективність лікарського засобу» (Стаття II.1.35 Наказу МОЗ № 426) | 39 |
| III.2.7. Визначення «подібний біологічний лікарський засіб (біосиміляр)» (Стаття II.1.40 Наказу МОЗ № 426) | 39 |
| III.2.8. Невизначені поняття | 39 |
| III.2.9. Інші розбіжності у визначеннях | 40 |
| III.3. ЗАСОБИ, ЩО ВХОДЯТЬ ДО СИСТЕМИ ДЕРЖАВНОЇ РЕЄСТРАЦІЇ | 42 |
| III.3.1. Можливі розбіжності в обсязі засобів, що підлягають реєстрації, що виникли в результаті невідповідності визначень деяких категорій | 42 |
| III.3.2. Засоби, що підлягають реєстрації в Україні та звільняються від реєстрації згідно із законодавством ЄС | 42 |
| III.3.3. Кров та плазма крові людини. Лікарські засоби, що походять з крові або плазми крові людини | 43 |
| III.3.4. Інші спостереження | 44 |
| III.4. СПРОЩЕНІ ПРОЦЕДУРИ ДЕРЖАВНОЇ РЕЄСТРАЦІЇ | 44 |
| III.4.1. Лікарські засоби, зареєстровані ЕМА | 44 |
| III.4.2. Лікарські засоби, зареєстровані окремими іноземними органами | 45 |
| III.4.3. Лікарські засоби, що підлягають закупівлі | 48 |
| III.5. ВИМОГИ ДО ЗАЯВИ ТА РЕЄСТРАЦІЙНОГО ДОСЬЄ | 50 |
| III.5.1. Вимоги до заяви | 51 |
| III.5.2. Вимоги до реєстраційного досьє | 51 |
| III.6. ЗАХИСТ ВЛАСНОЇ ІНФОРМАЦІЇ ТА ПАТЕНТНИХ ПРАВ | 63 |
| III.6.1. Захист даних | 63 |
| III.6.2. Патентний захист | 65 |
| III.7. ІНФОРМАЦІЯ ДЛЯ ПАЦІЄНТІВ ТА СПЕЦІАЛІСТІВ СФЕРИ ОХОРОНИ ЗДОРОВЯ (ЕТИКЕТКА, ІНФОРМАЦІЯ ДЛЯ ПАЦІЄНТА, SmPC) | 66 |
| III.7.1. SmPC | 66 |
| III.7.2. Інструкція для медичного застосування | 67 |
| III.7.3. Маркування лікарських засобів | 69 |
| III.8. ХІД ПРОЦЕДУРИ | 70 |
| ІІІ.9. СТРОКИ | 73 |
| ІІІ.10. ПІДСТАВИ ДЛЯ ПРИЙНЯТТЯ РІШЕННЯ ПРО РЕЄСТРАЦІЮ ТА ВІДМОВУ В РЕЄСТРАЦІЇ ЛІКАРСЬКОГО ЗАСОБУ | 74 |
| 74 | |
| ІІІ.11. ПРИЙНЯТТЯ РІШЕННЯ ПРО ДЕРЖАВНУ РЕЄСТРАЦІЮ ЛІКАРСЬКОГО ЗАСОБУ | 76 |
| ІІІ.11.1 Колективне рішення про реєстрацію/індивідуальне рішення про реєстрацію | 76 |
| ІІІ.11.2 Строк дії реєстраційного посвідчення | 78 |
| ІІІ.11.3. Реєстрація «під умовами» | 78 |
| ІІІ.12.ОБОВ’ЯЗКИ ВЛАСНИКА РЕЄСТРАЦІЙНОГО ПОСВІДЧЕННЯ | 79 |
| ІІІ.13. ПУБЛІЧНА ІНФОРМАЦІЯ | 80 |
| III.14. ПРОЦЕДУРА ПЕРЕРЕЄСТРАЦІЇ | 81 |
| III.15. ПІСЛЯРЕЄСТРАЦІЙНІ ЗМІНИ / ЗМІНИ ДО РЕЄСТРАЦІЇНИХ МАТЕРІАЛІВ | 82 |
| III.15.1. Загальні положення | 82 |
| III.15.2. Класифікація змін | 82 |
| III.15.3. Інші невідповідності | 83 |
| III.15.4. Процедура та строки | 84 |
| III.16. ФАРМАКОНАГЛЯД (ОКРЕМІ ПИТАННЯ) | 86 |
| IV. СПОСТЕРЕЖЕННЯ У СФЕРІ СТРАТЕГІЧНОГО РОЗВИТКУ | 88 |
| IV.1. ВІДСУТНІСТЬ ЧІТКОГО БАЧЕННЯ І СТРАТЕГІЇ СИСТЕМИ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ ЯК СКЛАДОВОЇ СТРАТЕГІЇ РОЗВИТКУ СИСТЕМИ ОХОРОНИ ЗДОРОВ’Я | 88 |
| IV.2. СТРАТЕГІЧНІ РІШЕННЯ НА ШЛЯХУ НАБЛИЖЕННЯ ДО СТАНДАРТІВ ЄС | 92 |
| IV.2.1. Бажаний рівень наближення законодавства | 93 |
| IV.2.2. Гармонізація досьє лікарських засобів, зареєстрованих в минулому | 96 |
| IV.3. ПИТАННЯ ЩОДО «Спрощеної» ПРОЦЕДУРИ РЕЄСТРАЦІЇ ЛІКАСЬКИХ ЗАСОБІВ, ДОЗВОЛЕНИХ В КРАЇНАХ, ЩО НЕ Є ЧЛЕНАМИ ЄС | 99 |
| IV.4. СПІВРОБІТНИЦТВО МІЖ УКРАЇНСЬКИМИ ОРГАНАМИ У СФЕРІ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ ТА ЕМА | 100 |
| V. СПОСТЕРЕЖЕННЯ В ОБЛАСТІ ЗАКОНОДАВСТВА | 102 |
| V.1. Загальні відомості | 102 |
| V.2. НЕЗРОЗУМІЛЕ СПІВВІДНОШЕННЯ МІЖ ЗАКОНОМ ПРО ЛІКАРСЬКІ ЗАСОБИ, ПОСТАНОВОЮ КМУ № 376 І НАКАЗОМ МОЗ № 426 | 102 |
| V.3. ПОВТОРЮВАНІ ПОЛОЖЕННЯ | 104 |
| V.4. СУПЕРЕЧЛИВЕ РЕГУЛЮВАННЯ СПРОЩЕНИХ ПРОЦЕДУР | 105 |
| V.5. НЕДОСТАТНІЙ РІВЕНЬ КООРДИНАЦІЇ ПРИ ВНЕСЕННІ ЗМІН ДО РІЗНИХ НОРМАТИВНИХ АКТІВ | 106 |
| V.6. МОНІТОРИНГ ЗМІН У ЗАКОНОДАВСТВІ ЄС | 106 |
| V.7. РЕКОМЕНДАЦІЇ | 107 |
| VI. ОРГАНІЗАЦІЯ ПРОЦЕСІВ | 108 |
| VI.1. ВСТУПНІ ЗАУВАЖЕННЯ | 108 |
| VI.2. СХЕМА ПРОЦЕДУРИ | 108 |
| VI.3. ВИЯВЛЕНІ ПРОБЛЕМИ ЩОДО ТРИВАЛОСТІ ПРОЦЕДУРИ | 114 |
| VI.3.1. Регламентування кінцевих строків (строків виконання) | 114 |
| VI.3.2. Відсутність ефективних механізмів для притягнення до відповідальності за затримки під час процедури реєстрації | 116 |
| VI.3.3. Тривала процедура перереєстрації як загроза безперервності ведення бізнесу | 116 |
| VI.4. СПОСТЕРЕЖЕННЯ У СФЕРІ ОРГАНІЗАЦІЇ ПРОЦЕСІВ | 116 |
| VI.4.1. Заявник подає документи в три етапи | 116 |
| VI.4.2. Повноваження МОЗ і ДЕЦ, що перетинаються під час процедури реєстрації | 117 |
| VI.4.3. Надмірне залучення колегіальних органів до процедури реєстрації | 118 |
| VІ.4.4 Зайва вимога щодо GMP сертифікації | 118 |
| VІ.4.5 Занадто ускладнена процедура після закінчення експертизи реєстраційних матеріалів | 118 |
| VІ.4.6 Низький рівень якості СОП ДЕЦ | 119 |
| VІ.4.7 Неефективне використання «Єдиного вікна» | 119 |
| VI.5. СПОСТЕРЕЖЕННЯ ЩОДО ПРИЙНЯТНОЇ МОДЕЛІ ПРОЦЕДУРИ | 120 |
| VI.5.1. Нечіткий розподіл повноважень | 120 |
| VI.5.2. Залучення зовнішніх експертів | 122 |
| VI.5.3. Неефективне залучення МОЗ в процедуру реєстрації | 122 |
| VI.5.4. Реєстрація шляхом прийняття наказу щодо декількох лікарських засобів | 123 |
| VI.5.5. Квазі-комерційний статус ДЕЦ | 123 |
| VI.6. РЕКОМЕНДАЦІЇ | 124 |
| VI.7. ОРГАНІЗАЦІЯ ПРОЦЕСІВ У ДЕРЖЛІКСЛУЖБІ | 129 |
| VI.7.1. Заміна процедури з використанням паперових носіїв електронною процедурою | 130 |
| VI.7.2. Скасування етапу первинної експертизи під час процедури підтвердження GMP-сертифікатів, виданих уповноваженими органами країн-членів PIC/S | 130 |
| VI.7.3. Функціонування Робочої групи | 131 |
| VII. СПОСТЕРЕЖЕННЯ У СФЕРІ КОМУНІКАЦІЇ ТА ПРОЗОРОСТІ | 133 |
| VII.1. ПРОГАЛИНИ В КОМУНІКАЦІЇ З БОКУ РЕГУЛЯТОРНИХ ОРГАНІВ | 133 |
| VII.1.1. Комунікація з громадськістю | 133 |
| VII.1.2. Обмежені функціональні можливості офіційних веб-сайтів | 133 |
| VII.1.3 Відсутність загальнодоступної інформації про поточні процедури, статистичні дані, результати щорічних внутрішніх і зовнішніх аудитів, звіти про результати оцінки | 134 |
| VII.2. КОМУНІКАЦІЯ МІЖ ДЕЦ ТА ЗАЯВНИКАМИ | 136 |
| VII.2.1. Обмежений обсяг інформації (рекомендацій), доступний заявникам, обмежена кількість документів-зразків | 136 |
| VII.2.2. Право на отримання консультації перед поданням документів не працює на практиці, платна наукова консультація не є доступною | 136 |
| VII.2.3. Неналежний рівень комунікації між ДЕЦ та заявниками | 138 |
| VII.3. ВІДСУТНІСТЬ ПРОЗОРОСТІ В РОБОТІ РЕГУЛЯТОРНИХ ОРГАНІВ | 139 |
| VII.3.1. Необґрунтовані обмеження доступу до інформації | 139 |
| VII.3.2. Нечіткий розподіл повноважень | 140 |
| VII.3.3. Відсутність інформації про експертів | 140 |
| VIII. СПОСТЕРЕЖЕННЯ У СФЕРІ ФІНАНСУВАННЯ | 142 |
| VIII.1. ВІДНОСНО НИЗЬКА ВАРТІСТЬ ДЕРЖАВНОЇ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ | 142 |
| VIII.2. ВАРТІСТЬ ЕКСПЕРТИЗИ НЕ РЕГУЛЮЄТЬСЯ ЗАКОНОМ | 143 |
| VIII.3. НЕЗНАЧНИЙ ОБСЯГ ІНВЕСТИЦІЙ В РОЗВИТОК СИСТЕМИ | 145 |
| IX. СПОСТЕРЕЖЕННЯ У СФЕРІ ЛЮДСЬКИХ РЕСУРСІВ В РАМКАХ ДЕЦ | 145 |
| IX.1. ПОТОЧНА ОРГАНІЗАЦІЙНА ТА HR СТРУКТУРА ДЕЦ | 145 |
| IX.1.1 Поточна організаційна структура ДЕЦ | 145 |
| IX.1.2. Запропоновані зміни в організаційній структурі ДЕЦ (пропозиція ДЕЦ) | 146 |
| IX.1.3. Оцінка пропонованих змін | 146 |
| IX.2. РЕКРУТИНГ, ПРОФЕСІЙНА ПІДГОТОВКА І АТЕСТАЦІЯ ЕКСПЕРТІВ ЦЕНТРУ | 147 |
| IX.2.1. Використання позаштатних експертів | 147 |
| IX.2.2. Використання колегіальних органів | 148 |
| IX.3. ВИНАГОРОДИ ТА ІНШІ ІНСТРУМЕНТИ МОТИВАЦІЇ В ДЕЦ | 149 |
| IX.4. ІНШІ КАДРОВІ ТА ОРГАНІЗАЦІЙНІ ПИТАННЯ | 150 |
| X. ВИКОРИСТАННЯ ІНФОРМАЦІЙНИХ СИСТЕМ У ПРОЦЕСІ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ В УКРАЇНІ | 151 |
| X.1. ВСТУПНІ ПРИМІТКИ | 151 |
| X.1.1. Застереження | 151 |
| X.2. ПОПЕРЕДНІ ВИСНОВКИ | 151 |
| X.3. ВИКОРИСТАННЯ ІНФОРМАЦІЙНИХ СИСТЕМ У ПРОЦЕДУРІ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ В УКРАЇНІ. СПОСТЕРЕЖЕННЯ, ВИСНОВКИ ТА РЕКОМЕНДАЦІЇ | 152 |
| X.3.1. Структура процесів реєстрації лікарських засобів. СОПи | 154 |
| (a) Спостереження | 154 |
| (b) Висновки, вплив та рекомендації | 155 |
| X.3.2. Система експертного супроводу лікарських засобів (MES) | 156 |
| (a) Спостереження | 156 |
| (b) Висновки, вплив та рекомендації | 158 |
| X.3.3. Система візуалізації інформації ДЕЦ (СВ) | 160 |
| (a) Спостереження | 160 |
| (b) Висновки, вплив та рекомендації | 161 |
| X.3.4. Державний реєстр лікарських засобів (ДРЛЗ) | 161 |
| (a) Спостереження | 161 |
| (b) Висновки, вплив та рекомендації | 163 |
| X.3.5. Управління розвитку інформаційних систем | 164 |
| (a) Спостереження | 164 |
| (b) Висновки, вплив та рекомендації | 164 |
| X.4. ЗАПРОПОНОВАНІ ЗМІНИ ТА ДІЇ | 165 |
| X.4.1. Структура майбутніх інформаційних систем | 165 |
| X.4.2. Переваги | 167 |
| X.4.3. Ризики та питання, на які слід звернути увагу під час впровадження структури майбутніх інформаційних систем | 167 |
| Х.4.4. Резюме результатів та рекомендацій | 169 |
| Х.4.5. Операційна сумісність інформаційних систем | 174 |
| Додаток 1. РЕЗУЛЬТАТИ ОПИТУВАННЯ | 175 |
СПИСОК АБРЕВІАТУР ТА СКОРОЧЕНЬ
ACC – Американська торговельна палата
ADR – Побічна реакція лікарського засобу
AFSSAPS – Французьке агентство з санітарного контролю за лікарськими препаратами
AIPM – Асоціація представників міжнародних фармацевтичних виробників України
AMS – Система моніторингу та контролю фармацевтичної діяльності
ANSM – Національне агентство з безпеки лікарських засобів і товарів медичного призначення (Франція)
ASMF – Мастер-файл на АФІ
BfArM – Федеральний інститут лікарських засобів та виробів медичного призначення Німеччини
BPMN – Система умовних нотацій для моделювання бізнес-процесів
CEP – Сертифікат відповідності монографії Європейської Фармакопеї
CESP – Загальноєвропейська платформа подання
ChMP – Комітет з лікарських засобів для застосування людиною
CMD (h) – Координаційна група з взаємного визнання та децентралізованих процедур – Людина
CPMP – Комітет із запатентованих лікарських засобів ЕМА
CTS – Інформаційна система клінічних досліджень
DMS – Система контролю хвороб
DrR – Реєстр лікарів
DSS – Система супроводу рішень для лікарів і фармацевтів
EDS – Система електронного документообігу в сфері охорони здоров’я
EHR – Електронна система медичних карток
EMA – Європейське агентство з лікарських засобів
EPAR – Європейський публічний звіт з оцінки лікарських засобів
EPR – Інформаційна система електронного призначення лікарських засобів
ESS – Інформаційна система надання електронних
FDA – Управління з санітарного нагляду за якістю харчових продуктів та медикаментів (Сполучені Штати Америки)
GMP – Належна виробнича практика
ICH – Міжнародна конференція з гармонізації технічних вимог до реєстрації фармацевтичних продуктів для використання людиною
IDEF0 – методологія функціонального моделювання та графічна нотація для формалізації процесів
INN – Міжнародна непатентована назва IT- Інформаційні технології
MBP – ІС планування бюджету на лікарські засоби
MPS – Інформаційна система закупівель лікарських засобів
OEM – Виробник обладнання
OTC – Безрецептурні лікарські засоби
PES – Система фармакоекономічного оцінювання
PhR – Реєстр фармацевтів
PMS – Система контролю ціноутворення на лікарські засоби
PVL- Електронний лист очікування пацієнтів
QRD- Групи перевірки якості документів
RIS – Система відшкодування витрат вартості лікарських засобів
SIS – Статистична інформаційна система
SmPC – Коротка характеристика лікарського засобу
SUSAR – Підозрювані непередбачені серйозні побічні реакції
URPL – Польський Офіс з реєстрації лікарських засобів, медичних виробів та біоцидних продуктів
VIS – Інформаційна система вакцинації
АПРаД – Асоціація фармацевтичних досліджень та розвитку
ВІЛ – Вірус імунодефіциту людини
ВООЗ – Всесвітня організація охорони здоров’я
ВРП – Власники реєстраційного посвідчення
ГМО – Генетично-модифіковані організми
Держлікслужба – Державна служба України з лікарських засобів та контролю за наркотиками
ДЕЦ – Державне підприємство «Державний експертний центр Міністерства охорони здоров’я України»
ДРЛЗ – Державний реєстр лікарських засобів України
ДФУ – Державна фармакопея України
ДЦП – Децентралізована процедура
ЄБА – Європейська бізнес асоціація
ЄБРР – Європейський банк реконструкції та розвитку
ЄЕЗ – Європейська економічна зона
ЄС – Європейський Союз
ЄФ – Європейська фармакопея
ЗТД – Загальний технічний документ
КЕГ – Консультативно-експертна група
КМУ – Кабінет міністрів України
КПЕ – Ключові показники ефективності
ЛЗ – Лікарський засіб
МІБП – Медичні імунобіологічні препарати
МКЯ – Методи контролю якості
МОЗ – Міністерство охорони здоров’я України
НЕР – Науково-експертна рада
НТР – Науково-технічна рада
ПВВ – Процедура взаємного визнання
ПІВ – Право інтелектуальної власності
ПУР – План управління ризиками
Регулярний звіт – Регулярно оновлюваний звіт з безпеки лікарського засобу
РП – Реєстраційне посвідчення
СВ – Система візуалізації інформації
Система Експертизи/ MES – Інформаційна Система Підтримки Проведення Експертизи Лікарських Засобів
СОП – Стандартні операційні процедури
ТЕК – Технічно-експертна комісія
ТРІПС – Угода про торговельні аспекти прав інтелектуальної власності
СПИСОК ВИКОРИСТАНИХ ЗАКОНОДАВЧИХ АКТІВ
Українське законодавство
Закон про лікарські засоби/Закон – ЗУ «Про лікарські засоби» від 04 квітня 1996 року
Право ЄС
Директива 85/374 – Директива Ради ЄС 85/374/ЄЕС від 25 липня 1985 року «Про наближення законів, постанов та адміністративних положень держав-членів щодо відповідальності за неякісну продукцію»
Директива 2001/20 – Директива 2001/20/ЄC Європейського парламенту та Ради від 4 квітня 2001 року «Про наближення законів, підзаконних актів та адміністративних положень держав-членів стосовно імплементації добросовісної клінічної практики при проведенні клінічних дослідів медикаментів, що застосовуються людиною»
Директива 2001/83 – Директива 2001/83/ЄС Європейського парламенту та Ради від 6 листопада 2001 року «Про Кодекс спільноти відносно лікарських препаратів, призначених для застосування людьми»
Директива 2002/98 – Директива 2002/98/ЄС Європейського парламенту та Ради від 27 січня 2003 р., «Щодо встановлення стандартів якості та безпеки збору, тестування, обробки, зберігання та розподілу людської крові та її складових», що вносить зміни до Директиви 2001/83/ЄС
Директива 2003/63 – Директива Комісії 2003/63/ЄС від 25 червня 2003 року, що вносить зміни до Директиви 2001/83/ЄС Європейського парламенту та Ради про Кодекс спільноти відносно лікарських препаратів, призначених для застосування людьми
Директива 2009/120 – Директива Комісії 2009/120/ЄС від 14 вересня 2009, що вносить зміни до директиви 2001/83/ЄС Європейського парламенту та Ради «Про Кодекс спільноти відносно лікарських препаратів, призначених для застосування людьми, стосовно лікувальних препаратів прогресивної терапії»
Директива 2010/84 – Директива 2010/84/ЄС Європейського парламенту та Ради від 15 грудня 2010 року, що вносить зміни щодо фармаконагляду до Директиви 2001/83/ЄС «Про Кодекс спільноти відносно лікарських препаратів, призначених для застосування»
Директива 2011/62 – Директива 2011/62/ЄС Європейського парламенту та Ради від 8 червня 2011 року, що вносить зміни до Директиви 2001/83/ЄС
Регламент 141/2000 – Регламент (ЄС) № 141/2000 Європейського парламенту та Ради від 16 грудня 1999 року «Про препарати-сироти»
Регламент 847/2000 – Регламент Комісії (ЄС) № 847/2000 від 27 квітня 2000, який встановлює положення для імплементації критеріїв позначень для лікарських засобів таких, як препарати-сироти та визначення термінів «подібний лікарський засіб» та «клінічна перевага»
Регламент 726/2004 – Регламент (ЄС) № 726/2004 Європейського парламенту та Ради від 31 березня 2004 року, який встановлює процедури Спільноти щодо видачі дозволу та контролю за лікарськими засобами, призначеними для застосування людям та у ветеринарії, та запроваджує Європейське Медичне Агентство
Регламент №1901/2006 – Регламент (ЄС) № 1901/2006 Європейського парламенту та Ради від 12 грудня 2006 року щодо лікарських засобів для використання у педіатрії, що вносить зміни до Регламенту (ЄЕС) №1768/92, Директиви 2001/20/ЄС, Директиви 2001/83/ЄС та Регламенту (ЄС) №726/2004
Регламент 1394/2007 – Регламент (ЄС) № 1394/2007 Європейського парламенту та Ради від 13 листопада 2007 року «Про лікарські засоби прогресивної терапії, та що вносить зміни до Директиви 2001/83/ЄС і до Регламенту (ЄС) № 726/2004»
Регламент 1234/2008 – Регламент Комісії (ЄС) №1234/2008 від 24 листопада 2008 щодо розгляду змін до умов дозволу на продаж лікарських засобів для людини та ветеринарних препаратів («Регламент щодо змін»), що регулює процедуру внесення змін до ліцензії на продаж. З правками, внесеними Регламентом (ЄС) 712/2012.
Регламент 1235/2010 – Регламент (ЄС) №1235/2010 Європейського парламенту та Ради від 15 грудня 2010 року, що вносить зміни щодо фармаконагляду за лікарським засобами для застосування людьми, до Регламенту (ЄС) №726/2004, який встановлює процедури Спільноти щодо видачі дозволу та контролю за лікарськими засобами, призначеними для застосування людям та у ветеринарії, та запроваджує Європейське Медичне Агентство; Регламенту (ЄС) №1394/2007 «Про лікарські засоби прогресивної терапії».
I. РЕЗЮМЕ
І.1 РЕЗУЛЬТАТИ ПОРІВНЯННЯ ЗАКОНОДАВСТВА УКРАЇНИ ПРОРЕЄСТРАЦІЮ ЛІКАРСЬКИХ ЗАСОБІВ ЗІ СТАНДАРТАМИ ЄС
(1) Українське законодавство у сфері державної реєстрації лікарських засобів вцілому було приведено у відповідність до законодавства ЄС у більшості аспектів.
(2) Водночас, у законодавстві наявні деякі суперечності і невідповідності. Найбільш суттєвими з них є наступні:
(a) спрощена процедура реєстрації лікарських засобів уповноваженими органами окремих країн, окрім ЄС, що дозволяє реєстрацію в Україні продукції, щодо якої не було проведено наукової оцінки відповідно до вимог Директиви 2001/83, не відповідає законодавству ЄС;
(b) Правила захисту реєстраційних даних (зокрема, ексклюзивності даних) істотно відрізняються від правил ЄС, причому не лише у частині строків захисту, але й суті механізму захисту;
(c) Існує одна інструкція для медичного застосування замість двох типів: інструкції для пацієнта, викладеної у зрозумілій і доступній формі, і резюме властивостей лікарського засобу (SPC) для фахівців в сфері охорони здоров’я;
(d) Відсутнє законодавче обмеження максимального строку всієї процедури реєстрації, водночас, встановлюються строки для окремих частин процедури;
(e) Строки реєстрації на практиці є більшими, ніж строки, передбачені законодавством ЄС;
(f) Не всі види реєстрації «під умовами» передбачені і застосовуються в Україні;
(g) Звіти про результати оцінки лікарських засобів у межах процедури реєстраціїне є загальнодоступними;
(h) Строки розгляду заяв на внесення змін до реєстраційних матеріалів занадто тривалі в порівнянні з законодавством ЄС;
(i) Нещодавня реформа законодавства ЄС у сфері фармаконагляду 2012 року не в повній мірі відображена в українському законодавстві.
І.2 СПОСТЕРЕЖЕННЯ У СФЕРІ СТРАТЕГІЇ І ПЛАНУВАННЯ
(3) Необхідним є чітке бачення і стратегія розвитку системи реєстрації лікарських засобів у рамках стратегії реформи охорони здоров’я.
(4) Україні слід визначитися щодо бажаного ступеня подальшого наближення українського законодавства до законодавства ЄС у сфері реєстрації лікарських засобів і приведення у відповідність зі стандартами ЄС реєстраційних матеріалів вже зареєстрованих лікарських засобів.
(5) У зв’язку з відносно загальним характером зобов’язань України в рамках Угоди про асоціацію в сфері лікарських засобів Україна має можливість обрати оптимальні для себе методи наближення.
(6) Спрощена реєстрація лікарських засобів, зареєстрованих в країнах, що не є членами ЄС, суперечить стандартам ЄС. Це питання потребує політичного рішення і законодавчого врегулювання.
(7) Відсутня реальна інституційна співпраця у сфері реєстрації лікарських засобів між відповідними установами України і ЄС.
Рекомендації:
(8) Включити питання щодо розвитку системи реєстрації лікарських засобів до Національної політики щодо забезпечення лікарськими засобами.
(9) Прийняти повністю нові законодавчі акти у сфері реєстрації лікарських засобів на заміну діючим для гармонізації українського законодавства із законодавством ЄС шляхом повної імплементації законодавства ЄС в українське законодавство.
Відтермінувати введення в дію нових законодавчих актів на строк, достатній для того, щоб дати можливість компетентним державним органам і заявникам підготуватися до змін.
(10) [Опціонально] Внести зміни до законодавства України з метою наближення його до європейського на рівні підзаконних актів без внесення змін до законів або прийняття нових законів.
(11) Розробити план гармонізації досьє лікарських, засобів зареєстрованих раніше, із європейськими стандартами. Визначити часові рамки і рівень гармонізації, з урахуванням можливостей українських органів у сфері державної реєстрації лікарських засобів та необхідності забезпечення безперервного доступу до ліків для пацієнтів України.
(12) Реформувати спрощенні процедури реєстрації лікарських засобів, зареєстрованих в країнах, які не є членами ЄС, для приведення їх у відповідність з європейським підходом до безпеки лікарських засобів. На наступному етапі -застосовувати спрощену процедуру для лікарських засобів, зареєстрованих в країнах ЄС.
(13) Ініціювати створення робочої групи або підкомітету з лікарських засобів на основі Угоди про асоціацію, включити до неї представників українських органів реєстрації лікарських засобів і ЕМА.
І.3 СПОСТЕРЕЖЕННЯ У СФЕРІ ЗАКОНОДАВСТВА
(14) Співвідношення між основними правовими актами, що регулюють процедуру реєстрації лікарських засобів, а саме Законом про лікарські засоби, Постановою КМУ № 376 та Наказом МОЗ № 426, є нелогічним і незрозумілим.
(15) Предмет регулювання актів законодавства різного рівня часто перетинається. Окремі регуляторні положення дублюються у текстах різних актів. При цьому всі зазначені акти містять унікальні положення, не відображені, а інколи і не відповідні іншим актам. Не виключена ситуація, коли Наказ МОЗ № 426 містить норми, відсутні у Законі України про лікарські засоби та Постанові КМУ № 376, Постанова КМУ № 376 необґрунтовано розширює положення Закону України про лікарські засоби тощо.
(16) Нормативно-правові акти, що регулюють спрощену процедуру реєстрації лікарських засобів, наразі містять чимало суперечностей.
(17) Процес внесення змін у систему законодавства про реєстрацію лікарських засобівне координується, що створює плутанину для заявників та службових осіб. Змінив нормативно-правових актах вищої юридичної сили автоматично непереносяться до актів нижчої юридичної сили. Наприклад, деякі положення щодо спрощеної процедури реєстрації, прийняті у 2016 році, спочатку були внесені до Постанови КМУ № 376, а же після цього – до Закону України про лікарські засоби.
(18) Деякі положення українського законодавства, які свого часу були гармонізовані ззаконодавством ЄС, наразі не приведені у відповідність із останніми змінами узаконодавстві ЄС.
Рекомендації:
(19) Всі основні елементи процедури державної реєстрації регулюються виключно Законом про лікарські засоби, в тому числі: визначення термінів, види продукції, що підлягають державній реєстрації, установи, які беруть участь у процедурі, види процедур реєстрації, підстави відмови або обмеження державної реєстрації, строк дії державної реєстрації, підстави для перереєстрації та відмови в перереєстрації, види процедур для внесення змін до реєстраційних матеріалів.
(20) Закон про лікарські засоби повинен чітко визначати сферу регуляторних повноважень, делегованих КМУ та/або Міністерству охорони здоров’я України.
(21) [Опціонально] Всі ключові елементи процедури державної реєстрації можуть бути відображені в шляхом внесення змін або викладення у новій редакції Постанови Кабінету Міністрів України, як органу відповідального за прийняття правил державної реєстрації лікарських засобів в Україні згідно з Законом про лікарські засоби.
(22) Законодавство має бути переглянуте для усунення повторень і суперечностей.
(23) Необхідно запровадити систему моніторингу і контролю відповідності законодавства України законодавству ЄС, наприклад, шляхом створення спеціального підрозділу в структурі МОЗ.
І.4 СПОСТЕРЕЖЕННЯ У СФЕРІ ОРГАНІЗАЦІЇ ПРОЦЕСІВ
І.4.1 Щодо тривалості процедури
(24) Строки в законодавстві та у внутрішніх процедурах установ, що беруть участь у процедурі реєстрації, часто встановлюються непослідовно і навіть нелогічно.
(25) Не встановлена допустима загальна тривалість всієї процедури державноїреєстрації, не визначена установа, яка б несла відповідальність за загальнутривалість процедури.
(26) Заявники не мають ефективних засобів притягнення до відповідальності зазатримки у державній реєстрації.
І.4.2 Щодо ефективності процесу
(27) Процедура перевірки відповідності заяви формальним вимогам є надмірно складною. Заявник подає документи в кілька етапів, є дві окремі стадії перевірки такої відповідності.
(28) Компетенції МОЗ і ДЕЦ перетинаються у межах процедури реєстрації.
(29) Колегіальні консультативні органи ДЕЦ відіграють невиправдано значну і часто вирішальну роль у процедурі реєстрації.
(30) Вимога про верифікацію українськими органами GMP-сертифікатів, що видані країнами-членами PIC/S (або хоча б країнами ЄС) для цілей державної реєстрації лікарських засобів, є не потрібною і часто порушує її хід.
(31) Процедура реєстрації, що передбачає участь МОЗ після отримання висновків експертизи, є надмірно складною.
(32) Стандартні операційні процедури ДЕЦ є недостатньо якісними.
(33) Використання формату “єдиного вікна” у процедурі державної реєстрації є неефективним та ніяким чином її не покращує.
І.4.3 Щодо прийнятої моделі процедури
(34) Розподіл повноважень в чинному законодавстві між різними органами, залученими до процесу реєстрації, не є чітким.
(35) В окремих сферах експертиза з використанням внутрішніх ресурсів ДЕЦ не є можливою, і ДЕЦ повинен повністю покладатися на зовнішніх експертів. Немає чітких правил щодо співпраці з зовнішніми експертами.
(36) Участь МОЗ в процедурі, як на початковій так і на кінцевій її стадії, є неефективною.
(37) Державна реєстрація шляхом видачі наказу, яким затверджується часто значний перелік різних лікарських засобів, є неефективною.
(38) Діючи на підставі договорів із заявниками, ДЕЦ функціонує як квазі-комерційна установа, що надає послуги заявникам. Такий підхід не узгоджується з реальною роллю ДЕЦ в системі і може викликати питання відповідності антимонопольному законодавству.
І.4.4 Щодо участі Держлікслужби
(39) Процедура підтвердження Держлікслужбою відповідності умов виробництва лікарських засобів вимогам GMP є паперовою і не здійснюється у електронній формі.
(40) Незважаючи на той факт, що попередня і спеціалізована експертиза при підтвердженні відповідності GMP повинна займати 2-5 і 3-15 робочих днів (в залежності від типу процедури), на практиці навіть просте підтвердження GMP-сертифікатів, виданих FDA або однією з європейських інспекцій, може зайняти 3-6 місяців і більше. Такі затримки можуть поставити під загрозу саму процедуру реєстрації. Більш того, сама наявність попередньої формальної експертизи поданих документів у межах процедури підтвердження відповідності вже виданих інспекторатами PIC/S GMP сертифікатів є зайвою.
(41) Повноваження Робочої групи, що функціонує в рамках Держлікслужби, не врегульовані чинним законодавством, незважаючи на її важливу роль у процедурі підтвердження відповідності GMP. Це робить вплив Робочої групи на процес прийняття рішень щодо відповідності GMP непередбачуваним і непрозорим.
(42) Відповідальність за прийняття рішень щодо відповідності GMP є розмитою.
Рекомендації
Наші рекомендації щодо можливих змін в системі повинні бути детально обговорені з представниками відповідних установ та інших зацікавлених сторін, щоб розробити такий спосіб їх реалізації, який би дозволив системі залишатися стабільною у перехідний період.
(43) Переведення всіх можливих завдань в сфері державної реєстрації до компетенції ДЕЦ (за умови врахування численних рекомендацій цього Звіту щодо зміни підходів до функціонування ДЕЦ).
(44) [Опціонально] Для підвищення рівня прозорості процесів та загального рівня довіри до системи реєстрації як у заявників, так і у представників громадськості, основні функції у сфері державної реєстрації, що на сьогодні виконуються ДЕЦ, можуть бути передані до компетенції спеціалізованої новоствореної структури. При цьому, обов’язковими умовами для передачі відповідних функцій є:
- збереження новоствореною структурою статусу державного підприємства у сфері управління МОЗ (новостворена структура не повинна мати статус органу державної влади);
- максимальна імплементація рекомендацій, що містяться у цьому Звіті, при розробці (і) організаційної структури новоствореної структури та (іі) основних засад її діяльності.
(45) [Опціонально] Переведення компетенції приймати рішення про державну реєстрацію до ДЕЦ/новоствореної структури, в той час як МОЗ зберігатиме повноваження загального нагляду за роботою ДЕЦ/новоствореної структури і розгляду адміністративних скарг на рішення ДЕЦ/новоствореної структури.Реалізація таких змін може вимагати внесення відповідних змін на рівні законів, а також зміни правового статусу ДЕЦ і його працівників.
(46) Поточну модель державної реєстрації через прийняття колективного наказу МОЗ щодо переліку лікарських засобів слід замінити системою індивідуальних рішень про державну реєстрацію.
(47) Необхідно запровадити інститут і процедуру адміністративного оскарженнярішення про державну реєстрацію.
(48) Процедура державної реєстрації повинна бути спрощена і консолідована, кількість етапів і подач/обмінів документами має бути максимально скорочена.
(49) [Опціонально] Вимога про підтвердження відповідності умов виробництва вимогам GMP, як обов’язкова частина державної реєстрації, повинна бути скасована по відношенню до заявників із діючими GMP сертифікатами, що видані в країнах-членах PIC/S (або хоча б в країнах ЄС). Питання сертифікації GMP повинні належати виключно до компетенції Держлікслужби. Водночас, Держлікслужба повинна бути виключена з процедури державної реєстрації.
(50) Час, необхідний для лабораторного тестування якості лікарських засобів і відтворюваності методів контролю, повинен включатися в загальний строк всієї процедури.
(51) Договір, як підстава для виконання ДЕЦ своїх обов’язків, повинен бути усунений або істотно доопрацьований. Здійснення експертизи ДЕЦ повинно розглядатися як виконання обов’язків держави в сфері реєстрації лікарських засобів, а не як надання комерційних послуг.
(52) Впровадження єдиного реєстраційного внеску, який повинен сплачуватися авансом. Сума платежу повинна бути врегульована у відповідному нормативно-правовому акті.
(53) Кваліфікаційна комісія1, Науково-експертна рада, Науково-технічна рада, Технічно-експертна комісія повинні бути замінені одним органом, що надаватиме консультації компетентному органу у сфері реєстрації лікарських засобів. Новостворений орган повинен мати виключно консультативні функції та надавати консультації лише з питань по суті, а не щодо процедури, і лише з питань, які є прецедентними або викликають сумніви з точки зору наукової оцінки.
(54) Рішення про передачу справи на розгляд консультативного органу повинно прийматися головою відповідного департаменту ДЕЦ або Директором ДЕЦ. В листі-направленні повинні вказуватися причини передачі справи в консультативний орган, а також точний обсяг питань, стосовно яких вимагається консультація.
(55) Експерти мають нести відповідальність тільки по відношенню до свого роботодавця. Це має захищати їх від відповідальності перед заявниками (за винятком випадків, коли експерт умисно порушив закон).
(56) Необхідно врегулювати співпрацю ДЕЦ із зовнішніми експертами.
(57) Залучення зовнішніх експертів повинно бути можливим у випадках, якщо виконання експертизи неможливе за рахунок власних ресурсів ДЕЦ.
(58) Офіційний список зовнішніх експертів повинен бути доступний громадськості.
(59) Зовнішні експерти повинні підписувати декларацію про відсутність конфлікту інтересів і підтверджувати відсутність такого конфлікту перед кожною експертизою. Не повідомлення про конфлікт інтересів має бути підставою для юридичної відповідальності.
(60) Повинні бути введені загальні обмеження по строкам процедури державної реєстрації (перереєстрації, внесення зміни в реєстраційні матеріали), починаючи з дати подання повної заяви і до дати винесення рішення про державну реєстрацію (перереєстрацію, внесення змін до реєстраційних матеріалів).
(61) Необхідно визначити єдиний компетентний орган в якості відповідального за здійснення державної реєстрації у встановлений законом строк. Цей орган повинен нести відповідальність за організацію процесу реєстрації у встановлені строки.
(62) Необхідно передбачити правовий механізм оскарження заявником затримок у процедурі реєстрації.
(63) СОПи в ДЕЦ, а також Інструкція з організації проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються з метою державної реєстрації, повинні бути переглянуті для того, щоб:
- виключити можливість їх неоднакового трактування працівниками ДЕЦ,
- забезпечити реалізацію процесів без використання додаткових джерел інформації,
- встановити розумні й ефективні внутрішні строки виконання.
(64) Планувати перехід до здійснення процедури підтвердження відповідності вимогам GMP в електронній формі, що сприяло би своєчасності, прозорості і більшій ефективності процедури. Це також полегшило би доступ до відповідної інформації для регуляторних органів, в тому числі і для МОЗ та ДЕЦ, а також зробило можливим громадський контроль за процедурою.
(65) Скасувати етап попередньої експертизи під час процедури підтвердження GMP-сертифікатів, виданих інспекціями країн-членів PIC/S. Забезпечити на рівні нормативно-правового акту, що повноваження щодо призупинення перебігу строку регуляторним органом можуть бути використані ним тільки один раз в тих випадках, коли посадові особи Держлікслужби запитують додаткові документи чи дані (за умови, що заявник належним чином відповідає на запит і надає всі запитувані дані).
(66) Припинити практику обов’язкового схвалення Робочою групою результатів експертизи під час процедури підтвердження GMP-сертифікатів, або чітко регламентувати її роль, повноваження і процес прийняття рішень на рівні Наказу МОЗ № 1130.
_________________
1 Не згадується в чинній редакції Наказу № 426, але передбачена внутрішніми документами ДЕЦ і може вливати на процедуру реєстрації, які почалися до внесення останніх змін до Наказу 426.
І.5 СПОСТЕРЕЖЕННЯ У СФЕРІ КОМУНІКАЦІЙ ТА ПРОЗОРОСТІ
(67) Інтерфейс веб-сайтів ДЕЦ, Держлікслужби і MОЗ не є зручним для користувача та має недосконалу систему навігації. Англійські версії веб-сайтів не оновлюються на постійній основі, а більша частина веб-сайтів не перекладається англійською взагалі.
(68) На даний час інформація про поточні процедури, звіти про результати оцінки, а також статистичні дані, які б демонстрували результати роботи і містили стисле викладення змісту внутрішніх і зовнішніх аудитів ДЕЦ і МОЗ, не є загальнодоступними.
(69) Офіційний сайт ДЕЦ містить дуже обмежену інформацію щодо особливостей подачі документів в ДЕЦ та інших аспектів процедури.
(70) Право на отримання консультації експерта ДЕЦ перед поданням документів на експертизу не працює на практиці.
(71) Інформація про внутрішні процедури ДЕЦ щодо реєстрації є конфіденційною без чітких причин чи підстав.
(72) Відсутня загальнодоступна інформація про розподіл повноважень між підрозділами ДЕЦ, про внутрішніх і зовнішніх експертів, які проводять експертизу реєстраційних матеріалів.
Рекомендації:
(73) Оновити офіційні веб-сайти МОЗ, ДЕЦ та Держлікслужби, щоб зробити їх зручними та доступними для користувачів, регулярно оновлювати в тому числі і англійські версії веб-сайтів.
(74) Внести зміни до Наказу МОЗ № 426 відповідно до останніх змін в Законі про лікарські засоби у частині публічної інформації про статус процедури реєстрації.
(75) Публікувати статус кожної поточної процедури реєстрації.
(76) Розміщувати у відкритому доступі статистику та підсумки внутрішніх та зовнішніх аудитів.
(77) Розміщувати у відкритому доступі звіти за результатами оцінки лікарських засобів.
(78) Беручи за зразок EMA, кожен лікарський засіб, який був зареєстрований, повинен мати власну сторінку з історією реєстрації засобу, перереєстрації та всіх внесених змін.
(79) Додати на офіційний веб-сайт рекомендації щодо здійснення кожної процедури, а також секцію FAQ та відповідні зразки всіх документів, які використовуються заявниками у межах процедури.
(80) Застосовувати на практиці положення Наказу МОЗ № 426 стосовно права отримання консультації від експертів ДЕЦ перед подачею документів на експертизу.
(81) Ввести можливість платної наукової консультації для заявників.
(82) Розробити і впровадити для заявників і їх фахівців тренінги/освітні курси ДЕЦ з фінальним екзаменом, що дозволить учасникам отримати сертифікат позавершенню (в результаті, сертифікат менеджера з регуляторних питань маєрозглядатися працедавцями як перевага).
(83) Доступ до документів, які визначають внутрішні процедури ДЕЦ, Держлікслужби, МОЗ та стосуються процедури державної реєстрації, неповинен бути обмеженим.
(84) Публікувати на веб-сайті ДЕЦ довідкову інформацію про внутрішніх та зовнішніх експертів, які проводять експертизу реєстраційних матеріалів, беручи за приклад EMA.
І.6 СПОСТЕРЕЖЕННЯ У СФЕРІ ФІНАНСУВАННЯ
(85) У порівнянні з європейськими країнами, вартість державної реєстрації лікарських засобів в Україні є відносно низькою.
(86) Ціни на експертизу не регулюються в нормативних актах, а встановлюються ДЕЦ на підставі внутрішнього наказу.
(87) Для пришвидшення розвитку реєстраційної системи України необхідно покращити систему фінансового менеджменту, бюджетування і планування, а також здійснити значні інвестиції в розвиток системи електронного урядування та електронного документообігу, інфраструктури, прозорості та комунікації, мотивації та розвитку персоналу.
Рекомендації:
(88) Спланувати поступове та обґрунтоване підвищення вартості експертизи.
(89) Знайти альтернативні джерела фінансування, окрім підвищення вартості експертизи (наприклад, за рахунок надання платних наукових консультацій).
(90) Ввести преференційні умови (зменшену вартість експертизи) з метою надання переваг певним видам лікарських засобів і/або заявнику, наприклад для малих та середніх підприємств, для препаратів-сиріт і лікарських засобів з педіатричними показаннями.
(91) Встановити максимальні розцінки на всі види експертизи на рівні нормативних актів.
І.7 СПОСТЕРЕЖЕННЯ У СФЕРІ ЛЮДСЬКИХ РЕСУРСІВ
(92) Середній дохід експерта ДЕЦ варіюється від близько 345 – 397 Євро на дату Звіту.
Ці суми є щонайменше вдвічі нижчими за ринкову заробітну плату та втричі меншими, ніж 3 роки тому (в зв’язку з девальвацією національної валюти). ДЕЦ не переглядав зарплати працівників останні 5 років.
(93) Близько 10% посад в ДЕЦ (50 з 482 посад) вакантні, включаючи посади для тимчасового заміщення в зв’язку з декретною відпусткою. У більшості структурних підрозділів ДЕЦ немає перевантаження через недостачу персоналу.
(94) У ДЕЦ відсутня розроблена та підтримувана система оцінки ефективності роботи.
Голови відповідних структурних підрозділів контролюють часові межі процедури реєстрації самостійно.
(95) Голови всіх департаментів самостійно визначають премії працівників департаменту відповідно до власних припущень щодо навантаження та вкладу в загальний результат за певний період. Відсутня система ключових показників ефективності.
(96) Керівництво ДЕЦ нечасто притягує працівників до дисциплінарної відповідальності. У випадку порушення процедури або серйозної затримки експерт зазвичай просто отримує зменшену премію та догану.
(97) ДЕЦ залучає велику кількість зовнішніх експертів, зазвичай добре відомих лікарів та науковців, які є спеціалістами в різних галузях медицини. Їх регулярно залучають до експертизи матеріалів досьє.
(98) Більшість посад у ДЕЦ заповнюються через рекомендації від зовнішніх експертів. У рідкісних випадках ДЕЦ отримує заявки від досвідчених кандидатів.
(99) Сьогоднішня форма організаційної структури призводить до концентрації контролю та відповідальності на рівні керівництва ДЕЦ, а саме Директора та Заступника Директора ДЕЦ.
(100) Всі основні структурні підрозділи підпорядковуються безпосередньо Директору ДЕЦ. Є два Заступника Директора: один відповідає за економічні та адміністративні функції, інший – за ІТ, інноваційні проекти та стандартизацію медичних послуг.
(101) Департамент інноваційних проектів виконує функції, які не притаманні ДЕЦ (розробка медичних стандартів, форм та інша технічна допомога МОЗ).
(102) ДЕЦ створив декілька дорадчих органів з метою розширення експертизи щодо різних видів лікарських засобів через залучення спеціалістів у сфері охорони здоров’я різних спеціалізацій. Вісімнадцять спеціалізованих комітетів у складі Експертної Консультаційної Групи залучаються до процесу експертизи матеріалів досьє. Наукова-експертна рада та Науково-технічна рада проводять фінальні засідання і вирішують, чи рекомендувати МОЗ зареєструвати лікарський засіб чи відмовити в реєстрації, а також низку інших питань.
(103) У штаті ДЕЦ присутні особи, які фактично працюють/надають послуги в МОЗ.
(104) ДЕЦ покриває деякі витрати МОЗ, як, наприклад, повсякденні офісні витрати, витрати на організацію заходів та відряджень.
Рекомендації:
(105) Здійснити обґрунтоване підвищення зарплат працівників та розробити компенсаційний пакет, який забезпечить періодичний перегляд заробітної плати.
(106) Прив’язати поступове підвищення зарплат експертів до підвищення рівня вартості експертизи.
(107) Здійснити детальний аналіз бізнес-ефективності ДЕЦ та можливості подальшої оптимізації у контексті перерозподілу бюджету.
(108) Запровадити прості інструменти для надання вичерпної інформації для управлінських рішень.
(109) Розробити систему ключових показників ефективності для структурних підрозділів та ключових співробітників ДЕЦ та прив’язати змінну частину компенсації працівників до поєднання індикаторів індивідуальної та групової продуктивності.
(110) Ввести чіткі правила залучення зовнішніх експертів, обмежити їх залучення лише тими випадками, коли не можна провести експертизу лише за допомогою ресурсів ДЕЦ.
(111) Ввести ефективні механізми набору персоналу та створити кадровий резерв наключові посади в ДЕЦ.
(112) Розробити організаційну структуру, яка підтримуватиме перерозподіл обов’язків від Директора до Заступників Директора та голів департаментів і підрозділів ДЕЦ, а також спростити процедуру прийняття рішень.
(113) Об’єднати основні функції в рамках декількох великих департаментів та закріпити їх за діючими Заступниками Директора (один – всі реєстраційні функції, інший – клінічні дослідження та похідні функції), в той час як допоміжні підрозділи підпорядковуватимуться безпосередньо Директору.
(114) [Опціонально] Перевести Департамент інноваційних проектів в МОЗ.
(115) Обмежити участь колегіальних органів в реєстрації лікарського засобу лише до унікальних та/або спірних випадків.
(116) Перевести фактичних працівників МОЗ, що знаходяться у штаті ДЕЦ, в окремий підрозділ з подальшим переведенням в МОЗ.
(117) [Опціонально] Обговорити можливість збільшення бюджету МОЗ для покриття витрат у сфері державної реєстрації.
1.8. СПОСТЕРЕЖЕННЯ У СФЕРІ ВИКОРИСТАННЯ ЕЛЕКТРОННИХ ІНФОРМАЦІЙНИХ СИСТЕМ
(118) Права інтелектуальної власності на Інформаційну систему експертного супроводу лікарських засобів ДЕЦ (MES) належать приватній компанії.
(119) Під час процедури державної реєстрації документація подається в основному у паперовому вигляді.
(120) Завдання для експертів призначаються через інформаційні системи лише частково.
(121) Система не має функціональної можливості реєстрації часу, витраченого експертами, або іншої інформації, збереження якої сприяло б кращій комунікації з ДЕЦ.
(122) Система MES має односпрямований інтерфейс з Системою візуалізації інформації (СВ). СВ не має можливостей, які б надали змогу заявниками взаємодіяти з ДЕЦ в системі.
(123) Обмін даними між MES та Державним реєстром лікарських засобів України (ДРЛЗ) проводиться вручну.
(124) Неповний процес обміну даними з іншими інформаційними системами. MES не пов’язана ні з Єдиним державним реєстром юридичних осіб, фізичних осіб-підприємців та громадських формувань, ні з іншими державними інформаційними системами для перевірки особи та статусу заявника.
(125) Функція відправки повідомлень заявникам не доступна в СВ.
(126) Всі користувачі в СВ реєструються вручну співробітниками ДЕЦ.
(127) Нинішній рівень кодування інформації (інформації про упаковку, дозування, концентрацію) обмежує використання ДРЛЗ для аналізу ринку, контролю цін, статистичних цілей та інших способів використання цієї інформації.
(128) Платформа розробки ДРЛЗ є застарілою.
(129) ДРЛЗ не містить інформації щодо цін і статистики використання лікарських засобів. Існує окремий реєстр цін на лікарські засоби, який утримується МОЗ.
(130) Відсутня функція резервного копіювання інформації. Це значно підвищує ризик втрати даних в разі проблем з мережею, у випадку пожежі, стихійного лиха тощо.
(131) Відсутня стратегія розвитку і використання інформаційних систем у сфері охорони здоров’я.
Рекомендації:
(132) Права інтелектуальної власності на програмне забезпечення повинні належати державі, в разі якщо розробка програмного забезпечення фінансується з державного бюджету або інших ресурсів, що належать державі. Такий підхід повинен реалізовуватися на основі загального законодавства про державні закупівлі та має застосовуватися до програмного забезпечення, розробленого на замовлення.
(133) Щодо прав інтелектуальної власності на існуюче програмне забезпечення, необхідно зрозуміти, чи є доцільним продовження відносин власності у старому форматі, чи необхідно організувати переведення цих прав на державну установу, або розробити новий формат та відмовитися від старої системи.
(134) Необхідно розглянути можливість введення правових та технічних заходів автентифікації і захисту електронної документації.
(135) Подання і обробка інформації повністю в електронному вигляді буде потребувати більш потужної ІТ-інфраструктури.
(136) Забезпечити функціональні можливості для реєстрації часу в рамках MES.
(137) Розробити і утримувати систему управління, яка сприятиме більшпродуктивній роботі експертів. Розробити відповідні ключові показникиефективності для експертів.
(138) Необхідно впровадити новий портал ДЕЦ для заявників. Портал повинен забезпечити всі необхідні функції для комунікації між заявниками та ДЕЦ:
- подачу реєстраційних досьє в електронній формі,
- можливості для надання запитів, відповідей, коментарів та надсилання повідомлень,
- електронні засоби зв’язку повинні переважно замінити існуючий документообіг на паперових носіях.
За допомогою двонаправленого інтерфейсу, ця інформація буде автоматично з’являтися на робочому місці експерта.
(139) Забезпечити автоматизований інтерфейс передачі даних між MES та ДРЛЗ.
(140) Схвалення реєстрації лікарських засобів повинно вводитися в систему посадовими особами МОЗ (якщо буде прийнято рішення залишити цей етап процесу). Після авторизації система повинна автоматично публікувати відповідну інформацію про лікарські засоби в ДРЛЗ.
(141) Створити інтерфейси для обміну даними з іншими інформаційними системами.
(142) MES та ДРЛЗ повинні бути доповнені відкритими інтерфейсами, що дозволяють інтегрувати інші інформаційні системи (третіх сторін), тобто системи лікарень, аптек, електронних рецептів та статистики. Електронна інформація може використовуватися іншими установами відповідно до їх рівня авторизації, наприклад, митницею, Держлікслужбою. Первинна інформація, яка має бути доступною автоматично в зазначених інтерфейсах, − це класифікатори та інформація щодо реєстрації лікарських засобів.
(143) MES повинна мати можливість автоматично отримувати інформацію, яка зберігається в інших державних інформаційних системах та є необхідною для її процесів, наприклад, дані з Реєстру юридичних осіб для перевірки правового статусу заявника.
(144) Забезпечити функцію електронного повідомлення для користувачів.
(145) Забезпечити самостійну реєстрацію користувачів або дворівневе адміністрування користувачів для заявників.
(146) Забезпечити вдосконалені кодування та опис інформації про лікарські засоби.
(147) Додати необхідну інформацію до реєстраційного номеру лікарського засобу. Розробити необхідні процедури для застосування нового кодування, які повинні застосовуватися до всіх нових реєстрацій. Необхідно визначити перехідний період, протягом якого будуть оновлені старі записи. У разі необхідності власники реєстраційних посвідчень повинні надати додаткову інформацію для оновлення реєстраційних записів.
(148) Забезпечення процедур та правової бази для використання реєстраційного номеру лікарських засобів в якості первинного ключа для ідентифікації лікарського засобу в рамках інтеграції ДРЛЗ до іншої інформаційної системи.
(149) Забезпечити контроль введення даних в інформаційну систему.
(150) Інші опції зміни технології повинні досліджуватися та оцінюватися.
(151) Перевірити та визначити вимоги до управління ціноутворенням на лікарські засоби. У майбутньому ціни можуть бути визначені в процесі реєстрації лікарських засобів. Тому інформацію стосовно ціноутворення необхідно включити в MES.
(152) Забезпечити резервні засоби обробки даних.
(153) Необхідно впровадити процес загального планування та прийняття рішень щодо інформаційних систем як між установами, так і всередині установ. Для впровадження такого процесу має бути чітке визначення понять «повноваження» та «прийняття рішень», які мають бути зафіксовані в єдиному документі планування (наприклад, «Стратегія розвитку інформаційних систем в галузі охорони здоров’я»).
II. ВСТУП
1. З метою покращення процедури реєстрації лікарських засобів в Україні, МОЗ звернулося до ЄБРР з пропозицією розпочати обговорення стратегії вдосконалення процедури реєстрації лікарських засобів та практики її застосування з метою їх приведення у відповідність зі стандартами/найкращими практиками ЄС. Обговорення стратегії має проводитись спільно з представниками індустрії, EMA, ВООЗ, Представництва ЄС в Україні, Американської торгової палати (ACC), Європейської Бізнес Асоціації (ЄБА), Асоціації виробників інноваційних ліків (АПРаД), Асоціації «Виробники ліків України» та інших галузевих асоціацій та відповідних державних органів.
2. ЄБРР розпочав проект «Консультування щодо регуляторних змін в фармацевтичному секторі України» («Проект») та обрав консорціум у складі п’яти компаній як консультантів Проекту: Tomasik JaworskiSp.p. (Лідер, Польща), Marchenko Danevych(Україна), APC Instytut Sp. ТОВ (Польща), Red Fox Consulting ТОВ (Латвія), Talent AdvisorsТОВ (Україна) («Консорціум»).
3. Консорціум запропонував комплексний підхід для оцінки діючої процедури реєстрації лікарських засобів. Відповідно до запропонованого підходу, оцінці підлягають юридичні, регуляторні аспекти, рівень прозорості та комунікацій, ІТ, HRта фінансові аспекти процедури реєстрації лікарських засобів.
4. Цей Звіт підготовлено за результатами Фази 1 Проекту.
II.1. ЦІЛЬ
5. Головна ціль Звіту – проаналізувати нинішній стан системи реєстрації лікарських засобів в Україні, визначаючи як позитивні аспекти, так і головні недоліки, а також запропонувати можливі шляхи покращення системи для приведення процедури у відповідність зі стандартами та практиками ЄС, а також підвищити її ефективність та прозорість.
6. У Звіті також приділена увага стратегічним аспектам, необхідним для обрання шляху реформування процедури реєстрації лікарських засобів та її розвитку.
II.2. МЕТОДОЛОГІЯ
7. Експерти Консорціуму проаналізували різні аспекти процедури реєстрації лікарських засобів на основі таких методів:
(a) Детальне порівняння законодавства та практики України та ЄС у сфері реєстрації лікарських засобів (перереєстрації, внесення змін).
(b) Онлайн опитування та особисті співбесіди з різними заявниками на реєстрацію, що представляють гравців на фармацевтичному ринку (українські та іноземні, інноваційні та генеричні, фармацевтичні та біотехнологічні компанії, а також провайдери регуляторних послуг, які працюють в Україні).
(c) Співбесіди з працівниками та експертами державних органів (МОЗ, Держлікслужба), а також працівниками ДЕЦ.
(d) Огляд внутрішніх документів ДЕЦ стосовно процедури експертизи, персоналу, ІТ інфраструктури, тарифів за послуги експертів і т. п.
(е) Огляд відповідних джерел інформації про процедуру реєстрації у вільному доступі, а також ролі МОЗ, Держлікслужби та ДЕЦ в процедурі.
(f) Огляд матеріалів щодо реєстрації (перереєстрації, внесення змін) та файлів певних лікарських засобів, що зберігаються в ДЕЦ та МОЗ.
(g) Співбесіди з експертами ВООЗ, Світового банку та Представництва ЄС в Україні, які брали участь у вже проведених оцінюваннях системи реєстрації лікарських засобів, а також огляд їх звітів та висновків.
(h) Обговорення початкових висновків з представниками заявників, а також з МОЗ, ДЕЦ та Держлікслужбою.
8. Робота в рамках Проекту, здійснена Консорціумом, постійно координувалася Комітетом, сформованим ЄБРР з представників МОЗ, ДЕЦ, Представництва ЄС, а також експертів різних галузевих та бізнес асоціацій.
II.3. ПРИПУЩЕННЯ
9. Фармацевтичний ринок України має один із найвищих показників росту в країні. Однак, регулювання індустрії і практика застосування такого регулювання потребують значних змін, які могли б потенційно стимулювати подальше зростання обсягів виробництва і продажів фармацевтичних компаній, а також залучення нових інвестицій, в тому числі від ЄБРР.
10. В той час як потенційний об’єм регуляторних змін доволі значний, реєстрація лікарських засобів виділена учасниками ринку для ЄБРР як одна із найбільш критичних проблем для нормального функціонування ринку. Процес реєстрації складний, включає в себе багато процедур та під-процедур, численні комунікації між заявниками та регуляторними органами і, як результат, невмотивовані затримки. Частина учасників ринку часто характеризує процес якнепрозорий, непередбачуваний і деколи недостатньо професійний.
11. Існуюча подвійна система реєстрації лікарських засобів в Україні складається з двох основних елементів:
Експертиза реєстраційних (перереєстрації, внесення змін) матеріалів експертами ДЕЦ як провайдера платних експертних послуг, за якою слідує рекомендація МОЗ затвердити чи відхилити реєстрацію (перереєстрацію, внесення змін).
Формальна реєстрація (перереєстрація, внесення змін), затверджена МОЗ, як державним органом, базуючись виключно на рекомендаціях ДЕЦ.
12. Консорціум припускає, що існуюча система реєстрації лікарських засобів була встановлена через декілька важливих факторів, а саме:
Впровадження певних рекомендацій різних європейських регуляторних установ в останні два десятиліття;
Нестача фінансування державних установ (включаючи МОЗ), яка була компенсована переданням певних регуляторних функцій державним підприємствам (як-то ДЕЦ);
Існуючі законодавчі положення стосовно адміністративних послуг та дозвільної системи, а також формулювання головного законодавчого акту в фармацевтичній сфері – Закону про лікарські засоби.
13. На основі отриманої інформації, Консорціум припускає, що на реєстрацію лікарських засобів історично впливали:
Певний рівень конкуренції між МОЗ та ДЕЦ, що в деяких випадках відіграє балансуючу роль, але в інших випадках міг бути деструктивним фактором розвитку процесу реєстрації;
Втручання правоохоронних органів та різні кримінальні розслідування стосовно окремих процедур реєстрації, що могло бути одним із факторів, які призвели до надто консервативного та необгрунтовано обачного підходу до прийняття рішень, а також розмитість відповідальності в МОЗ та ДЕЦ;
Політичний, соціальний влив, а також вплив бізнесу прискорили розвиток системи реєстрації у певних аспектах, але деякі аспекти системи все ще потребують значного доопрацювання.
ІІ.4. ЗАГАЛЬНІ ПРИЦИПИ
14. Проводячи оцінку нинішньої системи реєстрації лікарських засобів, Консорціум застосовував неупереджений та незалежний підхід. Експерти Консорціуму отримали та опрацювали відгуки та пропозиції стосовно процедури реєстрації лікарських засобів від представників індустрії, різних міжнародних організацій, а також від посадовців та експертів ДЕЦ/МОЗ/Держлікслужби.
15. У ході порівняльного аналізу, експерти Консорціуму враховували як позитивні приклади та помилки у вибраних європейських країнах на стадії приєднання до ЄС, так і попередні та наступні реформи систем реєстрації лікарських засобів.
16. Протягом всього процесу оцінювання, Консорціум застосовував такі критерії як обґрунтованість, істотність, вагомість, реальність виконання до всіх своїх висновків та пропозицій.
17. Зазначення всіх висновків та рекомендацій, наведених у Звіті, має на меті наближення системи реєстрації лікарських засобів в Україні до законодавства та найкращих практик ЄС, її покращення та розвиток шляхом трансформації у більш ефективну, прозору, професійну, передбачувану та безпечну систему.
ІІ.5. СТРУКТУРА ЗВІТУ
18. Звіт розділений на дві основні частини.
19. В першій частині Звіту (Розділ ІІІ) ми описуємо результати порівняння українського законодавства в сфері реєстрації лікарських засобів з відповідними нормативно-правовими актами ЄС та практику імплементації законодавства ЄС в певних країнах- членах ЄС.
20. В другій частині Звіту (Розділ ІУ-Х) експерти наводять основні спостереження в усіх сферах в частині реєстрації лікарських засобів та рекомендації щодо змін.
Розділ ІV описує спостереження щодо політики України в сфері реєстрації лікарських засобів.
Розділ V описує спостереження в сфері законодавства (якість законодавства законодавчий процес).
Розділ VІ описує спостереження в сфері організації просу реєстрації у ДЕЦ та Держлікслужбі.
Розділ VІІ описує спостереження щодо прозорості та комунікації між ДЕЦ та заявниками.
Розділ VIII описує спостереження в сфері фінансування системи реєстрації лікарських засобів.
Розділ IX описує спостереження в сфері ИЯ.
Розділ X описує спостереження в сфері інформаційних систем.
21. До Звіту додають результати опитування (Додаток 1).
II.6. ПОДЯКА
22. Від імені Консорціуму Консультантів висловлюємо подяку Європейському банку реконструкції і розвитку за надану можливість реалізовувати Проект «Консультування щодо регуляторних змін у фармацевтичному секторі України» та підтримку протягом Фази 1 Проекту.
23. Ми щиро вдячні МОЗ, ДЕЦ, Держлікслужбі та їх працівникам за надання необхідної інформації та плідну співпрацю.
24. Ми також дякуємо представникам індустрії та всім членам Координаційного Комітету за активну участь та підтримку під час Фази 1 Проекту.
III. АНАЛІЗ ВІДПОВІДНОСТІ УКРАЇНСЬКОГО ЗАКОНОДАВСТВА СТАНДАРТАМ ЄС
III.1. НОРМАТИВНО-ПРАВОВА БАЗА В УКРАЇНІ
25. Державна реєстрація лікарських засобів здійснюється МОЗ за участі Держлікслужби та ДЕЦ.
26. МОЗ відіграє головну роль на початковому та кінцевому етапах процедури реєстрації. До МОЗ надходить заява від заявника, що у подальшому передається в ДЕЦ для проведення експертизи. На основі висновків ДЕЦ щодо якості, безпеки та ефективності засобу та його рекомендацій, МОЗ приймає рішення про державну реєстрацію. Таке рішення виноситься у формі наказу (додатку до попередньої версії наказу), що розповсюджується одразу на декілька засобів. Після цього МОЗ видає реєстраційне посвідчення, підготоване ДЕЦ, та направляє його заявнику.
27. ДЕЦ проводить експертизу заяви та поданого досьє. Експертиза включає в себе попередню експертизу (щодо повноти реєстраційного досьє) та спеціалізовану експертизу (для винесення обґрунтованого висновку щодо ефективності, безпеки та якості лікарського засобу, що супроводжується рекомендаціями для МОЗ). Після видачі наказу МОЗ, ДЕЦ вносить лікарський засіб до ДРЛЗ та готує документацію, яку МОЗ має направити заявнику.
28. Держлікслужба проводить перевірку відповідності вимогам GМР протягом здійснення експертизи ДЕЦ.
29. Законодавство України про державну реєстрацію лікарських засобів складається із декількох нормативних актів, виданих державними органами різних рівнів. Сукупність великої кількості нормативних актів, які регулюють одні і ті ж питання, ускладнює орієнтування в цій системі. При цьому регулювання деяких питань у ряді законодавчих актах є різним – у більшості випадків, такі невідповідності спричинена різним ступенем деталізації, однак також наявні значні невідповідності, більшість з яких спричинені тим, що норми підзаконних актів не відповідають змінам, внесеним у акти вищої юридичної сили.
30. Основні законодавчі акти, які регулюють сферу державної реєстрації ЛЗ в Україні вказані нижче:
- Закон про лікарські засоби,
- Постанова КМУ № 376,
- Наказ МОЗ № 426,
- Наказ МОЗ № 220.
- Закон про лікарські засоби:
31. Закон про лікарські засоби визначає основні умови державної реєстрації ЛЗ.
32. Крім того, Законом встановлюються загальні норми, які регулюють систему, а саме:
- лікарські засоби дозволені до використання в Україні лише після їх державної реєстрації;
- державна реєстрація вимагає подання заяви до МОЗ;
- після винесення МОЗ рішення щодо реєстрації, видається окреме реєстраційне посвідчення, а відповідна інформація вноситься до ДРЛЗ;
- після закінчення строку дії реєстраційних посвідчень ЛЗ підлягають перереєстрації.
33. Закон містить комплекс основних положень, якими регулюється процедура, а саме:
- вимоги до заяв,
- кінцевий термін прийняття рішення МОЗ,
- конфіденційність даних, які вносяться заявником, виключне право та патентний захист,
- підстави для відмови від прийняття заяви,
- вимоги до маркування.
34. Крім цього, Закон визначає дві додаткові категорії лікарських засобів, які реєструються на спрощених умовах, а саме:
- лікарські засоби, зареєстровані реєстраційними органами відповідних країн (США, Канади, Австралії, Японії, Швейцарії, ЕМА) – початково, лише лікарські засоби, призначені для лікування окремих захворювань, після останнього доповнення ця норма розповсюджується на всі лікарські засоби ,зареєстровані відповідними регуляторними органами;
- лікарські засоби, що підлягають закупівлі за результатами закупівельної процедури, проведеної спеціалізованою організацією.
35. Законом передбачається, що процедура державної реєстрації та розмір збору за державну реєстрацію повинні визначатись КМУ.
Постанова КМУ № 376:
36. Видається, що КМУ вирішив лише частково врегулювати процедуру реєстрації, а повноваження щодо визначення більшої її частини залишити для делегувати безпосередньо МОЗ.
37. Постанова КМУ № 376 – це нормативний акт, у якому містяться норми, що:
- повторюють положення Закону без суттєвих змін,
- щодо деяких питань містять більш детальні положення, ніж Закон,
- містять положення, які не визначаються у Законі.
38. Що стосується загальних принципів системи державної реєстрації, Постанова КМУ № 376 містить новий принцип, який не передбачений в Законі, а саме: обов’язкову експертизу заяв, що проводиться спеціалізованою організацією – ДЕЦ.
39. Аналогічно до Закону, Постанова КМУ № 376 регулює такі елементи процедури:
- вимоги до заяв,
- граничний строк прийняття рішення МОЗ,
- конфіденційність даних, які подаються заявником, ексклюзивність даних та патентний захист,
- підстави для відмови від прийняття заяви,
- однак, рівень деталізації є різним залежно від конкретного предмету регулювання. Іноді, положення Постанови КМУ № 376 повністю повторюють Закон, в інших випадках – вони є більш детальними. Слід зазначити, що ці два акти законодавства не повністю збігаються щодо таких важливих питань, як ексклюзивність даних (Закон є більш конкретизованим).
40. Постанова КМУ № 376 враховує зміни до процедури, пов’язані з двома окремими категоріями лікарських засобів (ЛЗ, зареєстровані окремими іноземними агенціями та ЛЗ, які підлягають закупівлі за результатами закупівельної процедури, проведеної спеціалізованою організацією).
41. Цікаво, що останні зміни стосовно спрощеної процедури для цих лікарських засобів спочатку були внесені Постанова КМУ № 376, а потім у Закон – Постанова КМУ № 376 передбачала спрощену реєстрацію усіх ЛЗ, зареєстрованих ЕМА (Стаття 2.2) значно раніше, ніж відповідні зміни були внесені до Закону. Також, розширення категорій таких засобів (від ЛЗ для лікування окремих захворювань до усіх ЛЗ) було частково здійснено у Постанові КМУ № 376 (напр., Стаття 3.4) до того, як почали діяти відповідні зміни до Закону.
42. Окрім цього, Постанова КМУ № 376 більш детально регулює окремі питання, які лише побіжно згадуються у Законі, а саме:
- підстави можливого анулювання реєстраційного посвідчення та заборони використання лікарських засобів,
- збір за державну реєстрацію.
43. Постанова КМУ № 376 також регулює питання, які нормативно не врегульовані в жодному іншому нормативному акті, а саме:
- вимоги до експертів ДЕЦ;
- процедура внесення змін до реєстраційних матеріалів;
- граничні строки експертизи ДЕЦ для однієї з категорій лікарських засобів (засоби, зареєстровані окремими іноземними регуляторними органами).
44. На відміну від Закону, Постанова КМУ № 376 не містить визначень термінів та вимог до маркування.
Наказ МОЗ № 426:
45. Наказ МОЗ № 426 містить перелік визначень, який є набагато ширшим, ніж перелік, передбачений Законом. Очевидно, що деякі визначення, що містяться у Наказі МОЗ № 426, не збігаються з деякими визначеннями, що містяться у Законі та Постанові КМУ № 376. Це стосується, наприклад, визначення «конфіденційна реєстраційна інформація» (II. 1.29 Наказу МОЗ № 426) або «неправомірне використання реєстраційної інформації про безпеку та ефективність лікарського засобу».
46. Наказом МОЗ № 426 регулюються ті ж питання, що й Законом та Постановою КМУ № 376, однак таке регулювання є набагато більш детальним (а іноді воно відрізняється від положень актів вищої юридичної сили) в контексті процедури реєстрації, перереєстрації та внесення змін до реєстраційних матеріалів.
47. Наказ МОЗ № 426 містить детальні положення щодо порядку внесення змін до реєстраційних матеріалів, у тому числі класифікацію змін по різним типам ЛЗ.
48. Наказ МОЗ № 426 містить окремі положення про конфіденційність, ексклюзивність даних та патентний захист, що певною мірою відрізняються від норм актів вищої юридичної сили.
49. Наказ МОЗ № 426 також регулює питання, які жодним чином не врегульовані у Законі або Постанові КМУ № 376, у тому числі:
- безкоштовні консультації для заявників, реєстрація «під умовами»;
- вимоги до реєстраційного досьє для різних типів заяв (повне досьє, генеричне досьє тощо),
- процедура в рамках ДЕЦ, включаючи: порядок дій ДЕЦ, етапи попередньої та спеціалізованої експертизи, залучення консультативних органів ДЕЦ, взаємодія ДЕЦ с МОЗ та заявником після проведення експертизи,
- право на оскарження висновків ДЕЦ,
- строки експертизи у ДЕЦ, залежно від типу заяви,
- взаємодія ДЕЦ та МОЗ на початковому та кінцевому етапах процедури.
Наказ МОЗ № 220:
50. Наказом МОЗ № 220 визначається процедура та правила взаємодії між МОЗ та ДЕЦ на початковому (обробка заяв та матеріалів від заявника) та кінцевому (підготовка переліку ЛЗ, рекомендованих та не рекомендованих до реєстрації, видача наказу МОЗ про реєстрацію, надання документів заявнику) етапах процедури.
II.2. ВИЗНАЧЕННЯ
51. Основні визначення, що містяться у законодавстві України у сфері охорони здоров’я, є подібними до визначень, що містяться у законодавстві ЄС. Однак існують деякі розбіжності, найбільш суттєві з яких визначені нижче.
ІІІ.2.1. Визначення поняття «лікарський засіб»
52. Ми встановили деякі розбіжності у обсязі визначення поняття «лікарський засіб».
53. По-перше, визначення поняття «лікарський засіб» в законодавстві України (ст. 2.1 Закону) стосується лише загальних характеристик та цільового застосування речовини
(«будь-яка речовина або комбінація речовин (одного або декількох АФІ та допоміжних речовин), що має властивості та призначена для…»)
54. Подібне визначення у Директиві 2001/83 (ст. 1.2) стосується форми, у якій ЛЗ подається на розгляд («будь-яка речовина або комбінація речовин, що має властивості…»), що дозволяє здійснювати ефективний нагляд за засобами, які мають граничні властивості, та за продуктами, що не є лікарськими засоби, які умисно представлені на ринку як лікарські.
55. По-друге, Закон не містить загального правила про те, що його положення стосуються лише ЛЗ, що виготовлені промисловим шляхом і призначені для виходу на ринок (що виключило б зі сфери застосування Закону продукцію «inbulk» та АФІ), яке було б еквівалентним положенню Статті 2.1 Директиви 2001/83.
Критерії оцінки (ЄС)
Стаття 2.1 Директиви 2001/83
Положення цієї Директиви застосовуються до лікарських засобів для застосування людиною, які представлені на ринку держав-членів та виготовлені як промисловим шляхом, так і за допомогою методів, що включають виробничі процеси.
56. По-третє, Стаття 2.3 Закону містить перелік ЛЗ, які внесені до категорії «лікарських засобів».
Стаття 2 пункт 3 Закону
До лікарських засобів належать: АФІ, продукція «in bulk»; готові лікарські засоби (лікарські препарати, ліки, медикаменти); гомеопатичні засоби; засоби, які використовуються для виявлення збудників хвороб, а також боротьби із збудниками хвороб або паразитами; лікарські косметичні засоби та лікарські домішки до харчових продуктів.
57. Цей перелік є занадто широким у порівнянні з законодавством ЄС. За винятком категорії «готових лікарських засобів», всі інші типи ЛЗ не повинні підпадати (або, принаймні, не повністю підпадати) під категорію «лікарського засобу»:
a) АФІ та продукція «in bulk» повинні ліцензуватися щодо їх виробництва та обігу, але немає сенсу реєструвати їх як лікарські засоби (див. наші примітки в пунктах 80-82);
b) згідно із законодавством ЄС, гомеопатичні засоби не вважаються «лікарськими»;
c) засоби, які використовуються для виявлення збудників хвороб, а також боротьби зі збудниками хвороб або паразитами – можливість їх віднесення до «лікарських» залежить від їх властивостей, наприклад, засоби, які використовуються лише для виявлення, скоріше підпадають під категорію медичних діагностичних засобів in vitro(в лабораторних умовах);
d) косметичні засоби та домішки до харчових продуктів не можуть вважатись лікарськими засобами.
58. По-четверте, в Законі немає загального правила тлумачення, яке б визначало критерії віднесення продукції до категорії «лікарського засобу» у випадку виникнення сумнівів.
Критерії оцінки (ЄС)
Стаття 2.1 Директиви 2001/83
У разі виникнення ситуації, за якої певний продукт з урахуванням всіх його властивостей може підпадати і під визначення «лікарський засіб», і під визначення іншого виду, мають використовуватися положення цієї Директиви.
59. Існує лише одне конкретне правило, що стосується можливих протиріч між поняттями «лікарський засіб» та «медичний виріб» (Статті 3 та 4 Постанови КМУ № 753 від 2012 р. «Про затвердження Технічного регламенту щодо медичних виробів»). Такого ж правила, яке б застосовувалось у випадку протиріч з поняттями домішок до харчових продуктів, косметичних засобів та біоцидних продуктів, не існує.
Рекомендації
Застосовувати визначення «лікарського засобу», що міститься в Директиві 2001/83.
Визначити сферу застосування Закону про лікарські засоби в порядку, аналогічному до Статті 2.1 Директиви 2001/83.
Розробити загальне правило тлумачення для віднесення засобів, які мають граничні властивості, для їх класифікації як лікарських засобів, по аналогії зі Статтею 2.2 Директиви 2001/83.
Видалити перелік типів засобів в Статті 1.3 Закону.
III.2.2. Посилання на не Європейські фармакопеї при визначенні «гомеопатичних лікарських засобів»
60. Визначення, що міститься в Статті II.1.15 Наказу МОЗ № 426, є подібним до визначення Директиви 2001/83 (Стаття 1.5), однак, за відсутності процедури в Українській або Європейській фармакопеї, дозволяється посилатись на «Німецьку гомеопатичну фармакопею (GHP), Фармакопею Сполучених Штатів Америки(HPUS), Британську фармакопею (BHP), Гомеопатичну фармакопею Швабе».
61. Посилання на Фармакопею Сполучених Штатів Америки не відповідає положенням Директиви 2001/83.
Рекомендації
Вилучити зі Статті ІІ.1.15 Наказу МОЗ № 426 посилання на Фармакопею Сполучених Штатів Америки (HPUS).
III.2.3. Визначення «конфіденційної реєстраційної інформації» (Стаття II.1.29 Наказу МОЗ № 426)
62. Це визначення притаманне виключно законодавству України і не має аналогів в Директиві 2001/83.
63. Обсяг інформації з реєстраційного досьє, яка не кваліфікується як конфіденційна, видається достатньо вузьким та, в основному, включає в себе дані, що містяться в інструкціях для медичного застосування (які, в будь-якому випадку, є в публічному доступі, адже це передбачено іншими положеннями українського законодавства).
64. Такий широкий обсяг інформації, що визначається конфіденційною, виключає можливість публічного доступу до звіту про оцінку ЛЗ.
III.2.4. Визначення «незалежного експерта» (Стаття ІІ.1.35 Наказу МОЗ № 426)
65. Це визначення притаманне виключно законодавству України і не має аналогів в Директиві 2001/83.
66. Це визначення містить частину, у якій робиться припущення, що кожен експерт відповідає перед заявником за якість своєї роботи. Видається, що така особиста відповідальність змінює роль експерта в системі (незалежність від заявників).
Рекомендації
Змінити формулювання і вилучити з нього припущення про те, що експерт несе особисту відповідальність перед заявником.
III.2.5. Визначення «референтний лікарський засіб» (Стаття ІІ.1.53 Наказу МОЗ № 426) та «оригінальний (інноваційний) лікарський засіб» (Стаття ІІ.1.37 Наказу МОЗ № 426)
67. Згідно із законодавством ЄС, референтний лікарський засіб (Стаття 10.2а Директиви 2001/83) повинен бути зареєстрований в будь-якій країні ЄС, в порядку, що відповідає вимогам Директиви 2001/83.
69. У відповідному визначенні в українському законодавстві не встановлено, чи повинен «референтний лікарський засіб» бути зареєстрованим в Україні. Однак, в українському визначенні є посилання на «оригінальний (інноваційний) лікарський засіб», який в інших положеннях законодавства визначається як зареєстрований в будь-де «в світі». На основі цього можна зробити висновок, що референтний лікарський засіб, на який робиться посилання під час реєстрації генеричних засобів в Україні, може бути зареєстровані будь-де місці у світі, не обов’язково в Україні.
69. Таке широке визначення референтного лікарського засобу може призвести до реєстрації генеричних лікарських засобів на основі досьє низької якості, а також може підготувати всі умови для розміщення на ринку України генериків, подібних до оригінальних лікарських засобів, які ніколи не були представлені на ринку України.
Рекомендації
Доповнити визначення, додавши до нього вимогу реєстрації референтного лікарського засобу в Україні або, у якості альтернативи – в ЄС. В останньому випадку, компетентний орган України повинен мати доступ до документації щодо референтних засобів для того, щоб зареєструвати генерик.
III.2.6. Визначення «неправомірне використання реєстраційної інформації про безпеку та ефективність лікарського засобу» (Стаття ІІ.1.35 Наказу МОЗ № 426)
70. Це визначення притаманне виключно законодавству України і не має аналогів в Директиві 2001/83.
71. Мета визначення цього терміну є незрозумілою. Словосполучення «неправомірне використання реєстраційної інформації» не використовується в Наказі МОЗ № 426, за винятком Статті II. 1.35, де воно визначене. Разом із тим, це визначення не повністю відповідає Статті 9 Закону, у якій містяться положення щодо ексклюзивності даних. Якщо мета такого визначення полягає у встановленні протиправного характеру порушення ексклюзивності даних заявником на генеричний лікарський засіб, ДЕЦ або МОЗ, то воно має бути замінене положенням, в якому чітко б встановлювалися принципи відповідальності за порушення (хто несе відповідальність, які санкції передбачені за таке порушення).
Рекомендації
Вилучити визначення і замінити його чітким положенням про умови відповідальності за порушення режиму ексклюзивності даних.
III.2.7. Визначення «подібний біологічний лікарський засіб (біосиміляр)» (Стаття ІІ.1.40 Наказу МОЗ № 426)
72. Згідно із законодавством України, у визначенні поняття «біосиміляр» міститься застереження щодо необхідності спливу строку дії патенту на референтний ЛЗ.
73. Така умова реєстрації для реєстрації біосиміляру не передбачена в Директиві 2001/83. Директива 2001/83 не містить окремого визначення поняття «біосиміляр». Однак, існування такого типу лікарських засобів можна вивести зі Статті 10.4 Директиви 2001/83, у якій йдеться про «біологічний лікарський засіб, подібний до референтного біологічного засобу». В законодавстві ЄС можна знайти ще одне визначення «біосиміляру», а саме – в Посібнику ЕМА щодо подібних біологічних лікарських засобів, в Розділі 3.1 – який також не містить жодних посилань на патентний захист.
74. Така вимога законодавства України щодо необхідності спливу патентного захисту для реєстрації біосиміляру видається надмірною.
Рекомендації
Вилучити вимогу про необхідність спливу строку патентного захисту оригінального лікарського засобу для реєстрації біосиміляру.
III.2.8. Невизначені поняття
75. Деякі поняття, визначені в Директиві 2001/83, відсутні у Законі, а саме:
- «речовина» (Стаття 1.3 Директиви 2001/83);
- «генератор радіонуклідів» (Стаття 1.7 Директиви 2001/83);
- «зловживання лікарськими засобами» (Стаття 1.16 Директиви 2001/83);
- «рецепт на лікарський засіб» (Стаття 1.19 Директиви 2001/83);
- «ризики, пов’язані із використанням лікарських засобів» (Стаття 1.28 Директиви 2001/83), оскільки, термін «ризик», визначений в Статті 2.32.2 Наказу МОЗ № 898 не повністю відповідає вищенаведеному;
- мастер-файл (нормативно-довідковий архів) системи фармаконагляду» (Стаття 1.28е Директиви 2001/83).
Рекомендації
Внести відсутні визначення до відповідних нормативних актів.
III.2.9.Інші розбіжності у визначеннях
76. Низка визначень, які не мають першорядного значення, але можуть привести до невідповідності між законами України та ЄС при їх застосуванні на практиці, містять відмінності від визначень, що містяться у Директиві 2001/83. Такі відмінності включають:
a) потенційно звужене визначення поняття «допоміжна речовина» (ексципієнт) (Стаття 2.6 Закону про лікарські засоби, порівняно зі Статтею 2.3Ь Директиви 2001/83);
b) у визначенні поняття «активний фармацевтичний інгредієнт» наявне посилання на «фармацевтичну або іншу» дію, в той час як у ЄС здійснюється посилання на «фармацевтичну, імунологічну або метаболічну дію (Стаття ІІ.1.1 Наказу МОЗ № 426, порівняно зі Статтею 1.3а Директиви 2001/83);
c) Українське визначення «медичні імунобіологічні препарати» не повністю узгоджене з визначенням «імунологічних препаратів» в Директиві 2001/83 (Стаття II.1.33 Наказу МОЗ № 426 порівняно зі Статтею 1.4 Директиви 2001/83);
d) Українське визначення «лікарські засоби для передової терапії (біотехнологічні)» (Стаття II. 1.10 Наказу МОЗ № 426) є менш деталізованим, ніж визначення «лікарський засіб для передової терапії» ЄС;
e) Українське визначення «радіонуклідний набір» (Стаття П.1.46 Наказу МОЗ № 426) стосується «лікарського засобу», в той час як у визначенні ЄС згадується лише «препарат» (Стаття 1.8 Директиви 2001/83);
f) Українські визначення «первинна упаковка» і «зовнішня упаковка» (Статті II. 1.11 і П.1.39 Наказу МОЗ № 426) містять додаткові непотрібні вимоги, що стосуються функцій упаковки (у порівнянні зі Статтями 1.23 і 1.24 Директиви 2001/83);
g) Українське визначення «співвідношення користь/ризик» (Стаття 2.23 Наказу МОЗ № 898) є набагато складнішим, ніж визначення ЄС (Стаття 1.28а Директиви 2001/83), і потенційно суперечить йому;
h) в Директиві 2001/83 «план управління ризиками» визначається просто як опис «системи управління ризиками», при цьому останнє – це сукупність «заходів і втручань» (Стаття 1.28Ь і 1.28c Директиви 2001/83), в той час як в українському законодавстві «система управління ризиками» стосується «заходів», а «план управління ризиками» – лише «втручань» (Стаття 2.33 і 2.34 Наказу МОЗ № 898);
і) Українське визначення «фальсифікований лікарський засіб» (Стаття 2.2 Закону про лікарські засоби) є вужчим, ніж визначення у ЄС (Стаття 1.33 Директиви 2001/83), та сконцентроване на умисному невідповідному маркуванні або складі;
j) Українське визначення «біологічний лікарський засіб» (Стаття П.1.6 Наказу МОЗ № 426) не містить жодних даних щодо перевірки і контролю виробничого процесу, що передбачені пунктом 3.2.1.І.Ь Додатка до Директиви 2001/83);
k) Українське визначення «генеричний лікарський засіб» (Стаття II. 1.12 Наказу МОЗ № 426) стосується категорії «взаємозамінності» у медичному використанні, замість «біоеквівалентності» (яка передбачена у Статті 10.2 Директиви 2001/83);
i) Українське визначення «лікарський засіб генної терапії» (Стаття II. 1.13 Наказу МОЗ № 426) є більш загальним, ніж визначення у ЄС (пункт IV.2.1 Додатка до Директиви 2001/83);
m) У законодавстві ЄС не міститься визначення поняття «ефективність», проте, виходячи з вступного пункту 7 Директиви 2001/83, ефективність завжди залежить від терапевтичного ефекту, в той час як визначення «ефективності» в законодавстві України (Стаття II. 1.24 Наказу МОЗ № 426) не приймає до уваги елемент взаємозв’язку ефективності та терапевтичного ефекту;
n)Українське визначення «препарат обмеженого застосування (препарат-сирота)» (Стаття П.1.43 Наказу МОЗ № 426) не повністю відповідає визначенню ЄС (Стаття 2b Регламенту 141/2000), так як воно не поширюється на препарати, збут яких вимагає стимулювання незалежно від кількості відповідної популяції, і не вимагає доказів безальтернативності;
о) Українське визначення «фармацевтично еквівалентні лікарські засоби» (Стаття П.1.59 Наказу МОЗ № 426) є більш точним, ніж відповідне визначення у ЄС (Розділ П.1 Посібника Комітету із патентованих лікарських засобів), оскільки воно передбачає ще й один той самий метод застосування лікарських засобів;
р) Українське визначення «офіцинальна формула» лікарського засобу, що звільняється від державної реєстрації (Стаття 1.2 Постанови КМУ № 376) не відповідає визначенню ЄС (Стаття 3.2 Директиви), оскільки воно не містить посилань на вимоги фармакопеї і не визначає призначення цих засобів (для відповідних пацієнтів, що обслуговуються в аптеках).
Рекомендації
Внести зміни до відповідних нормативних актів, усунувши розбіжності між законодавством України та ЄС.
III.3. ЗАСОБИ, ЩО ВХОДЯТЬ ДО СИСТЕМИ ДЕРЖАВНОЇ РЕЄСТРАЦІЇ
77. Обсяг засобів, які підпадають під дію системи реєстрації лікарських засобів в Україні, в значній мірі подібний до відповідного обсягу, вказаного у Директиві 2001/83.
78. Зважаючи на мету цього Звіту, ми зосередилися на відстеженні розбіжностей у сферах застосування обох систем. Результати нашого аналізу наводяться нижче.
III.3.1. Можливі розбіжності в обсязі засобів, що підлягають реєстрації, що виникли в результаті невідповідності визначень деяких категорій
79. В українському законодавстві деякі категорії лікарських засобів визначенні таким чином (у зв’язку із тлумаченням), який не в повній мірі відповідає законодавству ЄС. Розбіжності, які можуть вплинути на визначення обсягу засобів, які підлягають державній реєстрації, включають в себе:
- визначення «лікарський засіб» (див. пункти 56-59 вище);
- визначення «офіцинальна формула » (див. пункт 76 р вище);
III.3.2. Засоби, що підлягають реєстрації в Україні та звільняються від реєстрації згідно із законодавством ЄС
80. Українським законодавством передбачена реєстрація двох видів ЛЗ, що не підлягають реєстрації відповідно до Директиви 2001/83, а саме:
активні фармацевтичні інгредієнти (АФІ) (за винятком АФІ, що виробляються в процесі виготовлення лікарського засобу, і не призначені для продажу іншому виробнику, а також біотехнологічні АФІ) та
продукція «in bulk».
81. Відповідно до законодавства ЄС, виготовлення і продаж АФІ або продукції «in bulk» іншому виробнику дозволяється на підставі ліцензії на виробництво, без необхідності отримання реєстраційного посвідчення.
82. Враховуючи мету законодавства, що регулює реєстрацію лікарських засобів, збереження вимоги про державну реєстрацію АФІ і продукції «inbulk» не є необхідним.
83. Звільнення таких категорій продукції від державної реєстрації не лише відповідає Директиві 2001/83, а також може вивільнити додаткові ресурси ДЕЦ і дозволить збільшити ефективність цієї установи при проведенні експертизи інших видів продукції. Згідно зі статистичними даними за 2013-2015 роки, наданими ДЕЦ, кількість справ в ДЕЦ, які стосуються АФІ і продукції «in bulk», була значною і складала 597 справ по АФІ і 45 – по продукції «in bulk».
84. Рішення про звільнення вищезазначених категорій виробів від державної реєстрації має прийматись по узгодженню із державними установами, що здійснюють нагляд за виробництвом і виробниками лікарських засобів, для того, щоб переконатися, що таке звільнення не зменшить ефективність контролю над ринком і не погіршить рівень захисту пацієнтів від доступу до незареєстрованих речовин.
Рекомендації
Обговорити з відповідними установами і виробниками можливе звільнення АФІ і продукції «in bulk» від державної реєстрації.
85. Інша категорія ЛЗ, що підлягають державній реєстрації в Україні і не мають аналогів в законодавстві ЄС – це «медичні вироби, які містять речовини, що у процесі використання надходять до системного кровотоку.» (1.1.2.5 Наказ МОЗ № 426). В ЄС всі види медичних виробів, в тому числі пристрої, що імплантуються, підпадають під процедури і вимоги, які відрізняються від вказаних в Директиві 2001/83. Деякі види лікарських засобів можливо зареєструвати в поєднанні з медичним виробом (так званий «комбінований лікарський засіб для передової терапії»), проте навіть в такому випадку медичний виріб розглядається лише як частина лікарського засобу, а не як окремий предмет реєстрації.
Рекомендації
Вилучити «медичні вироби» зі сфери державної реєстрації.
III.3.3. Кров та плазма крові людини. Лікарські засоби, що походять з крові або плазми крові людини
86. Між сферою застосування процедури реєстрації стосовно крові і плазми людини, а також препаратів, які походять з крові або плазми крові людини, існують розбіжності.
87. Препарати, які походять з крові і плазми крові людського походження, не підлягають реєстрації згідно із Директивою 2001/83, за винятком плазми, отриманої з використанням промислового процесу (Ст. 3.6). В українському законодавстві немає аналогічного положення стосовно цього питання. Наказ МОЗ № 426 виключає зі сфери своєї дії лише «кров і плазму, які використовуються для промислового виробництва готових препаратів крові». Таке виключення видається більш вузьким, ніж європейське.
88. Відповідно до законодавства ЄС, лікарські засоби, що походять з крові і плазми крові людини, загалом розглядаються як біологічні лікарські засоби, на які поширюється звичайна процедура реєстрації, із включенням деяких змін, які стосуються оптового розповсюдження (Ст. 83 Директиви 2001/83), самозабезпечення держав-членів (Ст. 110), безпеки (Ст. 109, 114 і 115) та вимог до досьє (Частина III Додатка до Директиви 2001/83). На відміну від цього, відповідно до законодавства України, значна частина цієї категорії засобів повністю звільняється від державної реєстрації. Звільнення стосується «засобів на основі крові і плазми крові, фракціонованих з крові людини- донора згідно із інструкціями виробника у відповідних акредитованих закладах».
89. Таке звільнення препаратів, які одержують з крові або плазми крові людини, від державної реєстрації не відповідає Директиві 2001/83.
Рекомендації
Змінити формулювання щодо обсягу звільнення від реєстрації препаратів, які походять з крові або плазми крові людини, що містяться в українському законодавстві.
Звузити обсяг звільнення препаратів, що одержані з крові або плазми крові людини, від процедури державної реєстрації, наприклад, додавши пункт про те, що виключення стосується лише засобів, призначених для переливання, на які поширюється дія стандартів, встановлених Директивою 2002/98 щодо якості і безпеки збору, тестування, обробки, зберігання і розподілу крові і компонентів крові людини.
III.3.4. Інші спостереження
90. У законодавстві України немає чіткого виключення щодо обов’язковості реєстрації для лікарських засобів, призначених для наукових досліджень і випробувань2 (як це передбачено в Статті 3.3 Директиви 2001/83). На практиці такі засоби не повинні реєструватись, однак ця практика повинна знайти відображення у чітких нормах законодавства.
91. До видів радіофармацевтичних засобів, які не підлягають обов’язковій реєстрації в Україні, також відносяться засоби, які зазначені в Директиві 2001/83 окрім «радіонуклідів у вигляді закритих джерел».
__________________________
2Наказ МОЗ № 237 лише дозволяє імпорт таких засобів.
III.4. СПРОЩЕНІ ПРОЦЕДУРИ ДЕРЖАВНОЇ РЕЄСТРАЦІЇ
92. Одночасно зі стандартною процедурою державної реєстрації, Українське законодавство містить спеціальні правила для виведення на ринок (державної реєстрації) певних категорій ЛЗ, що пройшли процедуру реєстрації в окремих іноземних юрисдикціях:
процедура, прийнята для лікарських засобів, зареєстрованих EMA за централізованою процедурою (надалі: «лікарські засоби, зареєстровані ЕМА»);
процедура, прийнята для лікарських засобів, зареєстрованих компетентними органами Сполучених Штатів Америки, Швейцарії, Японії, Австралії, Канади і ЄС за централізованою процедурою (надалі: «лікарські засоби, зареєстровані окремими іноземними органами»);
процедура, прийнята для лікарських засобів, що підлягають закупівлі за результатами закупівельної процедури, проведеної спеціалізованою організацією, яка здійснює закупівлі, на виконання угоди щодо закупівлі між центральним органом виконавчої влади України (МОЗ), що забезпечує формування та реалізує державну політику у сфері охорони здоров’я, та відповідною спеціалізованою організацією, яка здійснює закупівлі (надалі: «лікарські засоби, що підлягають закупівлі»).
III.4.1. Лікарські засоби, зареєстровані ЕМА
93. Спрощена реєстрація засобів, зареєстрованих ЕМА, була введена у Постанові КМУ № 376. При реєстрації таких лікарських засобів не застосовуються положення Наказу МОЗ № 426.
94. Відмінності від стандартної процедури вказані нижче:
a) Зменшений обсяг експертизи:
95. Рішення про державну реєстрацію повинно прийматись без експертизи ДЕЦ, а лише на підставі висновків ДЕЦ щодо дотримання інструкції для медичного застосування і методів контролю якості наданих матеріалів (в тому числі звіту про оцінку ЕМА).
b) Прискорені зміни і перереєстрація:
96. Відповідно до Наказу МОЗ № 426 експертиза співвідношення користь/ризик з метою перереєстрації, а також оцінка запропонованих змін (варіацій), повинна проводитись позачергово.
c) Висновки про відповідність до законодавства ЄС:
97. Прямого аналога цієї процедури в законодавстві ЄС немає. Однак, принцип спрощеної реєстрації ЛЗ, зареєстрованих ЕМА, можна порівняти з дією централізованої процедури в державах-членах ЄС. Згідно із Регламентом 726/2004, держави-члени ЄС мають певну роль під час процедури реєстрації (вони можуть висловити зауваження щодо проекту рішення ЕМА, що може викликати подальший розгляд справи, а також вимагати обговорення проекту рішення). Після закінчення реєстрації лікарського засобу ЕМА, реєстраційне посвідчення діє у всіх країнах ЄС, без необхідності подальшого розгляду місцевими органами та без будь-якої іншої форми реалізації результатів рішення ЕМА.
98. Виходячи із цього, впровадження в Україні процедури «визнання» продукції, зареєстрованої ЕМА, з прискореними термінами і обмеженим обсягом розгляду видається доцільним засобом наближення до законодавства ЄС в цій сфері, оскільки Регламент 726/2004 не має обов’язкової сили в Україні.
99. Для того, щоб прискорити наближення положень в законах України до положень ЄС, можна обговорити умови майбутньої участі України в централізованій процедурі, наприклад, право на отримання інформації щодо процедури, включення інструкцій для медичного застосування і маркування українською та російською мовами, що пов’язані з безпосередньою дією рішення ЕМА на території України.
Рекомендації
Підтримувати спрощену державну реєстрацію лікарських засобів, зареєстрованих за централізованою процедурою ЕМА.
Обговорити з партнерами ЄС можливість включити Україну в централізовані процедури ЕМА на спеціальних умовах.
III.4.2. Лікарські засоби, зареєстровані окремими іноземними органами
100. Обсяг продукції, що підлягає такій процедурі, був нещодавно розширений. Спочатку ця процедура застосовувалась до засобів, призначених виключно для лікування туберкульозу, ВІЛ/СНІД, вірусного гепатиту, раку і орфанних захворювань. Починаючи з травня 2016 року (набуття чинності Змін до Закону про лікарські засоби), спрощена процедура повинна застосовуватись до всіх засобів, зареєстрованих окремими іноземними органами.
101. Відмінності між стандартною процедурою і процедурою для засобів, зареєстрованих окремими іноземними органами, вказані нижче:
(a) Зменшення вимог до документації:
102. Закон про лікарські засоби вимагає від заявника подавати лише зазначені нижче документи:
- матеріали, що стосуються методів контролю якості,
- макети упаковки та інструкції для медичного застосування,
- підтвердження оплати реєстраційного збору,
- лист-зобов’язання щодо виробництва продукції, яка поставлятиметься в Україну на тих самих виробничих потужностях, які використовуються для виробництва для певних країн.
103. Згідно із останніми Змінами до Закону про лікарські засоби, заявник також повинен надати «майстер-файл» (нормативно-довідковий архів), однак сфера застосування цієї вимоги чітко не визначена і потребує деталізації у підзаконних актах.
(b) Зменшений обсяг експертизи та обмежено підстави для відмови в реєстрації:
104. Обсяг експертизи зменшується відповідно до обсягу наданої документації. ДЕЦ здійснює лише обмежену перевірку реєстраційних матеріалів. Згідно із Наказом МОЗ № 426, надана документація повинна бути перевірена на відповідність звіту про оцінку, що виданий відповідною іноземною організацією (IV.5.5 Наказу МОЗ № 426).
105. Окрім того, існують чіткі обмеження щодо обсягу експертизи:
a) Змінами до Закону про лікарські засоби передбачено, що «майстер-файл» (нормативно-довідковий архів) не підлягає експертизі в ході процедури,
b) лабораторне тестування не є можливим (Наказ МОЗ № 426, IV.5.5),
c) заборонено вимагати від заявника внесення будь-яких змін до наданих реєстраційних матеріалів (Наказ МОЗ № 426, IV.5.5).
106. Підстави для відмови в реєстрації є обмеженими і включають в себе:
- відсутність необхідних документів,
- надання недостовірної або неповної інформації,
- розбіжності у відомостях про виробника і місце виробництва.
107. Відповідно до Постанови КМУ № 376 (3.5), якщо у встановлений строк ДЕЦ не надав жодних висновків або рекомендацій, експертиза вважається проведеною, а МОЗ приймає рішення без висновків ДЕЦ.
(c) Оцінка відповідності законодавству ЄС:
108. Ми розуміємо, що спрощена процедура для засобів, зареєстрованих окремими іноземними органами, була введена у зв’язку з епідеміологічними потребами в Україні в останні роки. У цьому сенсі ми сприймаємо таку процедуру як надзвичайний захід. Розуміючи причини, які стояли за впровадженням цієї процедури, ми хотіли б висловити свою точку зору стосовно цієї процедури у контексті її відповідності законодавству ЄС.
109. По-перше, ця процедура допускає реєстрацію лікарських засобів без фактичної перевірки безпеки і ефективності засобу. Така спрощена процедура може бути прийнятною з точки зору законодавства ЄС, якщо вона стосується лише тих засобів, які вже пройшли відповідну (згідно із законами ЄС) перевірку іншим органом реєстрації лікарських засобів (ЕМА або місцевим органом держави-члена ЄС в порядку, що відповідає вимогам Директиви 2001/83). Проте, така процедура поширюється на засоби, що зареєстровані не лише ЕМА, а й декількома реєстраційними органами держав, які не є членами ЄС.
110. Законодавством ЄС не передбачено жодної форми визнання реєстрації або спрощеної реєстрації засобів, які пройшли реєстрацію за межами ЄС, навіть якщо така реєстрація здійснена визнаними реєстраційними органами США, Канади, Японії, Швейцарії чи Австралії. Засоби, зареєстровані в цих країнах, повинні проходити стандартну процедуру реєстрації в ЄС. Процедура в Україні, яка дозволяє спрощену реєстрацію та скорочену перевірку досьє (або відсутність перевірки) засобів, зареєстрованих за межами ЄС, не відповідає законодавству ЄС.
111. По-друге, залишаючи осторонь питання про відповідність законодавству ЄС, видається, що процедура містить ознаки дискримінації лікарських засобів, зареєстрованих в ЄС. Для засобів, зареєстрованих в США, Канаді, Японії, Австралії та Швейцарії, процедура (з внесеними Змінами до Закону про лікарські засоби) охоплює практично всі види продукції у всіх фармакотерапевтичних категоріях, в тому числі і генерики. Вироби, зареєстровані ЕМА, є лише оригінальними (інноваційними, з повним досьє) лікарськими засобами, і лише окремі типи з них перелічені у Додатку до Регламенту 726/20043. На практиці це означає, що такапроцедура може застосовуватися до пари сотень засобів, зареєстрованих ЕМА і до десятків тисяч засобів, зареєстрованих за межами ЄС. Для забезпечення рівнозначності реєстраційних посвідчень держав-членів ЄС і держав, які не є членами ЄС, було б логічним розширити сферу застосування спрощеної процедури для того, щоб вона охоплювала лікарські засоби, зареєстровані в порядку, що відповідає Директиві 2001/83, органами всіх держав-членів ЄС за процедурою взаємного визнання/децентралізованою процедурою та за національними процедурами, які відповідають вимогам Директиви 2001/83.
__________________________
3 Засоби, що підлягають централізованій процедурі реєстрації, включають:
1) Лікарські засоби, розроблені із залученням одного з наступних біотехнологічних процесів:
a. технологія рекомбінантних ДНК,
b. контрольована експресія генів, що кодують біологічно активні білки в прокаріоти і еукаріоти, у тому числі трансформовані клітини ссавців,
c. методи гібридом і моноклональних антитіл;
2) Лікарські засоби для передової терапії, як це визначено в Статті 2 Регламенту 1394/2007;
3) Лікарські засоби для ветеринарного застосування, призначені, в першу чергу, для використання в якості підсилювачів дії з метою сприяння зростанню тварин або для підвищення прибутку від тварин;
4) Лікарські засоби для застосування людиною, що містять нову активну речовину, яка, на дату набрання чинності цього Регламенту, не була зареєстрована Союзом, для яких терапевтичним показанням є лікування будь-якого з наступних захворювань:
a. синдром набутого імунодефіциту,
b. онкологічні захворювання;
c. нейродегенеративні розлади,
d. діабет,
е. аутоімунні захворювання та інші імунні дисфункції,
f. вірусні захворювання;
5) Лікарські засоби, які визначені як препарати-сироти відповідно до Регламенту (ЄС) № 141/2000.
_________________________________
112. По-третє, незважаючи на надзвичайний характер обговорюваної процедури, її дія не обмежена в часі (на відміну від спрощеної реєстрації продукції, що підлягає закупівлі). Більш того, сферу застосування процедури нещодавно було розширено – від засобів, призначених для лікування окремих захворювань (туберкульоз, ВІЛ/СНІД, вірусний гепатит, рак, орфанні захворювання) до всіх лікарських засобів, що викликає питання про те, чи є епідеміологічні потреби достатнім обґрунтуванням для підтримки такої узагальненої спрощеної процедури реєстрації.
Рекомендації
Поширити проведення процедури на лікарські засоби, зареєстровані державними органами країн-членів ЄС за національною процедурою (відповідно до Директиви 2001/83) або за процедурою взаємного визнання/децентралізованою процедурою.
Передбачити дату закінчення строку дії положень, що регламентують таку процедуру (за аналогією зі спрощеною реєстрацією продукції, що підлягає закупівлі) щодо лікарських засобів, не зареєстрованих в ЄС.
III.4.3. Лікарські засоби, що підлягають закупівлі
113. Цю процедуру було включено до Закону про лікарські засоби у серпні 2015 року, наскільки ми розуміємо, у відповідь на нестачу лікарських засобів певних категорій (вакцин, противірусних лікарських засобів) в Україні.
114. Ця процедура відрізняється від стандартної за наступними пунктами:
(a) Зменшення вимог до документації:
115. Закон про лікарські засоби вимагає від заявника подавати наступні документи:
- реєстраційне досьє, подане до відповідного іноземного реєстраційного органу або ВООЗ для прекваліфікації продукції,
- звіт про оцінку, виданий відповідним іноземним органом або ВООЗ, який провів здійснив прекваліфікацію продукції,
- методи контролю якості,
- оригінальна інструкція для медичного застосування і макет оригінальної упаковки,
- переклад інструкції для медичного застосування та маркування українською мовою,
- лист-зобов’язання щодо виробництво продукції, яка поставлятиметься в Україну на тих самих виробничих потужностях, які використовуються для виробництва для відповідної окремої юрисдикції
(b) Зменшений обсяг експертизи та обмеження підстав для відмови у реєстрації:
116. Матеріали, які подаються разом із заявою, повинні перевірятись лише на достовірність. Вони не оцінюються по суті. (Однак, обсяг оцінки видається розширеним Наказом МОЗ № 426, ІV.5.5, який вимагає, щоб представлена документація порівнювалась зі звітом про оцінку іноземних органів).
117. Підстави для відмови у реєстрації обмежені наступними:
- відсутністю необхідних документів,
- наданням недостовірної або неповної інформації,
- невірним перекладом маркування або інструкції для медичного застосування.
(c) Скорочення строків для ДЕЦ та МОЗ:
118. 7 робочих днів для ДЕЦ на проведення експертизи на автентичність та 7 робочих днів для МОЗ на прийняття рішення про реєстрацію.
119. Відповідно до Наказу МОЗ № 426 експертиза співвідношення користі і ризику з метою перереєстрації, а також оцінка запропонованих змін (варіацій), повинна проводитись позачергово.
(d) Інше:
120. Збір за реєстрацію не сплачується.
121. Термін дії реєстраційних посвідчень обмежений (до 31 березня 2019 року – дата закінчення запланованого терміну дії положень стосовно цього виду спрощеної процедури).
Оцінка відповідності законодавству ЄС:
122. Ми розуміємо, що прийняття цієї процедури також було реакцією на невідкладні епідеміологічні потреби і нестачу певних лікарських засобів в Україні. Однак, на відміну від процедури стосовно засобів, зареєстрованих окремими іноземними органами, існує чітке обмеження часу проведення процедури (до 31 березня 2019 року). Таким чином, надзвичайний характер процедури є очевидним і має чітке обґрунтування.
123. Наші примітки щодо відповідності процедури законодавству ЄС є аналогічними до тих, що стосуються процедури для засобів, зареєстрованих в окремих іноземних юрисдикціях. Проте, через надзвичайний і тимчасовий характер цієї процедури, ми розуміємо, що питання потенційної невідповідності цієї процедури до законодавства ЄС, не є суттєвим.
ІІІ.5. ВИМОГИ ДО ЗАЯВИ ТА РЕЄСТРАЦІЙНОГО ДОСЬЄ
124. Зміст заяви і реєстраційного досьє згідно із законодавством України в значній мірі подібний до вимог, встановлених у Директиві 2001/83.
III.5.1. Вимоги до заяви
125. Вимоги до заяви і змісту реєстраційного досьє, в основному частині, регулюються Додатками до Наказу МОЗ № 426.
126. Що стосується обсягу заяви, він є подібним до обсягу, передбаченого у Ст. 8 Директиви 2001/83. На відміну від Директиви 2001/83, законодавство України не вимагає подання наступних документів:
- копія посвідчень, отриманих в державах-членах ЄС або третіх країнах для розміщення лікарського засобу на ринку, короткий підсумок даних про безпеку, у тому числі дані, що містяться в періодичних звітах з безпеки, при їх наявності, і звіти про можливі побічні реакції, разом зі списком держав-членів ЄС, в яких розглядається заява на отримання реєстраційного посвідчення;
- опис характеристик продукції та інформація для пацієнта, запропоновані заявником або затверджені компетентними органами держави-члена ЄС;
- подробиці будь-якого рішення про відмову у реєстрації, як в ЄС, так і у третіх державах, а також причини такого рішення;
- заява про те, що клінічні випробування, які проводились за межами ЄС, відповідають етичним вимогам Директиви 2001/20.
127. Деякі з перерахованих вище вимог необхідні для сприяння проведенню процедури взаємного визнання/децентралізованої процедури і в цьому сенсі вони не мають відношення до України. Окрім цієї категорії, інші вимоги спрямовані на те, щоб гарантувати обізнаність компетентного органу про можливі проблеми з реєстрацією засобу в інших країнах. У зв’язку з цим, до законодавства України слід внести зміни, які б включали зазначені вище вимоги.
128. Загальні типи реєстрації відповідають типам реєстрації в ЄС (Директива 2003/63), а саме:
- лікарський засіб з повним досьє (окремим досьє) (є аналогом лікарського засобу, зареєстрованого відповідно до Статті 8 (3) Директиви 2001/83);
- лікарські засоби з повним досьє змішаного типу (що відповідає визначенню в Частині II Додатка 1 Директиви ЄС 2001/83). Наказ МОЗ № 426 містить додаткові вимоги до деяких документів (а саме: бібліографічні посилання, план управління ризиками), які не суперечать законодавству ЄС;
- генеричний лікарський засіб;
- гібридний лікарський засіб;
- подібний біологічний лікарський засіб (біосиміляр);
- засіб з добре вивченим медичним використанням;
- лікарський засіб з фіксованою комбінацією;
- засіб, зареєстрований на основі інформованої згоди власника даних;
- традиційний лікарський засіб (відповідає поняттю «традиційного рослинного лікарського засобу» в ЄС).
129. В ЄС не проводиться реєстрація АФІ і лікарського засобу з продукції «in bulk».
III.5.2. Вимоги до реєстраційного досьє
(a) Загальні примітки
130. Документи, необхідні для подання окремих типів заяв на зазначені вище лікарські засоби в значній мірі співпадають з документам, які вимагаються відповідно до законодавства ЄС (відмінності див. в розділі Додатків нижче).
131. Вимоги до змісту і складу реєстраційного досьє, як правило, відповідають стандартам ЄС.
(b) Детальні вимоги до реєстраційного досьє
132. В українському законодавстві такі вимоги, як правило, регулюються додатками до Наказу МОЗ № 426. Ми переглянули додатки до Наказу МОЗ № 426 і порівняли їх з відповідними документами ЄС. Результати нашого порівняння наведено нижче:
| Додаток до Наказу МОЗ № 426 | Відповідний документ ЄС |
| Додаток 1. Реєстраційна форма для лікарського засобу, що передається на державну реєстрацію | Пояснювальна записка для заявників. Лікарські засоби для медичного застосування. Розділ 2В. Модуль 1.2: Основні відомості. Форма заяви |
| Додаток 2. РЕЄСТРАЦІЙНА ФОРМА для гомеопатичного лікарського засобу, що передається на державну реєстрацію | Пояснювальна записка для заявників. Лікарські засоби для медичного застосування. Розділ 2В. Презентація та зміст досьє – Частина 1 Резюме досьє Частина 1А.
Модуль 1: Основні відомості. Форма заяви. Гомеопатичний лікарський засіб для медичного застосування |
| Додаток 3. РЕЄСТРАЦІЙНА ФОРМА для лікарського засобу, виробленого відповідно до затвердженої специфікації, що передається на державну реєстрацію | н/п |
| Додаток 4 РЕЄСТРАЦІЙНА ФОРМА для активного фармацевтичного інгредієнта, що передається на державну реєстрацію. | н/п |
| Додаток 5 Структура реєстраційного досьє (формат загального технічного документа – ЗТД) | Пояснювальна записка для заявників. Розділ 2В. Лікарські засоби для медичного застосування. Презентація та формат досьє. Загальний технічний документ (ЗТД). Модуль 4: Пункти 4.2.1, 4.2.2 та 4.2.3. |
| Додаток 6. Загальні вимоги до матеріалів реєстраційного досьє (в форматі Загального технічного документа) | Пояснювальна записка для заявників. Лікарські засоби для медичного застосування. Розділ 2В. Презентація та формат досьє. Загальний технічний документ (ЗТД). |
| Додаток 7. ОСОБЛИВІ ПОЛОЖЕННЯ, застосовні до гомеопатичних лікарських засобів і їх реєстраційного досьє | Директива 2001/83, Стаття 14 і 15 |
| Додаток 8 ОСОБЛИВІ ПОЛОЖЕННЯ, застосовні до лікарського засобу, виробленого відповідно до затверджених специфікацій і їх реєстраційного досьє | н/п |
| Додаток 9. Особливі положення, застосовні до лікарських засобів | Регламент 847/2000
Регламент 141/2000 |
| обмеженого використання (препаратів- сиріт) і їх реєстраційного досьє | |
| Додаток 10 Окремі лікарські засоби і спеціальні вимоги, пов’язані з їх реєстраційним досьє | Директива 2001/83 |
| Додаток 11 Лікарські засоби для генної терапії і терапії соматичними клітинами, а також для тканинно-інженерних препаратів і особливих вимог для їх реєстраційного досьє | Директива 2009/120 |
| Додаток 12 Перелік документів, необхідних для проведення експертизи реєстраційних матеріалів для державної реєстрації АФІ | н/п |
| Додаток 13 Критерії віднесення лікарського засобу для лабораторних випробувань | Документ Координаційної групи з взаємного визнання та децентралізованих процедур – Людина (CMDh): Макети, зразки і проби – Нова заява. Вих. док.: CMDh/260/2012, Ред.4 |
| Додаток 14 Реєстраційна форма для лікарського засобу, що передається на перереєстрацію | Пояснювальна записка для заявників. Заява на продовження терміну дії реєстраційного посвідчення |
| Додаток 15 Перелік документів, що надаються для перереєстрації лікарського засобу | Посібник CMDh з передової практики обробки поновлень під час процедури взаємного визнання та децентралізованої процедури. CMDh/004/2005/Ред.14 лютого2016 р. |
| Додаток 16. Перелік документів, що надаються для перереєстрації лікарських засобів, вироблених відповідно до затверджених специфікацій | н/п |
| Додаток 17 Вимоги до документів, що передаються на експертизу при внесенні змін до реєстраційних матеріалів протягом терміну дії реєстраційного посвідчення. | Керівні принципи від 16.5.2013 щодо відомостей про різні категорії змін, щодо функціонування процедур, викладених в Розділах II, IIa, III і IVРегламенту 1234/2008, а також на документи, які подаються відповідно до цих процедур.Інформація, отримана від установ Європейського союзу, органів і організацій, Європейської комісії (2013/с 223/01) |
| Додаток 18 Встановлення еквівалентності генеричних лікарських засобів | Комітет з лікарських засобів для медичного застосування (ChMP). Посібник із дослідження біоеквівалентності
Вих. док.: CPMP/EWP/QWP/1401/98 Ред. 1/ Corr |
| Додаток 19. Структура інструкції для медичного застосування лікарського засобу | Пояснювальна записка для заявників. Принципи пакування. Інформація про лікарські засоби для медичного застосування, зареєстровані ЄС
Директива 2001/83 CMDh Шаблон Групи перевірки якості документів (QRD) з коментарями для процедури взаємного визнання/децентралізованої процедури (на основі версії 10 шаблону QRD для CP), Вих. док.: cmdh/201/2005/rev.9 |
| Додаток 20 Вимоги до інструкцій для медичного застосування лікарського засобу | Пояснювальна записка для заявників. Принципи пакування. Інформація про лікарські засоби для медичного застосування, зареєстровані ЄС
Директива 2001/83/ CMDh Шаблон QRD з коментарями для процедури взаємного визнання/децентралізованої процедури (на основі версії 10 шаблону QRD для CP), Вих. док.: cmdh/201/2005/rev.9 |
| Додаток 21 Структура опису характеристик продукції для лікарського засобу | Пояснювальна записка для заявників.Принципи пакування. Інформація про лікарські засоби для медичного застосування, зареєстровані ЄС
Директива 2001/83 CMDh Шаблон QRD з коментарями для процедури взаємноговизнання/децентралізованої процедури (на основі версії 10 шаблону QRD для CP), Вих. док.: cmdh/201/2005/rev.9 |
| Додаток 22. Вимоги до опису характеристик продукції для лікарського засобу | Директива 2001/83
CMDh Шаблон QRD з коментарями для процедури взаємного визнання/децентралізованої процедури (на основі версії 10 шаблону QRD для CP), Вих. док.: cmdh/201/2005/rev.9 |
| Додаток 23. Вимоги до маркування на упаковці лікарського засобу | Пояснювальна записка для заявників. Принципи пакування. Інформація про лікарські засоби для медичного застосування, зареєстровані ЄС
Директива 2001/83, літера v CMDh Шаблон QRD з коментарями для процедури взаємного визнання/децентралізованої процедури (на основі версії 10 шаблону QRD для CP), Вих. док.: cmdh/201/2005/rev.9 |
| Додаток 24 Перелік допоміжних речовин, які обов’язково вказуються на упаковці, та інформація, яка повинна бути викладена в інструкції для медичного застосування лікарського засобу | Пояснювальна записка для заявників. Розділ 3b. Керівні принципи. Лікарські засоби для медичного застосування. Безпека, навколишнє середовище та інформація. Допоміжні речовини, вказані на етикетці та в листку-вкладиші лікарських засобів для медичного використання. |
| Додаток 25. Лист до Директора, Державний експертний центр МОЗ України | н/п |
| Додаток 26 Реєстраційна форма на внесення змін до реєстраційних матеріалів, які стосуються лікарського засобу | Пояснювальна записка для заявників. Заява на зміну реєстраційного посвідчення. |
| Додаток 27 Зміни до інструкцій для медичного застосування лікарського засобу | Повідомлення про зміну інформації про засіб відповідно до статті 61 (3) (не супроводжують зміни варіації) |
| Додаток 28 Перелік документів дляекспертизи зміни заявника (власникреєстраційного посвідчення) | Керівні принципи передачі Реєстраційногопосвідчення – державнівимоги.CMDv/GUI/031. Номер видання:02, Датавидання: 2 лютого 2016 р. |
Додаток 1 – Форма заяви
133. Українську форму заяви не оновлено до останнього формату (еАF), який є обов’язковим у ЄС з 1 січня 2016 року.
134. Формат еAF вимагає заповнення більшості полів у формі заявки нормативною лексикою (термінами, обраними з каталогів EUTCT, Індексовані терміни телематики Європейського Союзу, який є архівом ЄС і містить індексовані терміни на декількох мовах для постійного обміну даними між інформаційними системами та додатками по всій європейській мережі з регулювання лікарських засобів).
135. Рекомендується оновити український формат до еАF для підвищення рівня відповідності системи України стандартам ЄС.
136. В українській заяві можна позначити існуючі патенти і товарні знаки у формі заяви (аналогу цього у формі ЄС немає). Стосовно цього невідповідностей не виникає.
137. Українська форма заяви не передбачає внесення інформації, необхідної в ЄС для продукції, що підлягає Педіатричному регулюванню.
138. Перелік «документів, що додаються» відображає певні особливі вимоги згідно із законами України, напр.: копії патентів на винахід, корисну модель або промисловий зразок, які дійсні в Україні (при наявності), копії документів, пов’язаних із захистом торгової марки в Україні (при наявності).
139. Такі особливі вимоги відповідно до законодавства України не стосуються відповідності європейському законодавству.
Додаток 2 – Реєстраційна форма для гомеопатичного лікарського засобу, що передається на державну реєстрацію
140. Додаток відповідає законодавству ЄС.
Додаток 3 – Реєстраційна форма для лікарського засобу, виробленого згідно із затвердженим прописом, що передається на державну реєстрацію
141. В ЄС немає аналогу цього положення, в деяких країнах ЄС досі існують подібні «місцеві» спрощені процедури для деяких видів вітчизняної продукції. Такі засоби не можуть підлягати взаємній процедурі визнання в рамках ЄС, якщо їх досьє не буде гармонізовано.
142. Якщо немає суттєвих економічних причин, що перешкоджали б цьому, рекомендується повністю узгодити вказану процедуру зі стандартами ЄС і виключити можливість спрощеної реєстрації для даного виду продукції. Замість такої спрощеної процедури слід застосовувати гармонізовані правила реєстрації ЛЗ на основі добре вивченого медичного застосування, що прийняті у ЄС.
Додаток 4 Реєстраційна форма для АФІ, що подається на державну реєстрацію (перереєстрацію).
143. Додаток 4 стосується державної процедури реєстрації АФІ, яка не має прямого аналогу в ЄС.
144. В ЄС не існує вимоги окремої реєстрації АФІ. ASMFабо CEPдодаються до файлу готового лікарського засобу, який затверджується разом з кінцевим продуктом.
145. Збереження цієї процедури у законодавстві України не є необхідним з точки зору законодавства ЄС.
Додаток 5 – Структура реєстраційного досьє (формат загального технічного документа – ЗТД)
146. Структура ЗТД в Додатку 5 не відповідає структурі ЗТД в Розділі 2B, що використовується в ЄС. Виявлені відмінності вказано нижче:
147. Документи, що не передбачені в українському ЗТД (Примітка: деякі з них вимагаються іншими положеннями українського законодавства):
1.0. Супровідний лист
1.3.1. Україна: лише коротка характеристика лікарського засобу, в ЄС: коротка характеристика лікарського засобу, маркування та листок-вкладиш
1.3.2. макет
1.3.3. зразок
1.3.4. Консультації з цільовими групами пацієнтів
1.3.5. Інформація про засіб, раніше схвалений в державах-членах ЄС
1.3.6. Шрифт Брайля
1.5.3. (Розширена) ексклюзивність даних/ринкова ексклюзивність
1.5.4. Виняткові обставини
1.5.5. Реєстрація «під умовами»
1.6.1. Без ГМО
1.6.2. З ГМО
1.7.1. Подібність
1.7.2. Ринкова ексклюзивність
1.9. Інформація стосовно клінічних випробувань
1.10. Інформація стосовно педіатрії
2.7.5. Посилання на літературні джерела (документ не доступний в ЄС)
3.3. Посилання на літературні джерела (документ не доступний в ЄС)
148. Модулі від 2 до 5 є більш деталізованими у порівнянні з ЗТД, який використовується в ЄС (нумерація пунктів є більш детальною у ЗТД, що використовується у ЄС).
149. Рекомендується узгодити Додаток 5 із ЗТД, що застосовується в ЄС.
150. Виявлені помилки: В Додатку 5 міститься посилання на розділ 4 пункт 4, хоча там має бути посилання на розділ 5 пункт 1.
Додаток 6 – Загальні вимоги до матеріалів реєстраційного досьє
151. Вимоги до змісту досьє в ЗТД лише частково відповідають вимогам ЄС.
152. Розбіжності між Додатком 6 і документом ЗТД, який наразі використовується в ЄС, такі ж, як зазначено стосовно Додатка 5 (вище).
153. Розділи ЗТД не збігаються (особливо розділи Модулю 1), а Модулі 2-5 мають різний ступінь деталізації.
154. Вимоги носять занадто загальний характер і посилаються лише на Директиви і Регламенти ЄС, а не на настанови Комітету з питань патентованих лікарських засобів (CPMP), Міжнародної конференції з гармонізації (ICH), примітки до керівних принципів зведення правил та норм ЄС щодо лікарських засобів EudraLex, які є більш докладними і чіткими, ніж первинне законодавство ЄС.
155. Виявлені помилки: В Додатку 6 міститься посилання на розділ 4 пункт 4 хоча там має бути посилання на розділ 5 пункт 1.
156. В Додаток 6 необхідно внести правки для приведення його у відповідність до вимог ЄС.
Додаток 7 – Особливі положення, що застосовуються до гомеопатичних лікарських засобів і їх реєстраційних досьє
157. Додаток 7 загалом відповідає законодавству ЄС.
158. При посиланні на офіційні назви, Додаток 7 посилається спочатку на Державну фармакопею України або Європейську фармакопею, після цього – на Німецьку гомеопатичну фармакопею (GHP), гомеопатичну фармакопею США (HPUS), Британську гомеопатичну фармакопею (BHP), Гомеопатичну фармакопею Швабе). З метою забезпечення відповідності до законодавства ЄС, посилання на Фармакопею США слід виключити.
159. Додаток 7 вимагає включення інструкцій для медичного застосування, розроблених відповідно до вимог Додатку 20 Наказу МОЗ № 426, тільки якщо зовнішня упаковка містить недостатньо інформації. Аналога цього положення в законодавстві ЄС немає.
Додаток 8 Особливі положення, що застосовуються до лікарського засобу, що виготовляється згідно із затвердженим прописом і їх реєстраційних досьє
160. Див. примітки до Додатка 3.
Додаток 9 Особливі положення, що застосовуються до лікарських засобів обмеженого використання (препаратів-сиріт) і їх реєстраційних досьє
161. Додаток 9 відповідає Регламенту 141/2000.
162. Окрім того, (в Україні) зобов’язання щорічно представляти підтвердження препарату- сироти є обов’язковим. Така додаткова вимога не є необхідною з точки зору законодавства ЄС, однак у зв’язку з тим, що центральна процедура ЄС не поширюється на Україну, така вимога може бути корисною для українського компетентного органу для збереження оновленої інформації про статус засобу в ЄС.
Додаток 10 – Окремі лікарські засоби і спеціальні вимоги, пов’язані з їхнім реєстраційним досьє
163. Усі особливі вимоги до певних лікарських засобів відповідають вимогам Частини III Додатка 1 Директиви 2003/63.
Додаток 11 – Лікарські засоби для генної терапії і терапії соматичними клітинами, а також для тканинно-інженерних препаратів і особливих вимог для їх реєстраційного досьє
164. Відповідає Директиві 2009/120.
Додаток 12 – Перелік документів, необхідних для проведення експертизи реєстраційних матеріалів для державної реєстрації АФІ
165. Див. примітки до Додатку 4.
Додаток 13 – Критерії віднесення лікарського засобу для лабораторних випробувань
166. Зразки готової продукції для проведення лабораторних випробувань, які надаються разом із заявою на реєстрацію, не є обов’язковими в ЄС. Однак в деяких країнах ЄС надання зразків є обов’язковою умовою. Така вимога не суперечить законодавству ЄС.
167. Виявлені помилки: Додаток 13 має посилатись на пункт 5 (а не пункт 4) розділу IV.
Додаток 14 – Реєстраційна форма для лікарського засобу, що передається на перереєстрацію
168. Форма поновлення відповідає вимогам законодавства ЄС.
Додаток 15 – Перелік документів, що надаються для перереєстрації лікарського засобу
169. Перелік документів відповідає Керівним принципам Координаційної групи з взаємного визнання та децентралізованих процедур – Людина (CMDh) щодо поновлення за лютий 2016 року. Згідно з українським законодавством вимагаються також деякі додаткові документи з безпеки:
[1.8.] Зведені дані виробника/заявника про безпеку медичного застосування лікарського засобу в Україні протягом терміну дії реєстраційного посвідчення за формою, передбаченою законодавством.
[1.12.] Короткий опис системи фармаконагляду (якщо необхідно).
170. Він не подається для традиційних лікарських засобів, гомеопатичних лікарських засобів, що відповідають вимогам Додатка 7 Процедури та лікарських засобів, що стосуються «Медичних газів».
Додаток 16 – Перелік документів, що надаються для перереєстрації лікарських засобів, вироблених відповідно до затверджених специфікацій
171. Див. примітки до Додатка 3.
Додаток 17 – Вимоги до документів, що подаються для експертизи при внесенні змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення
172. Загалом, це копія директиви ЄС з деякими змінами відповідно до чинного законодавства України (напр., зміни, що, в першу чергу, стосуються ДФУ, потім – Європейської Фармакопеї, нова лікарська форма вже зареєстрованого лікарського засобу в Україні вимагає нової реєстрації).
173. Також існують деякі розбіжності у вимогах до змін з Директивою ЄС, які необхідно усунути:
- Б.ІІ.б.1 – в «Примітках» зазначається, що Кваліфікована особа має бути з ЄЕЗ, а не з третьої країни. Слід додати Україну.
- Б.ІІ.б.2 – «Умови» не відповідають вимогам розділу «Документація». У визначенні «Умови» виробник може розташовуватись лише в ЄЕП, а в розділі «Документація» зазначається виробник повинен бути з України. До «Умов» слід додати Україну.
- Б.ІІ.Б.2 – також згадується централізована процедура, яка не застосовується в Україні.
- Б.ІІ.б.3 – вимагається менше документів, ніж у ЄС. Пункт 5 відсутній.4
- Б.ІІ.г.1.) – пункт 9 відсутній. 5
- Б.ІІ.ф – пункт 4 документації згадується в таблиці, але відсутній опис в розділі документа.
Додаток 18 – Підтвердження еквівалентності генеричних лікарських засобів
174. Цей Додаток є дуже загальним стислим описом (3 сторінки) генеричної біоеквівалентності. без чіткого визначення поняття та умов біоеквівалентності (напр., параметрів прийнятності, плану дослідження і т.д.).
175. Деталізовані критерії біоеквівалентності встановлені у Настанові МОЗ СТ-Н МОЗУ 42-7.1:2014 «Лікарські засоби. Дослідження біоеквівалентності.
________________________
4«Уразі зміни параметра(ів) процесу, що не мають впливу на якість готового лікарського засобу, – заява про те, що підтвердження відсутності такого впливу отримано за попереднього схвалення оцінки ризику».
5«Параметри специфікації або пропозиції для певних лікарських форм не стосуються критичних параметрів, наприклад таких як:
- кількісне визначення;
- домішки (не стосується певного розчинника, який не використовується у виробництві);
- будь-які критичні фізичні характеристики (твердість або стійкість до роздавлювання таблеток без оболонки, розміри тощо);
- випробування не вимагається для певної лікарської форми згідно із загальними статтями ДФУ або Європейської фармакопеї;
- будь-яка вимога щодо вибіркового випробування.».
Додаток 19 – Структура інструкції для медичного застосування лікарського засобу
176. Інструкція для медичного застосування не відповідає ні вимогам ЄС до SmPC, ані до листка-вкладиша для пацієнта. Для детальної інформації див. наші примітки в розділі ІІІ.7.
177. Структура не відповідає шаблонам Групи перевірки якості документів (QRD) ЄС (розробленим Робочою групою з перевірки якості документів, яка сприяє ЕМА в лінгвістичних аспектах надання інформації про ЛЗ).
Додаток 20 – Вимоги до інструкції для медичного застосування лікарського засобу
178. Інструкція повинна пройти перевірку щодо її розбірливості для користувачів, що не є обов’язковим в Україні, але вимагається в ЄС.
179. В Україні також немає вимог щодо розміщення чорного трикутника у випадку, якщо лікарський засіб підлягає додатковому моніторингу.
180. В інструкції відсутня інформація про можливість повідомлення про побічні реакції.
Додаток 21 – Структура короткої характеристики лікарського засобу
181. Цей Додаток бере за основу структуру короткої характеристики лікарського засобу ЕС. Деякі пункти мають інше формулювання, ніж в QRD ЄС, але стосуються тих самих питань.
Додаток 22 – Структура короткої характеристики лікарського засобу
182. Див. примітки до Додатку 21.
Додаток 23 – Вимоги до маркування упаковки готового лікарського засобу
183. Цей Додаток бере за основу вимоги ЄС, окрім тих, що вказані нижче:
- вимога щодо розміщення заходів безпеки, що дозволяють перевірити справжність лікарського засобу, а також ідентифікувати окремі упаковки (що вимагається Директивою 2012/62 – зобов’язання виробників вступає в силу в лютому 2019 року);
- інструкції для медичного використання для безрецептурних ЛЗ.
Додаток 24 — Перелік допоміжних речовин, які мають бути обов’язково зазначені на упаковці, та інформація, що має бути наведена в інструкції для медичного застосування лікарського засобу
184. Перелік допоміжних речовин є ідентичним до відповідного переліку ЄС.
Додаток 25 – Лист – документ, який підтверджує дійсність патенту в Україні
185. Це локальна вимога, яка також застосовується деякими установами в країнах ЄС (напр., в Угорщині). Відповідного шаблону в ЄС немає.
Додаток 26 – Реєстраційна форма лікарського засобу, до реєстраційних матеріалів на який вносяться зміни
186. Зазвичай це копія форми реєстрації eAF ЄС на внесення змін з деякими відмінностями, притаманними українському законодавству (наприклад, зміни стосуються в першу чергу Національної Фармакопеї України, а вже після неї – Європейської Фармакопеї).
187. Крім того, існують деякі розбіжності у переліку змін, які необхідно усунути:
a) Помилка в номері класифікації;
b) Пункт Б.ІІ.ґ.6 має прив’язку лише до змін короткої характеристики лікарського засобу, в той час як eAF ЄС стосується всієї інформації про продукцію;
c) Відсутній Б.V.б.1 – Зміни щодо якості після застосування процедури реферування ЄС;
188. Об’єднання змін не допускається, як і в ЄС (на даний час лише деякі зміни, які перебувають у прямій залежності одна від одної, можуть бути представлені в одній заяві). Рекомендується забезпечити можливість об’єднання змін, як це передбачено у ЄЕЗ, оскільки це зберігає час, що витрачається на підготовку та дублювання адміністративної документації.
Додаток 27 – Зміни до інструкції для медичного застосування лікарського засобу
189. Положення Додатку є ідентичними до форми заяви ЄС для повідомлення про зміну назви відповідно до Статті 61 (3) Директиви.
190. В ЄС існує додаткова вимога про роз’яснення причин такої зміни, гарантію заявника про відсутність будь-яких інших змін, про відсутність впливу на коротку характеристику лікарського засобу, а також інформацію про дату внесення змін. Ці додаткові пункти слід додати до українського законодавства (а внесення зміни назви шляхом повідомлення необхідно зробити можливим на практиці, а не лише в теорії – див. наші примітки в пункті 298).
Додаток 28 – Перелік документів для проведення експертизи матеріалів при зміні заявника (власника реєстраційного посвідчення)
191. Цей Додаток є подібним до процедури зміни власника реєстраційного посвідчення в ЄС.
192. Перелік документів є локальною вимогою, а тому він не може оцінюватися на відповідність законодавству ЄС.
193. В Україні зміни до Системи фармаконагляду та Кваліфікованої особи, яка відповідальна за фармаконагляд внаслідок передачі реєстраційного посвідчення іншій особі, є частиною процедури зміни Власника реєстраційного посвідчення. В багатьох країнах ЄС зміни щодо фармаконагляду передбачені як наступна стадія після зміни власника реєстраційного посвідчення. Ми оцінюємо підхід, прийнятий в Україні, як прийнятний.
(c) Особливі положення законодавства України
194. В Наказі МОЗ № 426 (V.6) зазначено, що «При реєстрації готового лікарського засобу специфікації на діючу речовину (розділи реєстраційного досьє 3.2.S.4 ЗТД) можуть містити додаткові показники щодо технологічних параметрів діючої речовини, що обґрунтовані при фармацевтичній розробці (розділи реєстраційного досьє 3.2.Р.2 ЗТД), та доповнюють показники специфікацій, що містяться у МКЯ на діючу речовину та/або у сертифікаті якості, виданому виробником».
195. Аналогічних положень немає у законодавстві ЄС.
196. В Наказі МОЗ № 426 (У.9, У.10) міститься чітка вимога до ЛЗ, що підпадають під спрощену процедуру, щодо надання повного реєстраційного досьє (яке може подаватися у електронному вигляді).
(d) Подача реєстраційного досьє в електронному форматі
197. Згідно із законодавством України, подача реєстраційного досьє в електронному форматі вимагає попереднього узгодження з ДЕЦ. За загальним правилом, вимагаються друковані екземпляри документів. Законодавство ЄС не вимагає електронної форми подання, однак запровадження електронного документообігу, що відповідає стандартам ЄС, є неминучим для ефективної участі в процедурах взаємного визнання/децентралізованих процедурах.
Приклад (Польща)
У Польщі дозволяється подавати документацію в електронному вигляді (у форматі Nees або еСТD) без паперового примірника, з незначними винятками (такі документи повинні надаватись в оригіналі:супровідний лист, реєстраційна форма,затвердження заснування, довіреність, заява Кваліфікованої особи).
Платформа СЕSL (Загальноєвропейська платформа для подання документації, що використовується для здійснення зв’язку між заявниками та усіма реєстраційними органами ЄС) доступна для нових реєстрацій.
198. Для детальної інформації щодо подання в електронній формі див. наші примітки в розділі Х.4.1 нижче.
ІІІ.6. ЗАХИСТ ВЛАСНОЇ ІНФОРМАЦІЇ ТА ПАТЕНТНИХ ПРАВ
199. Положення українського законодавства щодо ексклюзивності даних та захисту патентних прав в процесі державної реєстрації лікарських засобів містяться в різних нормативних актах.
III.6.1. Захист даних
200. У законодавстві ЄС принципи захисту даних регулюються Директивою 2001/83. Основні принципи полягають в наступному:
a) Згідно із Директивою 2001/83, захист даних не обмежує положень законодавства стосовно захисту промислової та комерційної власності.
b) Заявник не зобов’язаний надавати результати доклінічних і клінічних випробувань, якщо він може довести, що лікарський засіб є генериком референтного лікарського засобу, що проходить реєстрацію або вже зареєстрований відповідно до Ст. 6 Директиви протягом не менше ніж 8 років в державі-члені ЄС або за централізованою процедурою (ексклюзивність даних).
с) Після отримання реєстраційного посвідчення, Власник реєстраційного посвідчення може відмовитись від захисту даних.
d) Генеричний препарат, зареєстрований на основі даних про референтний засіб, не може бути виведений на ринок, допоки не пройде 10 років з першої реєстрації референтного засобу (ринкова ексклюзивність); цей період може бути продовжений до 11 років, якщо протягом перших 8 років Власник реєстраційного посвідчення отримує терапевтичні показання, що вказують на значну клінічну користь.
е) Якщо референтний лікарський засіб був зареєстрований в іншій державі-члені ЄС, компетентний орган цієї держави, на вимогу, передає державі-члену ЄС, в якій реєстрація підлягає підтвердженню, повний склад продукту і всю відповідну документацію (в разі потреби).
201. В законодавстві України положення про захист даних розосереджені серед багатьох правових актів.
202. Принципи щодо захисту даних, передбачені законодавством України, зазначені нижче.
203. Після реєстрації в Україні на підставі повного досьє, реєстрація іншого лікарського засобу з такою ж діючою речовиною можлива не раніше, ніж через 5 років після дати першої реєстрації, за винятком випадків, коли заявник отримав згоду посилатися на досьє референтного засобу, або надав своє власне повне досьє (Ст. 9.12 Закону про лікарські засоби). Це формулювання значною мірою відповідає розумінню «ексклюзивності даних» у ЄС, однак період захисту відрізняється (5 років порівняно з 8 роками в ЄС). Існує також додаткова умова для забезпечення захисту даних – заява про державну реєстрацію референтного засобу повинна подаватись в Україні протягом 2 років з моменту його першої реєстрації в будь-якій країні. Така додаткова умова не відповідає законодавству ЄС.
204. Захист даних може бути продовжений до 6 років, якщо протягом перших 3-х років МОЗ реєструє нові показання, які вважаються такими, що мають суттєву перевагу у порівнянні з вже існуючими (ст. 9.13 Закону про лікарські засоби). В ЄС цей тип продовження терміну захисту поширюється на засоби з виключним положенням на ринку.
205. Будь-яке використання або посилання на реєстраційні дані щодо безпеки та ефективності референтного засобу іншого заявника з метою реєстрації генериків суворо заборонено («незаконне використання») і підлягає накладенню дисциплінарної, адміністративної, цивільної та/або кримінальної відповідальності (Ст. 9.17, Ст. 3 (2) Постанови КМУ № 376, Ст. П.36 Наказу МОЗ № 426). Законодавство ЄС не регулює санкції за порушення ексклюзивності даних – це питання національних законів.
206. Українське законодавство не містить положень щодо ринкової ексклюзивності, що передбачені Ст. 10.1.2 та 10.1.4 Директиви 2001/83. До того ж, законодавством України введено виключення Болар, в якому йдеться про те, що захист даних не виключає право здійснювати розробку лікарських засобів, в тому числі проводити дослідження еквівалентності. У Наказі МОЗ № 426 (11.36) прямо зазначено, що дозволяється подавати заяву в період захисту даних з метою реєстрації засобу після закінчення терміну захисту даних. Загалом, введення такого правила відповідає законодавству ЄС. Однак, видається, що воно не в повній мірі відповідає вимогам щодо суворої заборони використання або посилання на дані оригінального препарату протягом 5-річного періоду ексклюзивності даних (зокрема, Ст. 3 (2) Постанови КМУ № 376).
207. Передчасне посилання на реєстраційні дані оригінального засобу також є основою для надання ДЕЦ негативної рекомендації щодо державної реєстрації генеричного препарату (IV. 6.4 Наказу МОЗ № 426). В ЄС порушення ексклюзивності даних не входить в число підстав для відмови в прийнятті заяви. Насправді, відповідність заяви вимогам захисту даних має перевірятись на початковому етапі процедури. Якщо заявник не надав доклінічні та клінічні дані, а замість цього посилався на інший засіб, компетентний орган повинен перевірити, чи є таке посилання допустимим, і якщо ні, він повинен запросити від заявника подання повного досьє або відкликати заяву без подальшого розгляду. Загалом, така заява не повинна навіть пройти етап експертної оцінки.
III.6.2. Патентний захист
208. Директива 2001/83 не стосується патентного захисту (за винятком виконання виключення Болар в Ст. 10.6).
209. На відміну від цього, законодавство України щодо реєстрації лікарських засобів приймає до уваги патентний захист як один із факторів, що впливають на результат процедури реєстрації.
210. Українське законодавство вимагає, щоб деякі документи і заяви стосовно патентного захисту подавались разом із заявою, а саме: копія патенту або ліцензії, документ, що підтверджує чинність патенту в Україні, лист із зазначенням, що патентні права третіх сторін не порушуються внаслідок реєстрації (див. Статтю 9.17 Закону про лікарські засоби, Статтю 3(3) Постанови КМУ № 376, Статтю V.3 Наказу МОЗ № 426). Законодавство ЄС не вимагає такої додаткової інформації. З цього можна зробити висновок, що введення таких додаткових вимог має залишатись за рамками розгляду відповідності українського законодавства до Директиви 2001/83, оскільки це є однією з форм патентного захисту, яка входить в сферу, не гармонізовану на рівні ЄС, яка залежить від інших міжнародних зобов’язань України.
211. Порушення прав інтелектуальної власності, у тому числі патентів, є підставою для відмови в державній реєстрації МОЗ (ст. 9.25 з Закону про лікарські засоби). У випадку рішення суду про те, що реєстрація порушує патентний захист, ДЕЦ надає негативну рекомендацію (Наказ МОЗ № 426, IV.6.5). Що стосується дотримання законодавства ЄС, то умови щодо патентного захисту при наданні рекомендацій або рішення про реєстрацію лікарського засобу не є питанням, що регулюється Директивою 2001/83, а скоріше засобом забезпечення дотримання патентних прав. З цієї точки зору, такі додаткові підстави для відмови в реєстрації знаходяться поза питанням про відповідність до законодавства ЄС.
Приклад (Польща)
Фармацевтичне законодавство Польщі не містить положень про патентний захист в якості можливих підстав для відмови у видачі реєстраційного посвідчення.. Крім того, на практиці, Управління з реєстрації лікарських засобів Польщі не бере до уваги питання стосовно патентного захисту при обробці заявок на отримання реєстраційних посвідчень. Патентний захист сприймається як поняття, яке не стосується процедури видачі реєстраційних посвідчень. Відповідальність за порушення патентних прав бере на себе Власник реєстраційного посвідчення, який вирішує виготовити або продати продукт, порушуючи патентний захист, і розмістити його на ринку.
212. Окрім цього, Постанова КМУ № 376 свідчить, що КМУ може надати примусову ліцензію з метою державної реєстрації лікарських засобів. Детальні умови для введення процедури надання примусової ліцензії описані в Постанові КМУ № 877. Положення Постанови КМУ № 877 відповідають вимогам, викладеним в Угоді про торговельні аспекти прав інтелектуальної власності (ТРІПС).6
___________________
6 Угода ТРІПС є Додатоком 1С до Марракешської угоди про створення Світової організації торгівлі, підписаної у м. Марракеш, Марокко, 15 квітня 1994 року.
ІІІ.7. ІНФОРМАЦІЯ ДЛЯ ПАЦІЄНТІВ ТА СПЕЦІАЛІСТІВ СФЕРИ ОХОРОНИ ЗДОРОВЯ (ЕТИКЕТКА, ІНФОРМАЦІЯ ДЛЯ ПАЦІЄНТА, SМРС)
213. Директива 2001/83 передбачає три основні джерела інформації про використання лікарських засобів для громадськості – SmPC, інформація для пацієнта та інформація на етикетці лікарського засобу.
214. SmPC розглядається як основний документ та обов’язкове джерело інформації про властивості лікарського засобу. SmPC поширюється серед спеціалістів у сфері охорони здоров’я та медичних представників власників реєстраційного посвідчення на лікарський засіб. Інформація з SmPC може також бути використана в рекламі.
215. Інформація для пацієнта та етикетка мають відповідати SmPC. Інформація для пацієнта служить інструкцією для пацієнтів, є менш деталізованою, ніж SmPC, і повинна бути сформульована мовою, зрозумілою для пацієнтів.
216. Етикетка містить тільки основні відомості про лікарський засіб.
217. Пропоноване формулювання SmPC, інформації для пацієнта та етикетки є складовою частиною заяви на реєстрацію лікарського засобу. В подальшому таке формулювання підлягає експертизі, і затверджується за умови позитивного висновку експертизи.
218. Українське законодавство формально передбачає ідентичні джерела інформації про лікарський засіб: SmPC, інструкцію для медичного застосування та етикетку, однак тільки два останніх джерела містять повний обсяг інформації про лікарський засіб.
_________________
6 Угода ТРІПС є Додатоком 1С до Марракешської угоди про створення Світової організації торгівлі, підписаної у м. Марракеш, Марокко, 15 квітня 1994 року.
ІІІ.7.1. SmPC
219, SmPC не згадується ні в Законі про лікарські засоби, ні в Постанові КМУ № 376. Вона відсутня у списку матеріалів, які підлягають затвердженню наказом МОЗ про державну реєстрацію (пункт 5.3 Постанови КМУ № 376). Вимога надати запропоновану SmPC разом з заявою про реєстрацію, а також вимоги до змісту SmPC зазначаються лише в Додатках 21 та 22 до Наказу МОЗ № 426. Крім того, з окремих положень Наказу МОЗ № 426 можна зробити висновок, що розробка SmPC є радше опцією, аніж обов’язковим елементом процедури.
Наказ МОЗ № 426
III. 2.1
Реєстраційне досьє на традиційні лікарські засоби, що подаються на державну реєстрацію, містить докладні відомості, зазначені у модулях 1 – 3, а також:
(…) коротку характеристику лікарського засобу та/або інструкцію для медичного застосування без розгорнутих клінічних даних;
V.9
Якщо лікарський засіб був ліцензований ЕМА за централізованою процедурою, заявник може подавати інформацію … в електронному вигляді … за бажанням заявника – коротку характеристику лікарського засобу
VII.1
У разі складання умотивованих висновків щодо ефективності, безпеки та якості лікарського засобу (далі – висновки) Центр протягом 10 робочих днів готує та надає заявнику для редакційного узгодження проекти таких документів:
при державній реєстрації лікарського засобу – реєстраційного посвідчення, інструкції для медичного застосування лікарського засобу та короткої характеристики лікарського засобу (за наявності), МКЯ, тексту маркування первинної, вторинної (за наявності) упаковок готового лікарського засобу.
220. Як наслідок, в українській системі існує лише одне джерело інформації про лікарський засіб: інструкція для медичного застосування.
221. Нинішня роль SmPC в системі, відсутність чітко визначеної вимоги подавати SmPC під час процедури державної реєстрації лікарського засобу не відповідає вимогам законодавства ЄС.
222. Що стосується вимог до SmPC, зазначених в додатках до Наказу МОЗ № 426, вони в цілому відповідають вимогам законодавства ЄС. Для більш детальної інформації, просимо дивитися пункт 182 вище.
III.7.2. Інструкція для медичного застосування
223. Інструкція для медичного застосування грає роль інформації для пацієнта в розумінні Директиви 2001/83. В той же час, зважаючи на відсутність зобов’язання подавати та затверджувати SmPC, інструкція для медичного застосування слугує також джерелом інформації стосовно використання лікарського засобу і для спеціалістів у сфері охорони здоров’я. Така ситуація не сумісна з законодавством ЄС.
224. Інструкція має іншу структуру, ніж та, що є обов’язковою в ЄС. Вона більше схожа на скорочену версію SmPC, ніж на документ, призначений для пацієнта.
225. Основні відмінності включають:
a) структуру, яка відрізняється від нинішньої структури, затвердженої експертною комісією ЕМА;
b) відсутність змісту;
c) відсутня інформація щодо повідомлення про побічні реакції;
226. Подальші відмінності не є критичними, але повинні бути виправлені:
a) Українське законодавство вимагає надання МНН або загальноприйнятої назви для лікарського засобу з однією діючою речовиною, в той час як в ЄС таке зобов’язання існує для лікарських засобів, які включають в себе до трьох діючих речовин;
b) Згідно з законодавством ЄС, дозування та лікарська форма засобу повинні обов’язково бути вказані на зовнішній упаковці, в той час як згідно з українським законодавством лікарська форма та/або дозування зазначаються лише тоді, коли назва лікарського засобу використовується для декількох лікарських форм та/або має різне дозування;
c) Згідно з законодавством ЄС, шрифтом Брайля повинна бути зазначена назва лікарського засобу на зовнішній упаковці, в той час як в Україні це зобов’ язання поширюється не тільки на назву лікарського засобу, а й на його дозування та лікарську форму. Проте, МОЗ має визначити ті лікарські засоби, на які не поширюється це зобов’язання або для яких це зобов’ язання обмежене зазначенням тільки назви лікарського засобу.
d) Директива 2001/83 вимагає, щоб інформація для пацієнтів була доступною за запитом пацієнта у форматі, придатному для сліпих та слабозорих; подібне положення відсутнє в українському законодавстві;
e) Згідно з Директивою 2001/83, компетентний орган може дозволити вказувати менший обсяг даних у інформації для пацієнта або на етикетці, якщо продукт не призначений для отримання безпосередньо пацієнтом, або у випадку гострого дефіциту. Таке положення відсутнє в українському законодавстві. Для порівняння, стаття 12.3 Закону про лікарські засоби дозволяє МОЗ визначати додаткові вимоги щодо маркування етикеткою та пакування;
f) Інструкція та макет упаковки не підлягають тестуванню на легкість читання (консультаціям з цільовими групами пацієнтів);
g) Відсутня вимога зображення символу «чорного трикутника» на інструкції щодо лікарських засобів, які підлягають посиленому контролю;
h) Відсутня інформація для спеціалістів в сфері охорони здоров’я щодо необхідності повідомлення про побічні реакції.
227. До законодавства України необхідно внести відповідні зміни для приведення його положень щодо інструкції для медичного застосування у відповідність до вимог законодавства ЄС.
228. Перехід до моделі, що відповідає стандартам ЄС (два окремих документи для спеціалістів у сфері охорони здоров’я та пацієнтів) має бути ретельно спланований та повинен супроводжуватись перехідним періодом. Впровадження нової моделі потребує законодавчих змін та технічного забезпечення, яке дозволить спеціалістам у сфері охорони здоров’я отримати доступ до SmРС.
229. Ще одним питанням, яке потребує уваги, є застосування вимог українського законодавства до інформації для пацієнта та етикетки стосовно лікарських засобів, що закуповуються за результатами закупівельної процедури, проведеної спеціалізованою організацією, яка здійснює закупівлі. Стаття 12.7 Закону про лікарські засоби повністю звільняє такі лікарські засоби від необхідності дотримання зазначених вимог. З іншого боку, Наказ МОЗ № 426 не містить цього винятку і все одно вимагає, щоб заявник надав проект етикетки та інструкції. Українське законодавство містить суперечливі положення з цього приводу. У всякому разі ми розуміємо, що прийнятий підхід до цього питання (і спрощені правила реєстрації для цієї категорії лікарських засобів загалом) є лише тимчасовим рішенням. Якщо залишити це положення в чинному законодавстві, можуть виникнути сумніви щодо його відповідності критеріям статті 63.3 Директиви 2001/83.
III.7.3. Маркування лікарських засобів
230. Обсяг інформації, який повинен бути вказаний на упаковці лікарського засобу в Україні, є подібним до відповідного обсягу, що визначений законодавством ЄС.
231. Важлива відмінність стосується заходів безпеки. Після реалізації Директиви про фальсифікацію лікарських засобів7, в ЄС на упаковці лікарського засобу повинен бути відображений елемент захисту (у вигляді QR-коду), який дасть змогу дистриб’юторам та аптекам (і) перевіряти автентичність лікарського засобу та (іі) ідентифікувати окремі пакування. Упаковка повинна бути забезпечена спеціальним пристроєм, який дозволяє перевірити, чи не була вона пошкоджена ззовні. Нові вимоги будуть стосуватись більшості рецептурних лікарських засобів та окремих безрецептурних засобів, та набудуть чинності в ЄС 9 лютого 2019 р. Тим часом виробники адаптують свої виробничі лінії до нових умов. Індустрія та дистриб’ютори також створюють Європейську базу даних та національні архіви, за допомогою яких буде діяти система перевірки автентичності лікарського засобу шляхом сканування QR-коду в аптеках та з’єднання з базою даних для перевірки того, чи продукція не є фальсифікованою.
Рекомендація:
Україна повинна ініціювати обговорення зі своїми європейськими партнерами перспектив впровадження положень про боротьбу з контрафактною продукцією, включаючи можливу підтримку України з боку ЄС в цьому питанні.
__________________
7Директива 2011/62
232. Інші відмінності:
a) українське законодавство дозволяє зазначати менший обсяг інформації для невеликих пакувань (тільки найменування, склад та номер партії) – законодавством ЄС це не допускається;
b) в законодавстві України не існує особливих правил стосовно маркування етикеткою та інструкції щодо радіофармацевтичних препаратів, як того вимагає стаття 67 Директиви 2001/83;
c) відсутність вимоги маркування спеціальним символом лікарських засобів, які використовуються в педіатрії (як передбачено Директивою 1901/2006);
d) відсутність спеціальних вимог до маркування та інформації для пацієнта щодо лікарських засобів, що використовуються для прогресивної терапії, як передбачає Регламент 1394/2007;
e) можливість надавати певну інформацію у вигляді наклейки, за погодженням з МОЗ, що є особливістю українського законодавства (згідно Директиви 2001/83 це допустимо тільки щодо продукції від паралельного імпорту та спеціального маркування «в синій рамці» («blue box»).
III.8. ХІД ПРОЦЕДУРИ
233. Законодавство ЄС не регулює організацію процесу реєстрації та структуру компетентних органів в державах-членах. Незалежно від прийнятої моделі, вкрай важливо, щоб система гарантувала дотримання вимог Директиви щодо цілей та тривалості процедури.
Приклади:
Польща
До 2004 року обов’язки в сфері реєстрації лікарських засобів були розподілені між Міністерством охорони здоров’я (яке офіційно видавало рішення) та Президентом Бюро реєстрації лікарських засобів («URPL») (який приймав заявки, здійснював експертизу та готував проекти рішення для МОЗ):
Заявник подавав заяву в МОЗ через URPL.
МОЗ здійснювало формальний розгляд заяви протягом 30 днів від дати її подачі та (при необхідності) просило заявника надати додаткову документацію.
URPLготував звіт за результатами наукової оцінки лікарського засобу.
МОЗ видавало рішення про реєстрацію на основі звіту URPL.
З 20Н року практично всі функції в сфері реєстрації лікарських засобів були передані Президенту Бюро реєстрації лікарських засобів.
Повноваження щодо контролю якості та відповідності вимогам GMPнесе окремий державний орган – Головний фармацевтичний інспекторат.
Німеччина
Регуляторним органом Німеччини в сфері реєстрації лікарських засобів є Федеральний інститут лікарських засобів та виробів медичного призначення (BfArM) та Інститут Пауля Ерліха (PAI).
PAI відповідає за сироватки, вакцини, препарати крові, препарати кісткового мозку, препарати тканин, алергени, лікарські засоби перенесення генів, терапію соматичних клітин, терапію ксеногенних клітин та генетично виведених компонентів крові, а BfArM відповідальний за решту лікарських засобів для застосування людиною.
Ці установи несуть відповідальність за організацію клінічних досліджень, реєстрацію лікарських засобів та фармаконагляд.
Нагляд за обігом та якістю лікарських засобів здійснюють уряди 16 земель. На основі договору між урядами земель була створена координаційна група («Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten»), яка підпорядковується не федеральному уряду, а міністру охорони здоров’я Північного Рейну – Вестфалії.
Франція
Національне агентство з безпеки лікарських засобів та товарів для здоров ’я (ANSM) стало правонаступником Французького агентства санітарної безпеки товарів для здоров ’я8 (AFSSAPS) з 1 травня 2012р. та прийняло на себе відповідні завдання та повноваження.
Це державна установа знаходиться під керівництвом Міністерства охорони здоров’я. ANSM фінансується за рахунок грантів, отриманих від держави.
ANSM відіграє важливу роль для фармацевтичної галузі Франції. Сфера її повноважень включає в себе, зокрема, клінічні випробування, реєстрацію лікарських засобів, фармаконагляд, аналіз балансу співвідношення ризик-користь товарів для здоров ’я протягом строку їх придатності та питання їх якості.
Швеція
У Швеції один орган відповідає за клінічні випробування, реєстрацію лікарських засобів та питання виробництва, – Агентство лікарських засобів9. Воно уповноважено надавати дозвіл на здійснення клінічних випробувань лікарських засобів, приймати рішення про реєстрацію лікарського засобу, здійснювати моніторинг можливих побічних реакцій, лабораторний контроль якості, регулярні перевірки виробників та дистриб’юторів.
_____________
8 L’Agence française de sécurité sanitaire des produits de santé.
9 Läkemedelsverket.
234. Хід процедури реєстрації лікарських засобів детально не регулюється законодавством ЄС. Нижче викладені вимоги Директиви 2001/83 у цій сфері (разом з нашими висновками щодо відповідності українського законодавства цим вимогам).
235. Директива 2001/83 вимагає, щоб компетентні органи перевіряли заяви та відомості на відповідність її вимогам та умовам реєстрації лікарських засобів.
236. Законодавство України відповідає цим вимогам з точки зору обсягу експертизи, що стосується стандартної процедури реєстрації. Що стосується спрощених процедур (процедури, що стосується окремих юрисдикцій, а також закупівлі лікарських засобів за результатами закупівельної процедури, проведеної спеціалізованою організацією, яка здійснює закупівлі), то вони не відповідають законодавству ЄС в тій мірі, у якій вони застосовуються до лікарських засобів, зареєстрованих за межами ЄС. Європейське законодавство не дозволяє спрощеної реєстрації лікарських засобів, не зареєстрованих в ЄС. Див. також наші зауваження у пунктах 108-112 вище.
237. Директива надає право компетентним органам направляти лікарські засоби або їх складові на лабораторне випробування для підтвердження методів контролю якості виробником. Аналогічне положення існує у чинному законодавстві України. Критерії для направлення лікарського засобу на лабораторне випробування містяться у Додатку № 13 до Наказу МОЗ № 426.
238. Директива надає право компетентному органу витребувати у заявника додаткову документацію. Аналогічне положення є й у чинному законодавстві України.
239. Що стосується лікарських засобів, які походять з країн, що не є членами ЄС, Директива вимагає, щоб компетентні органи перевірили спроможність виробника та імпортера дотримуватися заявлених технологій та методів контролю якості.
240. Крім того, українське законодавство вимагає, щоб заявник пред’явив документ, який підтверджує відповідність умов виробництва лікарського засобу вимогам GMP. Для препаратів, вироблених в Україні, дійсна ліцензія на виробництво лікарських засобів є достатнім документом для виконання цієї вимоги. Звільняються від необхідності дотримання цієї вимоги лікарські засоби, подані на реєстрацію за спрощеною процедурою (раніше зареєстровані EMA чи компетентними органами США, Канади, Австралії, Японії та Швейцарії); в такому випадку гарантійного листа від виробника буде достатньо. На практиці, вимога про підтвердження відповідності сертифіката GMP застосовується до заявників з країн ЄС, чия продукція не пройшла реєстрацію за централізованою процедурою. Україна не визнає сертифікати GMP, видані в країнах ЄС, автоматично; Держлікслужба визнає такі сертифікати тільки після окремої доволі тривалої процедури їх підтвердження. Держлікслужба може використати своє право на проведення інспекції виробничої дільниці, що продовжить процес реєстрації.
241. На наш погляд, вимога про підтвердження відповідності сертифіката GMP стосовно заявників, що мають дійсні сертифікати, видані у країнах-членах ЄС, для цілей державної реєстрації не є обґрунтованою, і необхідно додатково проаналізувати те, чи повинна вона взагалі залишатися елементом процедури державної реєстрації. Підтвердження відповідності вимогам GMP повинне (якщо таке підтвердження залишиться необхідним) залишатися питанням, пов’язаним виключно з наглядом Держлікслужбою за умовами виробництва.
242. Що стосується фактичної організації процесу, ми не бачимо доцільності оцінювати її з точки зору відповідності законодавству ЄС. Організація цього процесу повинна аналізуватися з точки зору її ефективності та прозорості. Наші зауваження з цього приводу наведені у розділі VI стосовно організації процесів.
ІІІ.9. СТРОКИ
243. Директива 2001/83 вимагає, щоб процедура реєстрації лікарських засобів була завершена не більше ніж за 210 днів з дня подачі повної заяви. Якщо уповноважений орган вимагає додаткових пояснень від заявника, цей строк призупиняється на час, відведений для надання пояснень.
244. Українське законодавство не відповідає вимогам Директиви 2001/83 щодо граничної загальної тривалості процедури реєстрації.
245. По-перше, не встановлена гранична загальна тривалість всієї процедури державної реєстрації, починаючи з дати подання заяви до дати оприлюднення наказу про реєстрацію або отримання реєстраційного посвідчення. Законодавство передбачає граничні строки виконання лише певних етапів процедури:
- передача копії заяви з МОЗ до ДЕЦ для експертизи – 3 робочих дні;
- експертиза в ДЕЦ, залежно від типу заяви:
210 робочих днів на реєстрацію (і) на підставі повного досьє (автономного досьє) (іі) медичних імунобіологічних препаратів (МІБП), (ііі) біосимілярів;
7 робочих днів для реєстрації за спрощеною процедурою відносно (і) лікарських засобів, що підлягають закупівлі за результатами закупівельної процедури, проведеної спеціалізованою організацією, яка здійснює закупівлі (експертиза автентичності) та (іі) лікарських засобів, зареєстрованих певними іноземними компетентними органами. Однак, відповідно до Наказу МОЗ №426, строк складає 45 робочих днів для реєстрації в межах спрощеної процедури, включаючи строк (ііі) щодо всіх лікарських засобів, зареєстрованих ЕМА;
90 робочих днів на реєстрацію (і) інших типів лікарських засобів, (іі) АФІ, (ііі) лікарських засобів, що виробляються згідно із затвердженими прописами, (ту) МІБП, перекваліфікованих у категорію лікарських засобів, при реєстрації їх як лікарських засобів;
- передача ДЕЦ заявнику проектів документів для редакційного узгодження – 10 робочих днів;
- підготовка ДЕЦ переліків лікарських засобів, запропонованих до державної реєстрації (перереєстрації, внесення змін до реєстраційних матеріалів) та проектів документів для МОЗ – 5 робочих днів;
- прийняття рішення МОЗ на основі висновків та рекомендацій ДЕЦ – 10 робочих днів (в межах спрощеної процедури відносно лікарських засобів, зареєстрованих окремими іноземними компетентними органами, або тих, що закуповуються за результатами закупівельної процедури, проведеної спеціалізованою організацією, яка здійснює закупівлі, – 7 робочих днів) (3 робочих дня відповідно до Наказу МОЗ № 220);
- МОЗ видає реєстраційне посвідчення протягом 10 робочих днів після прийняття рішення.
246. Відповідно до вищезазначеного, вимога дотримання загального строку для проведення процедури реєстрації (210 днів) не виконується щодо препаратів з повним (автономним) досьє (за стандартною процедурою). Враховуючи строки, передбачені законом (не зважаючи на те, що певні строки довші у Наказі МОЗ № 426 та те, що Наказ МОЗ №426 встановлює додаткові процедури зі своїми строками виконання), загальна тривалість звичайної процедури реєстрації може зайняти 233 робочих дні (приблизно 330 календарних днів), за вирахуванням можливості призупинення перебігу строку компетентними органами (3 робочих дні для відправки листа- направлення в МОЗ + 210 робочих днів для експертизи ДЕЦ + 10 робочих днів для прийняття рішення МОЗ + 10 робочих днів для видачі реєстраційного посвідчення).
247. При цьому відповідне українське законодавство, як видається, прийняте на основі Регламенту 726/2004, що встановлює централізовану процедуру реєстрації лікарських засобів EMA, і де період у 210 днів встановлено для прийняття висновку Комітетом з лікарських засобів для застосування людиною. Необхідно наголосити, що довший строк для EMA обумовлений тим, що централізованій процедурі реєстрації підлягають лише окремі, найбільш складні та інноваційні типи лікарських засобів та тим, що під час цієї процедури мають враховуватися висновки експертів з різних держав-членів. Вибір європейської централізованої процедури в якості зразка для української національної процедури не видається обґрунтованим.
248. По-друге, загальна тривалість процедури реєстрації на практиці є довшою у зв’язку з доволі частими запитами регуляторних органів до заявників щодо надання додаткових документів або пояснень. Крім того, відповідно до українського законодавства строки призупиняються не лише на час отримання відповідей від заявника, але й на час отримання відповідей від третіх сторін (включаючи іноземні компетентні органи) та на час проведення лабораторних випробувань (п. VII.5 Наказу МОЗ № 426). Як видається, цей перелік підстав для призупинення строків реєстрації не відповідає положенням Директиви 2001/83.
ІІІ.10. ПІДСТАВИ ДЛЯ ПРИЙНЯТТЯ РІШЕННЯ ПРО РЕЄСТРАЦІЮ ТА ВІДМОВУ В РЕЄСТРАЦІЇ ЛІКАРСЬКОГО ЗАСОБУ
249. Згідно з українським законодавством, рішення про державну реєстрацію лікарського засобу приймається МОЗ на основі результатів експертизи заяви та поданих матеріалів (Закон про лікарські засоби). Постанова КМУ № 376 містить положення щодо того, що експертиза повинна проводитися ДЕЦ, який є спеціалізованим державним підприємством, що надає послуги з проведення експертизи. Залежно від результатів експертизи, ДЕЦ дає рекомендацію МОЗ щодо відмови в реєстрації лікарського засобу, або готує обґрунтовані висновки щодо якості, безпеки та ефективності лікарського засобу та надає рекомендацію МОЗ щодо його реєстрації.
250. Закон не встановлює, що рекомендації ДЕЦ є обов’язковими для МОЗ. На практиці, МОЗ слідує рекомендаціям ДЕЦ.
251. Законодавство ЄС чітко не регулює підстави для прийняття позитивного рішення щодо реєстрації лікарського засобу. Компетентний орган зобов’язаний перевіряти заяву на повноту та точність. З цієї точки зору, українське законодавство загалом відповідає законодавству ЄС.
252. Однак, відповідно до українського законодавства, МОЗ може прийняти рішення щодо реєстрації лікарського засобу без висновків експертизи ДЕЦ. Згідно із нещодавно прийнятим пунктом 3.5 Постанови КМУ № 376, в межах спрощеної процедури щодо лікарських засобів, зареєстрованих в певних іноземних юрисдикціях, експертиза вважатиметься проведеною, якщо ДЕЦ не надає свої висновки та рекомендації протягом 20 робочих днів. Такий підхід викликає серйозні сумніви щодо його відповідності законодавству ЄС, оскільки він застосовується до лікарських засобів, зареєстрованих за межами ЄС. Це може привести до допущення на ринок лікарських засобів, які попередньо не пройшли наукові дослідження згідно зі стандартами ЄС, без будь-яких подальших досліджень.
253. Директива 2001/83, як і українське законодавство, регулює підстави для відмови в реєстрації лікарського засобу.
254. Відповідно до Директиви 2001/83, в реєстрації лікарського засобу має бути відмовлено, якщо після перевірки заяви стало зрозуміло, що:
- співвідношення ризик-користь є недостатньо обґрунтованим;
- якісні та кількісні характеристики не відповідають заявленим;
- подані документи не відповідають вимогам.
255. У порівнянні з Директивою 2001/83, українське законодавство встановлює широкий перелік підстав для відмови у реєстрації. Знову ж таки, ті самі питання регулюються трьома різними нормативно-правовими актами, у кожному випадку дещо інакше. Підстави для відмови можна виділити в наступні групи:
під час експертизи реєстраційних матеріалів не підтвердилися відомості щодо ефективності, безпеки та якості такого лікарського засобу; незважаючи на інше формулювання, аніж те, що в Директиві 2001/83, ця підстава для відмови у реєстрації може вважатися сумісною з законодавством ЄС;
порушення прав інтелектуальної власності – як зазначено в 218-219 вище, на нашу думку, введення додаткового захисту прав інтелектуальної власності під час процедури реєстрації є прийнятним та не суперечить Директиві 2001/83.
256. Крім підстав для відмови у державній реєстрації, українське законодавство (пункт ІV.6 Наказу МОЗ № 426) окремо регулює підстави для надання рекомендації про відмову в реєстрації ДЕЦ. ДЕЦ не може рекомендувати лікарський засіб до реєстрації, «якщо за результатами спеціалізованої експертизи не підтвердилися висновки щодо його ефективності, безпеки та якості», а саме, якщо мають місце одна з 5 окремих підстав для ненадання позитивних рекомендацій ДЕЦ. Перші три з них відповідають Директиві 2001/83. Наступні дві стосуються порушення правил захисту реєстраційних даних (зокрема, ексклюзивності даних), та рішення суду щодо порушення прав інтелектуальної власності. Як вже зазначалося вище, ці дві підстави є прийнятними з точки зору законодавства ЄС, однак чинне регулювання цих питань повинне бути вдосконалене. Питання порушення правил захисту реєстраційних даних (зокрема, ексклюзивності даних) рекомендується досліджувати на ранній стадії процедури реєстрації. Порушення прав інтелектуальної власності як підстава відмови в державній реєстрації (ненадання рекомендації про державну реєстрацію) повинна бути сформульована однаково в усіх нормативно-правових актах.
257. В межах спрощеної процедури підстави для відмови в реєстрації є звуженими та включають:
(i) щодо лікарських засобів, зареєстрованих іноземними компетентними органами:
- відсутність необхідних документів,
- надання неправдивої чи неповної інформації,
- невідповідність інформації щодо виробника та місця виробництва.
(ii) щодо лікарських засобів, які підлягають закупівлі за результатами закупівельної процедури, проведеної спеціалізованою організацією, яка здійснює закупівлі, на виконання угоди щодо закупівлі між МОЗ та відповідною спеціалізованою організацією, яка здійснює закупівлі:
- відсутність необхідних документів,
- надання неправдивої чи неповної інформації,
- неточний переклад інформації на упаковці та інструкції.
258. Зменшення переліку підстав для відмови в державній реєстрації в межах спрощеної процедури є наслідком зменшеного обсягу документів, що підлягають експертизі. Як вже зазначалося вище, таке положення не відповідає законодавству ЄС у тій мірі, в якій воно дозволяє виведення на ринок лікарських засобів, не зареєстрованих в ЄС, без повної експертизи реєстраційного досьє згідно з вимогами Директиви 2001/83.
ІІІ.11. ПРИЙНЯТТЯ РІШЕННЯ ПРО ДЕРЖАВНУ РЕЄСТРАЦІЮ ЛІКАРСЬКОГО ЗАСОБУ
ІІІ.11.1 Колективне рішення про реєстрацію/індивідуальне рішення про реєстрацію
259. Найбільш очевидною особливістю української системи реєстрації в порівнянні з країнами ЄС є те, що в Україні рішення про реєстрацію приймається у формі наказу МОЗ, в якому зазначається перелік лікарських засобів, що підлягають реєстрації. Після прийняття наказу ці лікарські засоби включаються до Єдиного реєстру лікарських засобів України. До цього реєстру вносяться зміни у разі закінчення строку дії реєстраційного посвідчення, перереєстрації або внесення змін в реєстраційні матеріали.
260. Директива 2001/83 не встановлює вимог до форми рішення про державну реєстрацію лікарських засобів. Однак, на практиці в державах-членах ЄС воно має форму індивідуального рішення, яке стосується одного лікарського засобу одного заявника. Паралельно в цих країнах існують переліки зареєстрованих лікарських засобів, однак записи до цих переліків зазвичай вносяться на підставі попередніх індивідуальних рішень про реєстрацію лікарського засобу.
Приклад (Польща)
До 2002 року польська система реєстрації лікарських засобів в певній мірі нагадувала чинну українську систему, зокрема:
- Заявники подавали заяви не для індивідуальної реєстрації лікарського засобу, а для його внесення до офіційного реєстру.
- На відміну від чинного українського законодавства, рішення про реєстрацію приймалось компетентним органом (Комітетом з реєстрації лікарських засобів та виробів медичного призначення) в формі індивідуальної постанови. Ця постанова могла бути оскаржена нарівні з рішенням будь-якого іншого адміністративного органу, прийнятим щодо конкретної справи.
- На підставі постанови Міністр охорони здоров ’я вносив лікарський засіб до офіційного реєстру та підтверджував факт включення до реєстру шляхом видачі заявнику індивідуального посвідчення про реєстрацію.
- На підставі даних офіційного реєстру Міністерство охорони здоров ’я двічі на рік оприлюднювало перелік лікарських засобів, зареєстрованих в Польщі.
Польський фармацевтичний закон запровадив в 2002 році реєстрацію лікарських засобів на підставі індивідуального рішення про реєстрацію. Реєстрація здійснюється шляхом прийняття індивідуального адміністративного рішення, винесеного в результаті адміністративних розгляду в конкретній справі. На підставі винесення рішення вноситься відповідний запис до офіційного національного реєстру лікарських засобів, зареєстрованих в Польщі. Загальнодоступний перелік лікарських засобів, зареєстрованих в Польщі, оновлюється раз на рік.
Перехід на нову систему здійснювався на наступних умовах:
- індивідуальні посвідчення про реєстрацію стали реєстраційними посвідченнями лікарських засобів в розумінні нового закону та залишалися дійсними до закінчення строку їх дії,
- щоб продовжувати реалізацію лікарських засобів, власники попередніх посвідчень про реєстрацію (реєстраційних посвідчень на лікарський засіб в розумінні нового закону) повинні були привести свою документацію на лікарські засоби у відповідність до вимог нових стандартів та подати заяву про перереєстрацію.
261. Прийняття колективних рішень замість індивідуальних реєстрацій, на наш погляд, є більш обтяжливим для всіх зацікавлених осіб. Це зумовить затримки у винесенні рішень в окремих випадках, особливо щодо внесення змін. При цьому залучається експертна установа (ДЕЦ), яка зобов’язана підготувати проект наказу, що мало б бути виключною відповідальністю МОЗ. Крім того, такі рішення важко узгодити з правом індивідуального оскарження. Згідно з українською практикою, наказ про реєстрацію можна оскаржити тільки в судовому порядку (можливість оскарження в адміністративному порядку відсутня).
262. В такому випадку наказ МОЗ буде розглядатися як рішення державного органу, яке впливає на права заявника. Відповідно до Кодексу адміністративного судочинства України таке рішення може бути оскаржене в адміністративному суді. Незважаючи на це, судова практика оскарження заявниками наказів МОЗ, за якими їм було відмовлено у реєстрації, наразі відсутня.
Рекомендація:
Розглянути можливість заміну наказів про реєстрацію, ухвалених колективно, індивідуальними рішеннями про реєстрацію.
Затвердити порядок адміністративного оскарження індивідуальних рішень МОЗ щодо реєстрації/відмови у реєстрації.
III.11.2. Строк дії реєстраційного посвідчення
263. Відповідно до Директиви 2001/83 реєстраційне посвідчення лікарського засобу є дійсним протягом п’яти років, строк його дії може бути продовжено на основі переоцінки співвідношення користь/ризик. Після продовження реєстраційне посвідчення лікарського засобу буде дійсним на необмежений строк, але за наявності обґрунтованих підстав, що стосуються фармаконагляду, уповноважений орган може вирішити поновити строк дії посвідчення додатково на п’ять років.
264. Положення українського законодавства повністю відповідають вищезазначеним положенням Директиви 2001/83. Перша реєстрація є дійсною протягом 5 років, а перереєстрація є дійсною на необмежений термін, крім випадків, коли МОЗ прийняв рішення про додаткове продовження строку ще на п ’ять років за наявності обґрунтованих підстав, що стосуються фармаконагляду.
265. За інформацією, наданою представниками галузі, насправді МОЗ зазвичай здійснює перереєстрацію на 5 років замість перереєстрації на необмежений строк без належного обґрунтування. Для приведення українського законодавства у відповідність з законодавством ЄС важливо, щоб заявник мав право оскаржити необгрунтоване продовження строку на 5 років, а не видачу безстрокового посвідчення.
266. Директива 2001/83 містить так зване положення «sunset clause», відповідно до якого строк дії реєстраційного посвідчення лікарського засобу закінчується, якщо він фактично не був розміщений на ринку протягом 3 років з дати його реєстрації, або лікарський засіб відсутній на ринку 3 роки підряд, якщо тільки це не зумовлюється обґрунтованими причинами. Українське законодавство в цьому контексті не повністю відповідає законодавству ЄС, а тому воно має бути змінене і передбачати винятки для випадків, коли лікарські засоби не були розміщені на ринку протягом 3 років підряд з обґрунтованих причин.
ІІІ.11.3. Реєстрація «під умовами»
267. Директива 2001/83 передбачає три типи ситуацій, коли додаткові умови або обов’язки можуть бути покладені на власника реєстраційного посвідчення:
- додаткові обов’язки, що включаються в реєстраційне посвідчення лікарського засобу задля забезпечення безпеки (наприклад, обов’язок проведення дослідження безпеки чи ефективності лікарського засобу після реєстрації, надання звітності про негативні побічні реакції, удосконалення системи фармаконагляду) з встановленням граничного строку у разі необхідності (Ст.21а);
- додаткові обов’язки будь-якого характеру для власника реєстраційного посвідчення, який не в змозі надати вичерпні дані щодо ефективності та безпеки з об’єктивних причин, що можуть бути перевірені, коли продовження реєстраційного посвідчення лікарського засобу пов’язане зі щорічною переоцінкою цих умов (Ст.22);
- обов’язок післяреєстраційного проведення дослідження безпеки та ефективності лікарського засобу (з встановленням граничного терміну після видачі реєстраційного посвідчення лікарського засобу, обов ’язковим повідомленням про це заявника та внесенням змін у реєстраційне посвідчення) (Ст.22а).
268. Українським законодавством врегульовано лише другу ситуацію, коли заявник не в змозі надати певні дані. З такої позиції українському законодавству бракує вимоги, що продовження реєстрації є умовним у рамках щорічної переоцінки виконання заявником покладених зобов’язань.
269. В українському законодавстві також немає можливості включити умови в реєстраційне посвідчення лікарського засобу з причин, відмінних від відсутності детальних даних (Ст.21а Директиви 2001/83) та накладати додаткові обов’язки вже після реєстрації (Ст.22а Директиви 2001/83).
ІІІ.12.ОБОВ’ЯЗКИ ВЛАСНИКА РЕЄСТРАЦІЙНОГО ПОСВІДЧЕННЯ
270. Директива 2001/83 вимагає, щоб заявник був зареєстрований в ЄС та ЄЕЗ. В українському законодавстві відповідної вимоги немає. Для відповідності законодавству ЄС було б доречно додати вимогу щодо того, що заявник має бути зареєстрованим в Україні або ЄС/ЄЕЗ.
271. Директива 2001/83 не встановлює принципів відповідальності за якість лікарських засобів. Вона передбачає відповідальність заявника за точність даних у заяві та стверджує, що реєстрація «не повинна впливати на цивільну або кримінальну відповідальність виробника та, у відповідному випадку, власника реєстраційного посвідчення» (Ст.25). Враховуючи принципи відповідальності за якість лікарських засобів, передбачені у Директиві 85/374 щодо відповідальності виробника, відповідальність за якість лікарських засобів лежить переважно на виробнику або його імпортері.
272. Українське законодавство застосовує інший підхід до принципів відповідальності за якість лікарських засобів. Відповідно до нього заявник є відповідальним за ефективність, безпеку та якість лікарських засобів. Покладення відповідальності на заявника видається невмотивованим з урахуванням того, що часто заявник не виробляє і не імпортує лікарські засоби.
273. Директива 2001/83 встановлює наступні обов’язки заявника, пов’язані з отриманням реєстраційного посвідчення:
- враховувати науковий та технічний прогрес щодо методів виробництва та контролю якості;
- надавати компетентному органу будь-яку нову інформацію, яка може спричинити внесення змін до реєстраційних документів, зокрема інформацію про заборони або обмеження щодо лікарських засобів, представлених в інших країнах, інформацію, яка може вплинути на співвідношення ризик/ користь, результатів нових клінічних досліджень;
- забезпечувати актуалізацію інформації про лікарські засоби з сучасними науковими дослідженнями;
- надавати певну інформацію та документи на вимогу уповноваженого органу (дані, які демонструють, що співвідношення користь/ризик залишається сприятливим, копії мастер-файлу системи фармаконагляду, дані, які відносяться до обсягів продажів та рецептів).
274. Українське законодавство містить положення, які відповідають вищевикладеному. Однак ми не можемо визначити положення, які обґрунтовують причини вимагати додаткову інформацію та додаткові документи МОЗ у період дії реєстраційного посвідчення. До українського законодавства мають бути внесені зміни з цього питання.
275. Крім того, відповідно до Директиви 2001/83, власник реєстраційного посвідчення зобов’язаний надавати компетентному органу дані щодо фактичного розміщення лікарських засобів на ринку та інформувати про заплановані припинення поставок на ринок не менш ніж за 2 місяці. Відсутність відповідних положень в українському законодавстві ускладнює процес запровадження «sunset clause» МОЗ.
ІІІ.13. ПУБЛІЧНА ІНФОРМАЦІЯ
276. Директива 2001/83 гарантує, що певна інформація щодо лікарських засобів та процесу державної реєстрації є загальнодоступною. До такої інформації належить:
- реєстраційне посвідчення лікарського засобу, інструкція для медичного застосування, зведена характеристика лікарського засобу, умови, за яких було здійснено державну реєстрацію лікарського засобу (у випадку необхідності);
- звіт про оцінку (складається за результатами перевірки заяви про державну реєстрацію лікарського засобу), після того як інформація комерційного конфіденційного характеру була вилучена, та включає огляд, написаний у формі, зрозумілій для широкого загалу.
277. Українське законодавство відповідає цим вимогам лише частково.
278. В Україні існує загальнодоступна база даних зареєстрованих лікарських засобів (http://www.drlz.com.ua). База даних дозволяє отримати доступ до інформації про торгову марку, виробника, форму випуску, номер реєстраційного посвідчення, термін його дії, дату і номер наказу МОЗ, заявника, діючу речовину, синонімічні назви, склад активних речовин, АТХ-код (код по Анатомічній, Терапевтичній, Хімічній системікласифікації), умови відпуску лікарського засобу, термін придатності, інформацію про можливість здійснювати рекламування лікарського засобу та приналежність до наркотичних лікарських засобів, які закуповують відповідно до абзацу 17 статті 2 Закону України «Про публічні закупівлі». Доступна також інструкція для медичного застосування. В той же час відсутня повна інформація щодо державної реєстрації.
279. В Україні також не публікується звіт про оцінку лікарського засобу.
280. Нещодавно було внесені зміни до Закону про лікарські засоби, які зобов’язують ДЕЦ публікувати інформацію про подані заяви про державну реєстрацію, статус заяви та прийняті рішення (нова частина 7 статті 3 Закону про лікарські засоби). Втім, описані зміни досі не імплементовані.
281. Необхідним є подальше внесення змін до українського законодавства для його приведення у відповідність із законодавством ЄС.
282. На відміну від Директиви 2001/83, українське законодавство орієнтоване скоріше на захист конфіденційності реєстраційної інформації. В декількох випадках українські законодавці нагадали, що МОЗ і ДЕЦ зобов’язані захищати інформацію, яка міститься в заявах про державну реєстрацію та в реєстраційному досьє, від розголошення і недобросовісного комерційного використання. Наказ МОЗ № 426 надає дуже широке визначення поняття «конфіденційна реєстраційна інформація». Відповідно до Наказу МОЗ № 426 це «науково-технічна інформація, що міститься у реєстраційних матеріалах на лікарський засіб, включаючи частини I – IV або модулі 1 – 5 матеріалів реєстраційного досьє, методи контролю якості лікарського засобу», та, лише у виняткових випадках, вилучає зі змісту цього поняття певну інформацію, яка доступна в інструкції та інформації для пацієнта.
283 Зважаючи на суворий підхід до конфіденційності, ще більш виправданим є розширення об’єму інформації, яка має бути загальнодоступною, з метою посилення контролю з боку громадськості.
ІІІ.14. ПРОЦЕДУРА ПЕРЕРЕЄСТРАЦІЇ
284. Регулювання процедури перереєстрації лікарського засобу займає дуже незначне місце в Директиві 2001/83. Відповідно до положень Директиви 2001/83 лікарський засіб підлягає перереєстрації на підставі переоцінки співвідношення ризику/користь. У зв’язку з цим, власник реєстраційного посвідчення має представити зведений файл, який містить інформацію щодо якості, безпеки та ефективності, включаючи оцінку даних, що міститься у Звіті про підозрювані непередбачені серйозні побічні реакції та Періодичному звіті щодо оновлення інформації про безпеку, та інформацію про всі внесені зміни щонайменше за 9 місяців до закінчення строку дії реєстраційного посвідчення.
285. Українське законодавство в цілому відповідає вимогам Директиви 2001/83.
286. Важлива відмінність стосується строків подачі заяви про перереєстрацію. Відповідно до законодавства України заявник повинен подати заяву про перереєстрацію не раніше, ніж за 1 рік і не пізніше, ніж за 90 днів до закінчення строку дії реєстраційного посвідчення. Такий строк не відповідає вимогам Директиви 2001/83. Видається, що такі строки не узгоджені зі строками проведення експертизи заяв про перереєстрацію відповідно до Наказу МОЗ № 426 одна експертиза займає до 90 робочих днів (45 робочих днів для АФІ), а також відповідно до Постанови КМУ № 376 МОЗ має ще один місяць для прийняття рішення. Якщо додати ще декілька тижнів для здійснення перевірки на дотримання формальних вимог в МОЗ, на попередню експертизу та на заключний етап в МОЗ, то стає зрозуміло, що навіть правильно і своєчасно проведена процедура перереєстрації може зайняти близько півроку. Необхідно узгодити загальні строки та кінцеві терміни для вчинення послідовних дій органами влади.
287. Реєстраційна форма та перелік документів, необхідних для перереєстрації, як правило, відповідає Настановам CMDh (зі змінами від лютого 2016). Українське законодавство передбачає необхідність надання деяких додаткових документів:
- 1.8. Зведені дані виробника/заявника про безпеку медичного застосування лікарського засобу в Україні протягом строку дії реєстраційного посвідчення за формою, передбаченою законодавством;
- 1.12. Стислий виклад системи фармакологічного нагляду (за необхідності). Виклад не подається для перереєстрації традиційних лікарських засобів, гомеопатичних лікарських засобів, що відповідають вимогам додатку 7 до Наказу МОЗ № 426 та лікарських засобів, які належать до групи «Медичні гази».
III.15. ПІСЛЯРЕЄСТРАЦІЙНІ ЗМІНИ / ЗМІНИ ДО РЕЄСТРАЦІЇНИХ МАТЕРІАЛІВ
III.15.1. Загальні положення
288. Регулювання внесення змін до реєстраційних матеріалів в країнах ЄС сьогодні є сумішшю національних та загальноєвропейських норм. Єдині правила для внесення змін визначені Регламентом 1234/2008, і держави-члени могли самостійно вирішувати чи продовжувати застосовування норм національного законодавства щодо реєстрацій лікарських засобів, що були здійснені до 1 січня 1998 року. Подальші ремарки стосуються регулювання, визначеного Регламентом 1234/2008.
289. Українське законодавство значною мірою відповідає Регламенту 1234/2008.
III.15.2. Класифікація змін
290. Класифікація змін відповідає класифікації, прийнятій в ЄС. Проте існує інший підхід до «редакційних змін».
291. Українська категорія «технічні помилки» відповідає категорії ЄС «редакційні зміни». При цьому українська категорія ширша за змістом (охоплює також помилки в Короткій характеристиці лікарського засобу / інструкції для медичного застосування). В Україні вони не класифікуються як зміни, не вимагають заяви про внесення змін, але як і раніше повинні доводитися до відома МОЗ і (за винятком орфографічних / граматичних помилок) потребують затвердження Наказом МОЗ .
292. В ЄС, якщо зміни до реєстраційного досьє є виключно редакційними, такі зміни, як правило, не виносяться на розгляд окремо, але їх можна включити до змін, які стосуються тієї частини реєстраційного досьє, у якій відбулися редакційні зміни. Утаких випадках заявник повинен надати заяву про те, що зміст відповідної частини досьє не змінювався, окрім змін, вказаних самим заявником (Настанова ЄС щодо класифікації змін, Стаття 2). Видається, що в Україні немає такої можливості.
293. Щоб виправити технічну помилку, необхідно надіслати листа до МОЗ (єдина форма заяви не передбачена). Розгляд технічних помилок, які необхідно виправити, має здійснюватися в рамках попередньої експертизи (протягом 20 днів з моменту подання листа-заявки та необхідних документів для обґрунтування) шляхом порівняння виправлених реєстраційних матеріалів з оригінальними реєстраційними матеріалами заявника. Для деяких типів технічних помилок необхідно провести спеціалізовану експертизу поданих матеріалів (але вона не повинна тривати більше 20 робочих днів).
294. Дещо відмінний підхід також застосовується щодо внесення змін під час процедури перереєстрації. Він можливий як в ЄС, так і в Україні. Проте в ЄС власнику реєстраційного посвідчення рекомендовано розпланувати, за можливості, подання заяв про внесення змін таким чином, щоб їх подання не співпадало з періодом подання заяви про перереєстрацію лікарського засобу. Однак, якщо була виявлена необхідність внесення змін до реєстраційних матеріалів лікарського засобу, особливо якщо такі зміни стосуються питань безпеки, то власнику реєстраційного посвідчення рекомендовано звернутися до компетентного органу влади завчасно до подачі заяви про внесення змін для того, щоб узгодити особливості розгляду цих паралельних заяв. Було б доцільно внести аналогічне правило до українського законодавства.
III.15.3. Інші невідповідності
295. До інших невідповідностей належать:
a. Постанова КМУ №376 містить недосконале (надто вузьке) визначення змін до реєстраційного досьє. Відповідно до Постанови КМУ №376, зміни визначаються як «факти (обставини) щодо зареєстрованих лікарських засобів, які потребують внесення змін до реєстраційних матеріалів», проте в дійсності зміни можуть (і дійсно) виникають в результаті не лише виявлених фактів, але й у зв’язку з рішенням заявника (наприклад, назва ЛЗ).
b. В Україні відсутня норма, подібна до Статті 3.2 Регламенту 1234/2008, яка б визначала, що зміна, яку неможливо класифікувати, має за замовчуванням розглядатися як зміна типу ІВ.
c. Українське законодавство дозволяє заявникам звернутися за консультацією до ДЕЦ, проте – на відміну від Регламенту 1234/2008 – ДЕЦ не зобов’язаний надати заявнику рекомендацію щодо дотримання певних строків, обов’ язкову до виконання (45 днів у ЄС) та публікувати рекомендації.
d Для продовження реєстрації лікарського засобу в Україні необхідна нова реєстрація шляхом видачі нового реєстраційного посвідчення, в той час як в ЄС інформація про продовження реєстрації може включатися до оригінального реєстраційного посвідчення лікарського засобу (за такою ж процедурою, що і реєстрація).
e. У порівнянні з Регламентом 1234/2008, українське законодавство дозволяє групувати зміни (подавати заявку, яка охоплює кілька змін щодо реєстраційних матеріалів одного або декількох лікарських засобів) лише в обмеженій кількості ситуацій (ситуації відповідають пунктам 1 та 2 Додатку III до Регламенту 1234/2008; інші ситуації групування змін, які наведені в Додатку III, не передбачені законодавством України).
f. Українське законодавство не передбачає можливості тимчасового звільнення від зобов’язання надавати певні доклінічні або клінічні дані у випадку виникнення в пандемічної ситуації в якості винятку.
g. Українське законодавство не визнає потенційну серйозну небезпеку для навколишнього середовища у якості підстави для внесення термінових змін, пов’язаних з безпекою.
296. Крім того, наявні певні невідповідності в українській реєстраційній формі (див. наші зауваження викладені у пунктах 172-173 вище).
III.15.4. Процедура та строки
297. Що стосується процедури внесення змін, Регламент 1234/2008 не регулює це питання для національної реєстраційної процедури, а лише для процедури взаємного визнання та децентралізованої процедури. Незважаючи на це, норми цього акта в тій частині, що стосується «національної» частини процедури взаємного визнання та децентралізованої процедури можуть слугувати як приклад для українського законодавства, з урахуванням наступних зауважень.
298. Відповідно до Регламенту 1234/2008, про зміни типу IA/IAIN необхідно повідомити протягом 12 місяців після їх внесення (або відразу після зміни в певних випадках) – цей принцип має назву «do-and-tell» («зроби та повідом»). Після цього компетентний орган влади повідомляє заявника протягом 30 днів про те, чи він приймає, чи відмовляє у прийнятті зміни. Якщо зміна схвалена компетентним органом, то останній повідомляє заявника про те, чи необхідно вносити зміни в реєстраційне посвідчення.
299. В Україні таке повідомлення є лише одним із варіантів для заявника, а альтернативою є стандартна заява на внесення змін. Законодавство не заохочує заявників вдаватися до цього варіанту. Навпаки, в Україні відсутня процедури, яка б визначала порядок дій органів влади після отримання такого повідомлення. На практиці заявники не користуються перевагами принципу «do-and-tell» для внесення змін типу IA, а віддають перевагу стандартним заявам.
300. Що стосується змін типу ІБ, Регламент 1234/2008 вимагає, щоб заявники надсилали повідомлення до внесення змін. Зміни вважаються прийнятими, якщо протягом 30 днів відповідний компетентний орган влади не виступає проти їх внесення («tell-and-do») («повідом та зроби»).
301. Українське законодавство використовує той же термін, що і європейське, – «повідомлення», але українська модель не передбачає принципу «мовчазної згоди» органу влади, що призводить на практиці до тієї ж (стандартної) процедури для змін типу ІБ та типу ІІ.
302. Що стосується змін типу II, Регламент 1234/2008 передбачає строк тривалістю у 60 днів для підготовки звіту про результати експертизи та рішення за результатами розгляду заяви. Зазначений строк продовжується до 90 днів лише у разі внесення змін, що стосуються нових показань до застосування лікарського засобу.
303. В Україні базовий строк для внесення змін такого типу становить 60 календарних днів. На перший погляд можна зробити висновок, що українське законодавство відповідає стандартам ЄС для змін типу II. На практиці ж строки, необхідні для розгляду заяви про внесення зміни типу II в Україні є набагато більшими, ніж 60 днів. Що стосується обмеження в 60 днів для змін типу Ш, то його можна вважати надмірним (у порівнянні з 30-денним періодом, протягом якого компетентний орган може надати свої заперечення за законодавством ЄС).
304. Для ілюстрації можливих строків процедури внесення змін в Україні нижче наводиться інформація щодо строків деяких процедур:
- 7 робочих днів для оформлення рахунку-фактури,
- 14 робочих днів для попередньої експертизи,
- зупинка процедури (максимум 30 днів),
- 3 дні на затвердження попередніх недоліків,
- експертиза (максимум 60 календарних днів)
- зупинка процедури (максимум 60 днів)
- 10 робочих днів на передачу проекту документа власнику реєстраційного посвідчення,
- 15 робочих днів на затвердження/редагування проекту документа власником реєстраційного посвідчення,
- 5 робочих днів на підготовку офіційного списку,
- 3 дні на прийняття рішення МОЗ про затвердження змін.
305. Таким чином, процес може зайняти 147 робочих днів, навіть для незначних змін типу ІА (в ЄС це займе не більше 37 календарних днів).
306. Українське законодавство передбачає скорочені строки проведення експертизи в ДЕЦ заяв про внесення змін до реєстраційних матеріалів, а саме:
- 90 робочих днів – зміни, які вимагають нової реєстрації;
- 45 робочих днів – зміни, пов’язані зі змінами у діючій речовині сезонної, передпандемічної або пандемічної вакцини проти грипу людини (ЄС: 30 календарних днів);
- 20 робочих днів – виправлення технічних помилок.
307. Як і у випадку з багатьма іншими строками, пов’язаними із процедурами державної реєстрації, до них не включаються строки дій, що вчиняються регуляторними органами до і після експертизи. Крім того, зазначення строків через посилання на «робочі дні» може збити з пантелику та призвести до нелогічних результатів, наприклад, в теорії прискорена процедура експертизи змін до реєстраційних матеріалів вакцини триває довше, ніж стандартна процедура для внесення змін (відповідно 45 робочих днів = приблизно 63 календарних дні проти 60 календарних днів).
308. Пріоритет віддається продукції, зареєстрованій за спрощеними (прискореними) процедурами (ЕМА, відповідними органами інших країн, що застосовують високі регуляторні стандарти, або прекваліфіковані ВООЗ). Внесення змін до реєстраційних матеріалів у таких випадках здійснюється позачергово.
III.16. ФАРМАКОНАГЛЯД (ОКРЕМІ ПИТАННЯ)
309. Питання, пов’язані з фармаконаглядом в принципі виходять за рамки цього Звіту. Ми коротко проаналізували поточний стан регулювання в цій галузі, а також зосередили свою увагу на відповідності законодавства України положенням законодавства ЄС щодо вимог до заяв про реєстрацію (перереєстрацію) лікарських засобів.
310. З аналізу законодавства можна зробити висновок, що в Україні вимоги до документів, пов’язаних з фармаконаглядом, в процесі реєстрації (перереєстрації), є аналогічними вимогам ЄС.
311. Україна уже імплементувала більшість нормативних вимог ЄС щодо фармаконагляду. Положення Наказу МОЗ № 898 загалом відповідають положенням законодавства ЄС про здійснення нагляду за ADR, складання списків ADR та критеріїв для встановлення частоти повторення ADR, невиконання обов’язків щодо подання звітності (форми, терміни), подання періодично оновлюваних РОЗБЛЗ.
312. Невідповідності між українським законодавством та законодавством ЄС обумовлені тим, що в українському законодавстві ще не в повній мірі імплементовано нові положення, запроваджені Регламентом 1235/2010 та Директивою 2010/84.
313. В Україні для досягнення повної відповідності законодавству ЄС необхідним є запровадження наступних змін:
a) Національний інтернет-портал про лікарські засоби (нинішня відкрита база даних, доступна за посиланням www.drlz.com.ua. не містить загальнодоступних звітів про результати експертизи, висновків про плани щодо управління ризиками;
b) Зобов’язання подавати інформацію про ADR в електронному вигляді;
c) Громадські слухання;
d) Єдину оцінку періодичних звітів про безпеку лікарських засобів;
е) Більш суворий контроль за неінтервенційними пост-реєстраційними дослідженнями безпеки;
f) Додатковий контроль за певними лікарськими засобами;
g) Додаткове регулювання питань щодо змін частоти і дати подання періодичних звітів про безпечність лікарських засобів та щодо зобов’язання розглядати дії, що стосуються реєстраційного посвідчення лікарського засобу.
IV. СПОСТЕРЕЖЕННЯ У СФЕРІ СТРАТЕГІЧНОГО РОЗВИТКУ
ІV.1. ВІДСУТНІСТЬ ЧІТКОГО БАЧЕННЯ І СТРАТЕГІЇ СИСТЕМИ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ ЯК СКЛАДОВОЇ СТРАТЕГІЇ РОЗВИТКУ СИСТЕМИ ОХОРОНИ ЗДОРОВ’Я
314. Основною функцією кожної системи реєстрації лікарських засобів є забезпечення їх належної якості, безпеки та ефективності.
315. Крім основної ролі, належним чином побудована система реєстрації лікарських засобів може сприяти досягненню й інших цілей у сфері охорони здоров’я або соціального і економічного розвитку. Наприклад, прискорені процедури для певних видів лікарських засобів можуть допомогти оперативно реагувати на епідеміологічні загрози; прозора та ефективна системи реєстрації може сприяти залученню вітчизняних та іноземних інвестицій у фармацевтичний сектор.
316. З іншого боку, розробка системи реєстрації без узгодження її з іншими елементами системи охорони здоров’я може позбавити державу ефективного інструменту або навіть перешкоджати стратегічним цілям всієї системи охорони здоров’я. Наприклад, незадовільний рівень захисту прав інтелектуальної власності під час процедури реєстрації може стати перешкодою для виходу інноваційних компаній із своїми ЛЗ на ринок або затримати вихід на ринок деяких ЛЗ.
317. З урахуванням вищевикладеного, система реєстрації лікарських засобів не повинна сприйматися ізольовано від інших елементів системи охорони здоров’я. Ефективна система реєстрації лікарських засобів – це не тільки система, яка забезпечує належний рівень експертизи лікарських засобів до їх виведення на ринок, але і система, яка сприяє досягненню інших стратегічних цілей держави у сфері охорони здоров’я.
318. Для того, щоб оцінити ефективність системи реєстрації в ширшому контексті, описаному вище, стратегія держави у сфері розвитку системи охорони здоров’я, включаючи розробку системи реєстрації лікарських засобів, повинна бути прийнята до уваги.
319. В ході нашого огляду загальнодоступних документів і актів для цілей підготовки Звіту, ми знайшли декілька ініціатив на державному та неурядовому рівнях, автори яких представили своє бачення стратегії розвитку системи реєстрації лікарських засобів в Україні.
1) «Програма економічних реформ в Україні на 2010-2014 роки» (Україна)
Програма, прийнята Президентом України, передбачає розробку і прийняття комплексного документа, який визначатиме політику держави в сфері охорони здоров’я. Однак такий документ не було прийнято.
2) «Національна стратегія побудови нової системи охорони здоров’я в Україні на період 2015 – 2025» (Стратегічна дорадча група з питань реформування системи охорони здоров’я в Україні, листопад 2014 року)
Документ підготовлено Стратегічною дорадчою групою з питань реформування системи охорони здоров’я в Україні, за спільною ініціативою української влади іМіжнародного фонду «Відродження». Документ був представлений для публічного обговорення в листопаді 2014 року. Ця стратегія не була прийнята в якості офіційного документа.
Стратегія представляє собою ряд пропозицій, сформульованих українськими та міжнародними експертами щодо майбутнього системи охорони здоров’я України. Деякі з пропозицій стосуються реформування системи реєстрації лікарських засобів в Україні:
- пропозиція реформувати МОЗ таким чином, щоб перейти від здійснення оперативних функцій до розробки політики – наприклад, МОЗ здійснюватиме контроль за діяльністю автономного агентства, відповідального за реєстрацію лікарських засобів, через своїх представників у керівному органі агентства;
- забезпечення відповідності стандартам, правилам і нормам, викладеним в нормативних актах ЄС;
- запровадження спрощеної процедури реєстрації – визнання Україною результатів експертизи реєстраційних досьє лікарських засобів, зареєстрованих у країнах із суворими регуляторними вимогами (ЄС, США, Швейцарія, Японія, Австралія, Канада).
3) «Здоров’я – 2020: український вимір» (проект програми, 2013 рік)
У цьому документі описується стратегія розвитку системи охорони здоров’я України. Стратегія так і не набрала чинності. Проект закону «Про затвердження Національної програми «Охорона здоров’я 2020: Український вимір» був відкликаний з Верховної Ради в лютому 2014 року.
4) «Бачення 2020: План дій у сфері розвитку фармацевтичного сектору в Україні» (Американська торгівельна палата, АПРАД, PwC, 2014 рік)
Документ, розроблений галузевими організаціями, що представляють інноваційні фармацевтичні компанії (Американська торгівельна палата, АПРАД) спільно з PriceWaterhouseCoopers, представив бачення розвитку інноваційної фармацевтичної промисловості в Україні. Мета цього документу полягала в розробці плану дій, для розвитку конкурентоспроможності українського фармацевтичного сектору, з акцентом на інноваційній промисловості та біологічній продукції.
Автори доповіді вказали на необхідність подальшого підвищення ефективності і прозорості системи реєстрації лікарських засобів як на один з ключових моментів для підвищення конкурентоспроможності фармацевтичної промисловості.
5) «Концепція дерегуляції обігу фармацевтичної продукції в Україні» (Пацієнти України, Arzinger, Всеукраїнська мережа ЛЖВ, квітень 2016 року)
У документі, підготовленому неурядовою організацією, містяться пропозиції про скасування або зміну існуючих процедур ліцензування, включаючи процедуру реєстрації лікарських засобів, для спрощення бізнесу і збільшення конкуренції на фармацевтичному ринку, що, на думку авторів, призведе до зниження цін, залучення інвестицій і збільшення асортименту та допоможе розвивати економіку.
Автори документа запропонували такі стратегічні завдання у сфері дерегуляції реєстрації лікарських засобів: 1) підвищити рівень доступу пацієнтів до лікування в Україні шляхом збільшення числа ефективних і безпечних зареєстрованих лікарських засобів і швидкості їх виведення на ринок України; 2) зниження адміністративних витрат і часових ресурсів щодо процедур ліцензування, які включаються в ціну лікарських засобів на ринку, таким чином, зменшити кількість бар ‘єрів для виходу лікарських засобів на український ринок.
6) «Ефективне управління ринком лікарських засобів в Україні» (Світовий банк, 2016 рік)
У доповіді обговорюється поточний стан нормативного регулювання фармацевтичного ринку України і викладаються пропозиції щодо його покращення.
У сфері реєстрації лікарських засобів автори доповіді визначають такі напрями для вдосконалення:
Зміцнення інституційного потенціалу регуляторних органів, в тому числі чіткий розподіл функцій між ними та ліквідація дублювання функцій.
Подальше спрощення регуляторних процедур, покращення їх адміністрування, для того, щоб зробити їх ефективними та привабливими для бізнесу.
Підвищення прозорості регуляторних процедур та діяльності регуляторних органів.
7) «Законодавча оцінка українського фармацевтичного сектору на відповідність стандартам ЄС в перспективі імплементації Угоди про асоціацію між Україною та ЄС», Представництво ЄС, червень 2016 року
Метою дослідження, проведеного ECORYS для Представництва ЄС в Україні, було запропонувати схему реформування фармацевтичної галузі України відповідно до законодавства ЄС, з особливим наголосом на реформування системи реєстрації лікарських засобів.
Автори доповіді дали такі рекомендації у сфері реєстрації лікарських засобів:
- здійснювати моніторинг результатів спрощеної процедури для прискореної реєстрації лікарських засобів, що підлягають закупівлі через міжнародні організації, і в довгостроковій перспективі усунути спрощену процедуру, що не відповідає нормативно-правовій базі ЄС;
- реорганізувати МОЗ і ДЕЦ з метою розширення повноважень ДЕЦ;
- посилити допомогу ЄС Україні в реформуванні української системи реєстрації лікарських засобів.
8) «Програма Кабінету Міністрів України» (затверджена постановою Верховної Ради України від 11.12.2014 № 26-VIII)
Дерегуляція фармацевтичного ринку, значне зниження кількості ліцензій і дозволів (2015-2016), а також реалізація наступних кроків:
- визнання реєстрації лікарських засобів і виробів медичного призначення, які виробляються в країнах ЄС, США, Канаді, Австралії, Японії, з подальшим застосуванням принципу взаємності;
- розробка та прийняття Закону України «Про внесення змін до Закону України «Про лікарські засоби»;
- розробка та прийняття Закону України «Про обов’язкове медичне страхування».
9) Національна політика щодо забезпечення лікарськими засобами
Ведеться робота над текстом документу. Остаточний текст проекту поки не доступний.
320. Незважаючи на ряд ініціатив, в Україні до сих пір немає документа, прийнятого на урядовому рівні, який міг би окреслити комплексну стратегію розвитку системи реєстрації лікарських засобів в контексті розвитку системи охорони здоров’я, а також слугувати орієнтиром для законодавців і посадових осіб.
321. Сама по собі відсутність такого документа не перешкоджає можливості проведення реформ. Насправді, за останні роки система знаходиться під впливом постійних реформ. Однак видається, що ці реформи, не реалізували жодну комплексну модель реєстрації лікарських засобів. Деякі з них, за попередніми висновками, навіть суперечать одна одній.
Приклад
У 2013 році процедура реєстрації була значно ускладнена: за новими нормами заяви про реєстрацію мали подаватись до МОЗ замість ДЕЦ, була введена формальна верифікація заяв у МОЗ, після якої досьє мало подаватися в ДЕЦ відповідно до рекомендацій МОЗ і т.д.
У той же час, з метою спрощення процедури були введені положення, що стосуються функціонування «єдиного вікна».
322. Без розробки і прийняття стратегічного документа існує ризик того, що результатом реформи системи реєстрації лікарських засобів в Україні стане поєднання випадкових рішень, що просуваються різними зацікавленими сторонами, а не узгоджений порядок дій, спрямованих на побудову системи на основі комплексного бачення.
323. Рішення, яким визначаються стратегічні цілі, які повинні бути досягнуті за рахунок розвитку системи реєстрації лікарських засобів, є політичним. Пропозиції експертів або неурядових/галузевих асоціацій можуть братися до уваги під час вирішення питання щодо стратегії розвитку системи реєстрації, проте фінальне рішення, що збалансує інтереси різних зацікавлених сторін, буде реалістичним і найкраще підходитиме для реальних потреб української системи охорони здоров’я, належить прийняти саме українській владі.
324. Роботи з підготовки Національної політики щодо забезпечення лікарськими засобами створюють можливість для визначення бажаної моделі системи реєстрації лікарських засобів в Україні, визначення ролі, яку система реєстрації лікарських засобів повинна відігравати у всій системі охорони здоров’я і виокремлення засобів для досягнення стратегічних цілей.
Рекомендація
Включити до порядку денного переговорів в процесі підготовки Національної політики щодо забезпечення лікарськими засобами питання, пов’язані з розробкою системи реєстрації лікарських засобів.
325. Прикладом стратегічної цілі є наближення законодавства і практики реєстрації лікарських засобів до європейських стандартів. Ця стратегічна ціль підтримується і просувається законодавцями і чиновниками, а також має широку підтримку серед зацікавлених сторін, які безпосередньо мають справу із системою реєстрації лікарських засобів. Тим не менш на шляху до цієї цілі необхідно прийняти ряд стратегічних рішень (див. розділ ІV.2 нижче).
ІV.2. СТРАТЕГІЧНІ РІШЕННЯ НА ШЛЯХУ НАБЛИЖЕННЯ ДО СТАНДАРТІВ ЄС
326. Угода про асоціацію містить ряд зобов’язань, що стосуються наближення українського законодавства і практики до стандартів ЄС.
327. Однак, окрім ексклюзивності даних/ ринкової ексклюзивності (стаття 222), Угода про асоціацію не містить конкретних положень, які б безпосередньо стосувалися функціонування системи реєстрації лікарських засобів.
Угода про асоціацію – відповідні положення
Ст. 1
2. Цілями цієї асоціації є:
(…) (d) запровадити умови для посилених економічних та торговельних відносин, які вестимуть до поступової інтеграції України до внутрішнього ринку ЄС, у тому числі завдяки створенню поглибленої і всеохоплюючої зони вільної торгівлі, як це визначено у Розділі IV («Торгівля і питання, пов ’язані з торгівлею») цієї Угоди, та підтримувати зусилля України стосовно завершення переходу до діючої ринкової економіки, у тому числі шляхом поступової адаптації її законодавства до acquis ЄС.
Стаття 426
Сторони розвивають співробітництво в галузі охорони здоров’я з метою підвищення рівня його безпеки та захисту здоров ’я людини як передумови сталого розвитку та економічного зростання.
Стаття 427
1. Співробітництво, зокрема, охоплює такі сфери:
a) зміцнення системи охорони здоров ’я України та її потенціалу, зокрема шляхом впровадження реформ, подальшого розвитку первинної медико-санітарної допомоги та навчання персоналу; (…)
2. Із цією метою Сторони обмінюються інформацією та найкращими практиками і здійснюють інші спільні заходи, в тому числі в рамках підходу «охорона здоров’я у всіх політиках» та поступової інтеграції України в європейські мережі охорони здоров’я.
Стаття 428
Україна поступово наближує своє законодавство та практику до принципів acquis ЄС, зокрема у сфері інфекційних хвороб, служб крові, трансплантації тканин і клітин, а також тютюну. Перелік відповідних актів acquis ЄС визначено у Додатку ХLІ до цієї Угоди.
328. Підсумовуючи вищевикладене, реєстрація лікарських засобів є однією зі сфер, у яких Україна повинна поступово наближати своє законодавство і практику до принципів законодавства ЄС, однак ані рівень такого наближення, ані його терміни так і не були встановлені.
329. Загальний характер зобов’язань України у сфері реєстрації лікарських засобів за Угодою про асоціацію залишає відносно багато місця для визначення масштабів необхідних реформ та їх кінцевої мети.
330. Ми визначили два основних стратегічних питання: по-перше, яким є запланований рівень подальшого наближення українського законодавства до законодавства ЄС у сфері реєстрації лікарських засобів, по-друге, в якій мірі лікарські засоби, зареєстровані до імплементації нового законодавства, повинні підлягати перереєстрації відповідно до нових процедур у відповідності до нормативних актів ЄС.
ІV.2.1. Бажаний рівень наближення законодавства
331. На основі огляду законодавства України можна зробити висновок, що за останні 16 років спостерігається значний прогрес у наближенні українського законодавства до норм законодавства ЄС. Українська система реєстрації лікарських засобів в її нинішньому вигляді спирається на ті ж принципи і поняття, які передбачені у Директиві 2001/83, в тому числі основні визначення, обсяг необхідної документації для різних видів процедур. Незважаючи на це, в українському законодавстві все ж наявні прогалини у нормативно-правовій базі, а також деякі сфери, які потребують внесення змін (як зазначено в розділі ІІІ вище).
332. З урахуванням виправлення перерахованих недоліків, нинішній рівень відповідності українського законодавства законодавству ЄС у сфері реєстрації лікарських засобів створює міцну основу для подальшого наближення.
333. Необхідно визначити, чи вже досягнутий рівень наближення є задовільним на нинішньому етапі реформування системи, чи українське законодавство потребує подальших змін.
334. Угода про асоціацію не визначає ані порядок, в якому повинно бути досягнуто «поступове наближення» законодавства, ані граничних строків такого наближення. З преамбули Угоди про асоціацію випливає, що «прогрес в наближені з ЄС у (…) правовій сфері» є однією з умов для «політичної асоціації та економічної інтеграції» України з ЄС (див. пункт 8 декларативної частини Угоди про асоціацію).У цьому контексті можна зробити висновок, що в інтересах інтеграції з ЄС, Україна повинна прагнути до чіткого наближення законодавства у сфері реєстрації лікарських засобів, наскільки це можливо з урахуванням поточного статусу України (Україна не є членом ЄС).
335. На наш погляд, подальше наближення українського законодавства до законодавства ЄС є можливим і доцільним, особливо якщо стратегічні цілі розвитку системи реєстрації лікарських засобів включають майбутню участь України у процедурі взаємного визнання реєстрацій між ЄС та Україною.
336. Метою подальшого наближення є досягнення рівня відповідності, який є рівносильним імплементації Директиви 2001/83 і відповідних документів до законодавства України (у максимально можливій мірі для країн, що не є членами ЄС).
Приклад для порівняння (Польща)
В Угоді про асоціацію 1991 року Польща взяла на себе зобов’язання наблизити своє законодавство до законодавства ЄС.
У відповідності до статті 68 Європейської угоди про створення асоціації між Європейськими співтовариствами та їх державами-членами, з одного боку, і Республікою Польща, з іншого боку, від 16 грудня 1991 року (набрала чинності 1 липня 1994 року): «Договірні Сторони визнають, що основною умовою для економічної інтеграції Польщі у Співтовариство є наближення існуючого і майбутнього законодавства цієї країни до законодавства спільноти в цілому. Польща докладе всіх зусиль для забезпечення того, щоб майбутнє законодавство відповідало законодавству Співтовариства».
На момент набрання чинності Угодою про асоціацію між ЄС та Польщею система реєстрації лікарських засобів в Польщі регулювалась Законом від 10 жовтня 1991 року про інспекцію фармацевтичних продуктів, медичних матеріалів, аптек та оптових фармацевтичних підприємств.
При виборі методу наближення польського законодавства до законодавства ЄС, Польща вирішила підготувати і прийняти абсолютно нову нормативну базу, що мала на меті імплементувати Директиву 2001/83. Новий Польський фармацевтичний закон був прийнятий 6 вересня 2001 року і набув чинності 1 жовтня 2002 року. Формулювання нового закону, а також його структура, повністю відповідали тексту Директиви 2001/83.
На дату набрання чинності Польським фармацевтичним законом дата вступу Польщі в ЄС ще не була відома. Проте в новий закон також було включено набір положень, які повинні були застосовуватися після вступу Польщі в ЄС (наприклад, щодо участі Польщі в процедурі взаємного визнання). Набрання чинності цими положеннями було відкладено до моменту вступу Польщі в ЄС.
337. Для досягнення рівня наближення, рівносильного імплементації законодавства ЄС, українські законодавці повинні розглянути можливість розробки нового законодавства, що базуються виключно на положеннях Директиви 2001/83 і відповідних нормативних актів ЄС, з метою повної заміни чинного законодавства (як це було зроблено, наприклад, в Польщі), а не надалі вносити безсистемні зміни до чинних нормативних актів. Для того, щоб бути ефективною, імплементація повинна охоплювати всі нормативні акти у сфері реєстрації лікарських засобів, а не тільки окремі фрагменти законодавства. Крім того, необхідно передбачити чіткі часові рамки імплементації нового законодавства з метою уникнення можливих колізій між новими та старими нормами. Особливого значення це набуває в контексті моделі українського законодавства (поєднання нормативних актів, що приймаються різними органами державної влади).
338. У той самий час, процес внесення змін до законів в Україні є затяжним та потребує наявності політичної волі на рівні Парламенту. Таким чином, з метою прискорення процесу наближення, альтернативно до прийняття нових законів, які мають бути затверджені Парламентом, положення права ЄС можуть бути імплементовані на рівні КМУ шляхом внесення відповідних змін до Постанови КМУ № 376, або ж шляхом прийняття нової Постанови.
339. Відповідно до положень статті 9 Закону про лікарські засоби, КМУ відповідає за встановлення правил державної реєстрації лікарських засобів в Україні. Таким чином, КМУ уповноважений встановлювати правила державної реєстрації лікарських засобів у тих випадках, коли такі правила відповідають загальним положенням Закону про лікарські засоби.
340. Введення в дію нового законодавства повинно бути відкладено на період, достатній для заявників та органів у сфері реєстрації лікарських засобів для пристосування до нових регуляторних вимог (проведення тренінгів, підготовка нових шаблонів, застосування нових СОПів, налагодження внутрішньої організації органів у сфері реєстрації лікарських засобів і т.д.).
Нам відомо про законодавчі ініціативи, спрямовані на приведення різних аспектів законодавства України у сфері реєстрації лікарських засобів у повну відповідність до Директиви 2001/83.
Проект Закону про особливості імплементації окремих положень законодавства Європейського Союзу щодо обігу лікарських засобів (реєстраційних номер 4465), який на час підготовки Звіту перебуває на розгляді у Верховній Раді України, спрямований на впровадження нових правил щодо реєстрації лікарських засобів у суворій відповідності до Директиви 2001/83.Згідно розділу XIV, цей проект передбачає набрання ним чинності 1 січня 2017 року.
Проект закону про лікарські засоби № 2162-д є ще одним проектом закону, що наразі знаходиться на розгляді у Верховній Раді України. Як і попередній проект, цей акт ставить собі за мету подальше наближення українського законодавства у сфері реєстрації лікарських засобів до законодавства ЄС, однак у дещо меншій мірі.
Ми не вдавалися до детального аналізу вищевказаних проектів, однак ми припускаємо, що їх метою є домогтися ще більшого рівня імплементації законодавства ЄС, у тому числі з використанням термінології Директиви 2001/83 і пов’язаних з нею актів.
Рекомендація:
Прийняти повністю нові законодавчі акти у сфері реєстрації лікарських засобів на заміну діючим для гармонізації українського законодавства із законодавством ЄС шляхом його повної імплементації.
[Опціонально] Всі ключові елементи процедури державної реєстрації можуть бути відображені шляхом внесення змін або викладення у новій редакції Постанови КМУ, як органу відповідального за прийняття правил державної реєстрації лікарських засобів в Україні згідно з Законом про лікарські засоби. Відкласти введення в дію нових законодавчих актів на строк, достатній для того, щоб дати можливість компетентним державним органам та заявникам підготуватися до змін.
ІV.2.2. Гармонізація досьє лікарських засобів, зареєстрованих в минулому
341. При введенні нових правил щодо реєстрації лікарських засобів виникає питання, пов’язане з тим, чи будуть такі правила застосовуватися до лікарських засобів, що зареєстровані до їх введення в дію.
342. Здійснені нещодавно реформи українського законодавства у сфері реєстрації лікарських засобів не враховують цей аспект.
343. Якщо нове законодавство буде застосовуватися тільки до майбутніх реєстрацій, то вплив наближення українського законодавства до законодавства ЄС буде досить обмеженим. На нашу думку, реальне наближення системи реєстрації лікарських засобів в Україні до законодавства та стандартів ЄС повинне частково стосуватися і реєстрацій, здійснених до введення в дію нового законодавства.
344. Поширення нового законодавства на минулі реєстрації матиме на меті: по-перше, сприяння покращенню доступу українських пацієнтів до лікарських засобів, що відповідають європейським стандартам якості, безпеки та ефективності – причому не тільки зареєстрованих вперше, але й вже виведених на ринок, і по-друге, створення підґрунтя для вільного обігу лікарських засобів з України в ЄС.
345. Процес оновлення повинен бути ретельно спланований з метою недопущення порушення нормальної роботи реєстраційних органів, а також уникнення проблем з поставками лікарських засобів на ринок.
346. Ключові рішення, які повинні бути прийняті у зв’язку з оновленням законодавства, стосуються строків, сфери застосування та процесуальної моделі оновлення.
a) Строки:
347. Рішення щодо строків повинно передбачати остаточну дату, до якої реєстраційна документація лікарських засобів, що підлягає процедурі оновлення, повинна бути приведена у відповідність до нових вимог, а також проміжні дати – для подання документів заявниками і для проведення експертизи документів органами з реєстрації лікарських засобів.
348. Ці строки повинні бути скориговані відповідно до можливостей регуляторних органів у системі реєстрації лікарських засобів в Україні. Відповідні стратегічні рішення повинні прийматися після проведення консультації з заявниками та з урахуванням фінансових та організаційних зусиль, яких потребуватиме від останніх процес оновлення.
b) Сфера застосування:
349. Сфера оновлення може бути дуже широкою (зобов’язання оновлювати кожну реєстрацію, за прикладом Польщі) або обмеженою (виключення певних видів лікарських засобів, наприклад, традиційні лікарські засоби або лікарські засоби, що виробляються згідно із затвердженим прописом).
350. Приймаючи рішення про масштаби оновлення необхідно брати до уваги вплив цього процесу на доступність лікарських засобів. Варто виходити з того, що заявники можуть відмовитися від оновлення деяких реєстрацій (наприклад, через низьку рентабельність у порівнянні з витратами на оновлення, незадовільні результати дослідження безпеки старих лікарських засобів, або через нездатність продемонструвати біоеквівалентність відносно певних оригінальних лікарських засобів. Цей процес повинен бути спланований таким чином, щоб звести до мінімуму ризик дефіциту лікарських засобів певного виду на ринку.
c) Процесуальна модель:
351. З точки зору процедури, оновлення повинне включати або надання регуляторним органам повного переліку документації (хімічна, фармацевтична, доклінічна, клінічна) у відповідності до європейських вимог, або лише доповнення вже існуючої документації з метою приведення реєстраційних досьє у відповідність до цих вимог. Вибір оптимальної моделі залежить від ступеня відповідності реєстрацій, здійснених до цього часу, європейським вимогам. Вибір оптимальної моделі можна залишити на розсуд заявників. У будь-якому випадку завдання органу з реєстрації лікарських засобів – розглянути документацію щодо лікарського засобу, перевірити її повноту і оцінити, чи відповідає лікарський засіб вимогам безпеки, якості та ефективності.
352. Оновлення реєстраційних матеріалів може бути здійснене або шляхом нової реєстрації для кожного лікарського засобу, який підлягає процедурі оновлення (повна процедура), або шляхом перереєстрації лікарського засобу.
Приклад для порівняння (Польща)
Оновлення національних реєстрацій лікарських засобів вимагалося від Польщі в рамках зобов’язань держави-члена, з метою виконання Директиви 2001/83 (див статтю 6 Директиви: «Жодний лікарський засіб не може бути розміщений на ринку держави – члена, доки компетентними органами цієї держави – члена не буде видано дозвіл на його продаж відповідно до цієї Директиви або не буде надано дозвіл відповідно до Регламенту (ЄС) № 726/2004 …»).
У порядку виключення, Польща і ЄС у Договорі про вступ від 16 квітня 2013 року домовилися про перехідний період:
«Додаток XII. У подяку часткового відступлення від вимог щодо якості, безпеки та ефективності, викладених у Директиві 2001/83, реєстраційні посвідчення для фармацевтичних продуктів, включених до переліку (в Доповненні А до цього Додатку), що видані у відповідності до польського законодавства до дати приєднання, залишаються в силі, доки вони не будуть оновлені у відповідності до законодавства ЄС у визначені у вищезазначеному документі строки, або до 31 грудня 2008 року, в залежності від того, що настане раніше. Незважаючи на положення розділу III, глави 4, Директиви 2001/83,на реєстраційні посвідчення, про які йде мова у цьому виключенні, не користуватися перевагами процедури взаємного визнання у країнах- членах ЄС».
Таким чином, у випадку Польщі було встановлено граничний термін для оновлення реєстраційних посвідчень, встановлений на 31 грудня 2008 року, а також індивідуальні дедлайни, встановлені для окремих видів лікарських засобів, в залежності від дати закінчення строків дії їх реєстраційних посвідчень (з січня 2004 року по 31 грудня 2008 року).
Обсяг процесу оновлення (названий в Польщі «гармонізація») був доволі широким – він охоплював всі лікарські засоби, дозволені для збуту на території Польщі (7349 лікарських засобів).
Власники реєстраційних посвідчень вирішили не оновлювати реєстраційну інформацію щодо більш ніж 500 лікарських засобів. Такі лікарські засоби могли бути легально розміщені на ринку до закінчення строку дії їх неузгоджених (негармонізованих) реєстраційних посвідчень, і залишатися на ринку до закінчення строків використання для кожної окремої партії.
6771 лікарський засіб було узгоджено (гармонізовано). 177 заявок на узгодження (оновлення) було відхилено.
Технічно модель гармонізації була побудована навколо концепції поновлення реєстраційних досьє. На дату набрання чинності нового Фармацевтичного закону (що в повній мірі імплементував Директиву 2001/83) старі посвідчення про реєстрацію стали «реєстраційними посвідченнями» в сенсі нового закону, дійсними до дат закінчення строку дії, зазначеного у них, але не довше, ніж до 31 грудня 2008 року.
Для того, щоб поновити реєстраційне посвідчення його власник зобов’язаний був «доповнити документацію щодо лікарського засобу і привести її у відповідність до вимог нового Закону». На практиці, як правило, заявники надавали повністю нову документацію, замість внесення доповнень до старих реєстраційних досьє. Післярозгляду документації відповідними реєстраційними органамиПольщі,адміністративне рішення про поновлення видавалось на п’ять років. У рішенні реєстраційний орган міг накласти зобов’язання додатково доповнити документацію, з зазначенням дати подання такої додаткової документації.
Рекомендація:
Розробити план гармонізації лікарських засобів, зареєстрованих раніше,доєвропейських стандартів.
Визначити часові рамки і рівень гармонізації з урахуванням можливостей українських органів у сфері реєстрації лікарських засобів та необхідності забезпечення безперервного доступу до ліків для пацієнтів України.
IV.3. ПИТАННЯ ЩОДО «СПРОЩЕНОЇ» ПРОЦЕДУРИ РЕЄСТРАЦІЇ ЛІКАСЬКИХ ЗАСОБІВ, ДОЗВОЛЕНИХ В КРАЇНАХ, ЩО НЕ Є ЧЛЕНАМИ ЄС
353. Прискорена процедура реєстрації, яка була введена Законом «Про внесення змін до статті 9 Закону України «Про лікарські засоби» щодо спрощення державної реєстрації лікарських засобів», застосовується до лікарських засобів, які вже були зареєстровані в ряді країн.
354. Українське законодавство передбачає спрощену реєстрацію лікарських засобів, зареєстрованих відповідними реєстраційними органами США, Канади, Японії, Австралії, Швейцарії та лікарських засобів, зареєстрованих за централізованою процедурою EMA. Жодна наукова експертиза цих ЛЗ в Україні не проводиться. Експертиза ДЕЦ обмежується порівнянням звіту про оцінку іноземного агентства з представленим досьє, а також перевіркою автентичності наданих матеріалів. ДЕЦ заборонено вимагати внесення змін до реєстраційних матеріалів для цих ЛЗ.
355. З урахуванням нинішніх проблем, пов’язаних з доступом до лікарських засобів в Україні і можливостями регуляторних органів, мотиви, які стояли за введенням «спрощеної» реєстрації, є загалом зрозумілими. Однак, у довгостроковій перспективі, модель «спрощеної» процедури може потребувати змін, виходячи з двох причин.
356. По-перше, законодавство ЄС не передбачає механізму визнання реєстраційних посвідчень, виданих у третіх країнах. Для розміщення на ринку ЄС, лікарський засіб в обов’язковому порядку повинен пройти процедуру реєстрації. У довгостроковій перспективі, ЛЗ, зареєстровані за «спрощеною» процедурою у країнах, що не є членами ЄС, повинні бути перереєстровані у процесі оновлення українського законодавства (його гармонізації).
357. По-друге, в результаті застосування «спрощеної» процедури для ЛЗ, зареєстрованих не в ЄС, можливою є ситуація, за якої лікарські засоби на ринку (наприклад, через використання наданої речовини, або через зареєстроване дозування, показання або недостатні обмеження щодо безпеки) не будуть відповідати вимогам ЄС щодо показань безпеки у зв’язку з різними науковими підходами, прийнятими органами з регулювання обігу лікарських засобів у всьому світі.
358. Як наслідок, лікарські засоби з різними показаннями безпеки або різним дозуванням однієї й тієї ж діючої речовини за тими ж показаннями, можуть одночасно бути присутніми на українському ринку.
359. Така ситуація навряд чи прийнятна з точки зору українських регуляторних органів, які повинні мати змогу забезпечити дотримання єдиного наукового підходу, поєднаного з підходом, що застосовується EMA і агентствами країн ЄС. Питання можуть виникнути, наприклад, у випадку перереєстрації лікарських засобів, які не мають еквівалентів в ЄС або реєстрація яких в ЄС має різні строки. Який підхід варто застосовувати для оцінки балансу ризику/користі – підхід іноземного агентства країни, що не входить до ЄС, та у якій було зареєстровано лікарський засіб, чи підхід ЕМА (або іншого національного агентства країни-члена ЄС), яке не приймає показники безпеки лікарського засобу? На нашу думку, чіткий вибір повинен бути зроблений на користь підходу ЕМА. Було б доцільно вказати, що науковий підхід ЕМА є основним для органу з реєстрації ЛЗ в Україні в разі суперечливих наукових підходів серед різних агентств.
Рекомендація:
Реформувати «спрощену» процедуру реєстрації ЛЗ, зареєстрованих в країнах, які не є членами ЄС, з метою її приведення у відповідність з європейським підходом до безпеки лікарських засобів.
На наступному етапі – застосовувати спрощену процедуру для продуктів, зареєстрованих у країнах ЄС.
ІV.4. СПІВРОБІТНИЦТВО МІЖ УКРАЇНСЬКИМИ ОРГАНАМИ У СФЕРІ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ ТА ЕМА
360. Зобов’язання України у сфері наближення її системи реєстрації лікарських засобів до стандартів ЄС повинні бути підтримані відповідним рівнем співпраці з боку установ ЄС, серед них, зокрема, ЕМА.
361. В Угоді про асоціацію сторони зобов’язалися розвивати співробітництво у сфері охорони здоров’я (стаття 426), зокрема, в області зміцнення системи охорони здоров’я і її потенціалу в Україні, в тому числі шляхом здійснення реформ, подальшого розвитку первинної медико-санітарної допомоги, а також навчання персоналу (стаття 427 пункт 1 підпункт (а)). З цією метою сторони домовилися обмінюватися даними і передовими практиками, а також здійснювати інші спільні заходи, в тому числі шляхом поступової інтеграції України в Європейські мережі в області суспільної охорони здоров’я (пункт 2 статті 427).
362. Наскільки нам відомо, наразі не існує платформи для систематичного обміну інформацією та знаннями між ЕМА та органами з реєстрації лікарських засобів в Україні.
Рекомендація
Ініціювати створення робочої групи або підкомітету з лікарських засобів на основі Угоди про асоціацію, включити до неї представників українських органів реєстрації лікарських засобів і ЕМА.
363. Ми вважаємо, що створення можливості для офіційного діалогу у сфері регулювання може сприяти налагодженню контактів між фахівцями з обох сторін.
364. Залежно від політичної волі обох сторін, сфера співпраці може охоплювати:
- підтримку в імплементації нового законодавства у сфері реєстрації лікарських засобів для забезпечення його повної відповідності законодавству ЄС;
- тренінги для українських фахівців і посадових осіб для більш повного розуміння особливостей застосування нормативно-правової бази ЄС на практиці;
- створення систем зв’язку та обміну інформацією;
- участь представників українських реєстраційних органів у науково-технічних нарадах ЕМА в якості спостерігачів.
V. СПОСТЕРЕЖЕННЯ В ОБЛАСТІ ЗАКОНОДАВСТВА
365. У розділі ІІІ ми оцінили українське законодавство з точки зору його відповідності законодавству і стандартам ЄС. У цьому розділі ми хотіли б зосередити увагу на проблемах українського законодавства, що не є прямо пов’язаними з законодавством ЄС, але мають важливе значення для підвищення ефективності процедури реєстрації лікарських засобів.
V.I. ЗАГАЛЬНІ ВІДОМОСТІ
366. Методи регулювання, в тому числі методи законодавчої діяльності, не підлягають узгодженню в ЄС і залишаються виключною компетенцію держав-членів ЄС. Останні застосовують різні підходи побудови законодавства, в залежності від власних законодавчих традицій і конституційних принципів. З цієї причини наші подальші зауваження, що стосуються методів регулювання, що застосовуються в Україні, не засновані на будь-якій єдиній європейській моделі регулювання, а скоріше орієнтовані на універсальні вимоги ефективності та прозорості.
367. Українське законодавство у сфері державної реєстрації лікарських засобів складається з низки нормативних актів, прийнятих органами державної влади різних рівнів. Основними нормативними актами, що регулюють сферу державної реєстрації лікарських засобів в Україні, є Закон про лікарські засоби, Постанова КМУ № 376 і Наказ МОЗ № 426. Аналіз цих актів, а також останні зміни до них, свідчать про низку недоліків нинішньої моделі регулювання.
368. З теоретичної точки зору українське законодавство у сфері реєстрації організоване за ієрархічним принципом, де Закон про лікарські засоби є актом вищої юридичної сили, а Постанова КМУ № 376 та Наказ МОЗ № 426 регулюють деталі процедури реєстрації. На практиці ж регулювання процедури реєстрації на різних рівнях та ступінь деталізації цих актів видаються непослідовними.
369. Ідея регулювання процедури реєстрації як на законодавчому рівні, так і на рівні підзаконних нормативних актів є загалом зрозумілою. Такий метод законодавчої діяльності може бути ефективним за умови, що розроблені чіткі правила щодо співвідношення між нормативними актами різної юридичної сили. В українському законодавстві у сфері державної реєстрації лікарських засобів немає таких чітких правил. Український законодавець надає можливість впроваджувати підзаконними актами нові елементи процедури та навіть нові типи процедури, що не передбачені законом. Як результат, в Україні діє система, яка зазнає постійних змін, що не зрозумілі ані посадовим особам, ані представникам індустрії. Така ситуація може призвести до проблем з інтерпретацією окремих положень цих актів.
370. Найбільш очевидні приклади такого неефективного регулювання представлені нижче.
VI. НЕЗРОЗУМІЛЕ СПІВВІДНОШЕННЯ МІЖ ЗАКОНОМ ПРО ЛІКАРСЬКІ ЗАСОБИ, ПОСТАНОВОЮ КМУ № 376 І НАКАЗОМ МОЗ № 426
371. Закон про лікарські засоби як нормативний акт, що має основоположне значення для системи реєстрації лікарських засобів, повинен описувати всі ключові елементисистеми. На практиці це не завжди так. Закон описує систему державної реєстрації занадто загально і залишає завдання формулювання процедури органам виконавчої влади. Формально, установою, що має повноваження визначати порядок державної реєстрації, є КМУ; на практиці ж КМУ делегував значну частину регулювання МОЗ без будь-яких особливих обмежень щодо кінцевої структури системи.
Закон України про лікарські засоби, стаття 9 частина 26
Порядок державної реєстрації (або перереєстрації) лікарського засобу і суми зборів за державну реєстрацію (або перереєстрацію) лікарського засобу, встановлюється КМУ.
Постанова КМУ № 376, Розділ 2
Державна реєстрація лікарського засобу здійснюється МОЗ на підставі заяви та результатів експертизи реєстраційних матеріалів на такий засіб, проведеної ДЕЦ у визначеному МОЗ порядку.
372. Такий спосіб регулювання викликає певні сумніви. У поточній моделі рішення про фактичну форму процедури приймають органи виконавчої, а не законодавчої влади. Це також означає, що на практиці імплементація законодавства ЄС в Україні знаходиться в руках МОЗ та КМУ, а не Верховної Ради України. В нинішній законотворчій моделі система державної реєстрації може бути радикально змінена за одну ніч без внесення змін до жодної норми в Законі про лікарські засоби та без розгляду цих змін у парламенті.
373. Приклади, наведені нижче, яскраво ілюструють цю проблему.
374. Закон про лікарські засоби не згадує ДЕЦ в якості установи, відповідальної за експертизу. Насправді в Законі про лікарські засоби немає посилання на експертизу як на окремий етап процедури державної реєстрації, і немає жодних часових обмежень для цього етапу процедури. З точки зору Закону, процедура в цьому відношенні може бути сформована КМУ на власний розсуд10, без жодних обмежень. З точки зору Закону про лікарські засоби, КМУ також може замінити вимоги до процедури експертизи в ДЕЦ іншими вимогами, може продовжити граничні строки експертизи без жодних обмежень і т.д.
375. Деякі питання, що мають основоположне значення для функціонування всієї системи державної реєстрації, не регулюються Законом; замість цього вони регулюються актами нижчої юридичної сили. Наприклад, Закон не визначає строку дії реєстраційного посвідчення, не визначає основних положень щодо здійснення перереєстрації та внесення змін у процесі державної реєстрації. Ці процедури визначені тільки у Постанові КМУ № 376 і деталізовані у Наказі МОЗ № 426.
_________________
10 Із незначними виключеннями – після нещодавніх змін, Закон визначає максимальні строки для експертизи в спрощеному порядку.
376. Іншим прикладом є реалізація виключення Болар, що дозволяє проводити дослідження без порушення патентних прав. В українському законодавстві виключення Болар було реалізовано не в Законі про лікарські засоби, а у Наказі МОЗ № 426, в якості елементу визначення «незаконного використання реєстраційної інформації про безпеку та ефективність лікарського засобу».
377. Державна реєстрація «під умовами» є ще одним прикладом. Ані Закон, ані Постанова КМУ № 376 не передбачають можливість такої реєстрації – вона ґрунтується виключно на Наказі МОЗ № 426.
378. Підстави для відмови у державній реєстрації визначаються по-різному у різних нормативних актах. Згідно з Законом про лікарські засоби у реєстрації має бути відмовлено, якщо безпечність або ефективність лікарського засобу не підтверджена. У Постанові КМУ № 376 також згадується про якість лікарського засобу як підставу для відмови у реєстрації. Наказ МОЗ № 426 розширює можливі підстави для відмови (містить підстави для надання негативних рекомендацій ДЕЦ щодо державної реєстрації ЛЗ) також за недотримання вимог до документації згідно з вимогами законодавства та порушення режиму ексклюзивності даних.
379. Внутрішні правила (СОПи ДЕЦ, Інструкція ДЕЦ) можуть виходити далеко за межі простої імплементації положень актів вищої юридичної сили, шляхом введення нових елементів до процедури.
Приклад для порівняння (ЄС)
Директива 2001/83 регулює всі важливі аспекти процедури, включаючи визначення, сферу регулювання, елементи заяви, підстави для відхилення зави і строки.
Докладні вимоги, що стосуються складу досьє, регулюються в додатку, який є невід’ємною частиною Директиви 2001/83.
Приклад для порівняння (Польща)
Польський фармацевтичний закон відповідає структурі Директиви 2001/83, і додатково регулює національну процедуру реєстрації більш детально.
Всі ключові елементи регулювання присутні в самому законі. Тільки технічні питання, які не стосуються суті регулювання (наприклад, питання детальної процедури, збори, технічні вимоги), що делеговані відповідним міністрам (головним чином міністру охорони здоров’я) повинні регулюватися наказами (так званими «правилами»). У кожному випадку, коли Польський фармацевтичний закон делегує законодавчі повноваження міністру, він чітко визначає сферу делегованих повноважень і керівні принципи щодо змісту регулювання.
V.3. ПОВТОРЮВАНІ ПОЛОЖЕННЯ
380. Предмет регулювання актів законодавства різної юридичної сили частково співпадає та в значній мірі дублюється.
381. Наприклад, визначення термінів має важливе значення для всієї системи державної реєстрації, і в принципі визначення повинні міститися у акті вищої юридичної сили. Наприклад, Директива 2001/83 містить більше 40 визначень в основному тексті та у Додатку. Польський фармацевтичний закон містить близько 80 визначень. Закон про лікарські засоби містить 19 визначень (деякі з них стосуються інших сфер регулювання). Постанова КМУ № 376 взагалі не містить визначень, але використовує поняття, не визначені ані Законом, ані Постановою КМУ № 376 (наприклад, «генеричний лікарський засіб»). Переважна частина визначень міститься у Наказі МОЗ № 426 (близько 60 визначень). Деякі з визначень, передбачених Наказом МОЗ № 426 повторюють визначення з Закону, а деякі додають нові елементи до вже визначених в Законі понять.
382. Так, наприклад, Закон містить дуже широке визначення поняття «лікарський засіб», що підлягає державній реєстрації. Винятки до цього визначення (наприклад, стосовно АФІ, що використовується для отримання готового лікарського засобу, щодо деяких продуктів, що базуються на людській крові та плазмі), містяться у Наказі МОЗ № 426.
383. Ще одним прикладом повторюваного і непослідовного регулювання є вимоги до заяви про реєстрацію ЛЗ. Деякі вимоги до зави визначаються актами всіх рівнів – Законом, Постановою КМУ № 376 та Наказом МОЗ № 426. Деякі з вимог дублюються у всіх актах, а деякі додаються одним із них. На практиці застосовуються вимоги, що містяться у Наказі МОЗ № 426 та додатках до нього. Видається, що чим більшу юридичну силу має нормативний акт, тим менше вимог до заяви він містить, що на практиці означає, що фактична форма процедури і обсяг вимог, що пред’являються до заявників, повністю залежать від органів виконавчої влади (КМУ і, зокрема, МОЗ).
V.4. СУПЕРЕЧЛИВЕ РЕГУЛЮВАННЯ СПРОЩЕНИХ ПРОЦЕДУР
384. Закон, Постанова КМУ № 376 і Наказ МОЗ № 426 визначають різні типи спрощених (прискорених) процедур. Наприклад, відповідно до Закону (після останніх змін) спрощена процедура стосується всіх ЛЗ, зареєстрованих компетентними органами США, Швейцарії, Японії, Австралії, Канади і EMA за централізованою процедурою. Видається, що спрощена процедура щодо вищезазначених ЛЗ, наявність якої передбачена відповідно до останніх змін до Закону та Постанови КМУ № 376, потребує розробки нового наказу МОЗ, що деталізував би цю процедуру. Проект відповідного наказу МОЗ наразі перебуває на стадії публічного обговорення та не набув чинності на дату підготовки Звіту. У відповідності до поточної версії Наказу МОЗ № 426 (V.10.1) застосування спрощеної процедури обмежене оригінальними (інноваційними) лікарськими засобами (молекула не представлена на ринку України) для лікування соціально небезпечних хвороб (туберкульоз, ВІЛ/СНІД, вірусні гепатити), а також лікарськими засобами з оригінальною молекулою для лікування орфанних та онкологічних захворювань, що були зареєстровані в країнах, регуляторні органи яких застосовують високі стандарти якості, або прекваліфіковані ВООЗ, зокрема: FDA (США), EMA (за централізованою процедурою), Swissmedic (Швейцарія), PMDA (Японія), MHRA (Великобританія), TGA (Австралія).
385. Існують також істотні відмінності між вищезгаданими актами щодо сфери застосування спрощених процедур. Постанова КМУ № 376 і Наказ МОЗ № 426 (проте не Закон про лікарські засоби) розрізняють тип спрощеної процедури, що стосується виключно лікарських засобів, зареєстрованих EMA за централізованою процедурою. На ту ж категорію лікарських засобів поширюється інший тип спрощеної процедури, що стосується лікарських засобів, що дозволені окремими іноземними агентствами (в тому числі, але не обмежуючись, EMA), проект наказу МОЗ щодо якого перебуває на стадії публічного обговорення.
386. Різними нормативними актами також встановлені відмінні строки експертизи у разі застосування спрощеної процедури. Законом (після внесення останніх змін) було введено строк 7 робочих днів для проведення експертизи у ДЕЦ. У відповідності до Постанови КМУ № 376 такий строк становить 20 робочих днів.
V.5. НЕДОСТАТНІЙ РІВЕНЬ КООРДИНАЦІЇ ПРИ ВНЕСЕННІ ЗМІН ДО РІЗНИХ НОРМАТИВНИХ АКТІВ
387. Беручи до уваги підходи до регулювання процедури державної реєстрації, прийняті в Україні (декілька нормативних актів різної юридичної сили, що регулюють один і той самий предмет), будь-які реформи системи повинні бути скоординовані таким чином, щоб охопити всі нормативні акти у сфері реєстрації лікарських засобів. Виходячи з нашого аналізу українського законодавства видається, що це не завжди так.
388. Найбільш релевантний приклад стосується нещодавніх змін у Закон (Закон України «Про внесення змін до статті 9 Закону України «Про лікарські засоби» щодо спрощення державної реєстрації лікарських засобів»). Зміни розширили сферу застосування спрощеної процедури реєстрації, змінили вимоги до заяви та скоротили строки реєстрації. Ці зміни набули чинності у червні 2016 року. Однак на практиці Вони не застосовуються у зв’язку з тим, що до новий наказ, що деталізував би відповідні спрощені процедури, до цих пір перебуває на стадії публічного обговорення.
389. Такий метод регулювання створює плутанину для заявників, а також співробітників установ, що беруть участь у процедурі реєстрації.
V.6. МОНІТОРИНГ ЗМІН У ЗАКОНОДАВСТВІ ЄС
390. Виходячи з результатів нашого аналізу, представленого в розділі ІІІ вище, українське законодавство вже досягло значного рівня гармонізації з законодавством ЄС у сфері реєстрації лікарських засобів. Тим не менш, в ході нашого аналізу ми виявили, що деякі положення українського законодавства, які вже гармонізовані з законодавством ЄС, наразі не приведені у відповідність з останніми змінами у законодавстві ЄС. Наприклад, український законодавець не в повній мірі взяв до уваги реформу законодавства з фармаконагляду в ЄС, що відбулася в липні 2012 року (Регламент 1235/2010 та Директива 2010/84), або нові вимоги, введені Директивою 2011/62 (вступила в силу в січні 2013 року). ЗТД формат реєстраційного досьє, що є обов’язковим в Україні, також не в повній мірі відповідає формату, що застосовуються в ЄС.
391. Для досягнення відповідності стандартам ЄС важливо також стежити за змінами до норм, що не носять обов’язкового характеру, особливо до Пояснювальної записки для заявників та інших настанов EMA або Європейської Комісії. Це важливо також у зв’язку з тим, що Україна імплементувала деякі положення законодавства ЄС, що не носять обов’язкового характеру, до Наказу МОЗ № 426 (наприклад, додатки до Наказу МОЗ № 426 частково ґрунтуються на Пояснювальній записці для заявників, яка є настановою, що була опублікована Європейською Комісією).
392. Крім того, ряд положень українського законодавства прямо посилаються на нормативні акти ЄС, зазначаючи їх точний номер та версію (наприклад, у Наказі МОЗ № 426 у визначенні генеричного лікарського засобу є посилання на документ ЕМА CPMP/QWP/EWP/1401/98 Rev.1). Якщо документ EMA буде оновлено, то це положення Наказу може також потребувати оновлення.
393. Через вказані вище причини, рекомендується запровадити певні форми інституційного моніторингу змін до законодавства ЄС (нормативних актів, настанов, прецедентної практики), для того, щоб в майбутньому забезпечити збереження відповідності українського законодавства до стандартів та нормативних актів ЄС.
V.7. РЕКОМЕНДАЦІЇ
Всі основні елементи процедури державної реєстрації повинні регулюватися виключно Законом України про лікарські засоби, в тому числі:
- визначення термінів,
- види ЛЗ, що підлягають державній реєстрації,
- установи, які беруть участь у процедурі,
- види процедур реєстрації,
- підстави для відмови або обмеження державної реєстрації,
- строк дії державної реєстрації,
- підстави для перереєстрації та відмови в перереєстрації,
- види процедур внесення змін.
Закон повинен чітко визначити сферу регуляторних повноважень, делегованих КМУ та/або МОЗ.
Законодавство має бути переглянуте з метою усунення повторень і суперечностей.
[Опціонально] Усі основі елементи процедури державної реєстрації повинні бути визначенні у новій Постанові Кабінету Міністрів України або шляхом внесення відповідних змін до Постанови КМУ № 376
Необхідно запровадити систему моніторингу і контролю відповідності законодавства України законодавству ЄС, наприклад, шляхом створення спеціального підрозділу в структурі МОЗ.
VI. ОРГАНІЗАЦІЯ ПРОЦЕСІВ
VI.1. ВСТУПНІ ЗАУВАЖЕННЯ
394. Ми розглянули процедуру державної реєстрації, через призму правових актів, внутрішніх документів відповідних установ, огляд темплейтів, інформації, отриманої в ході зустрічей з представниками ДЕЦ і представниками індустрії, а також опитувань регуляторних представників фармацевтичних компаній.
395. В ході нашого дослідження різні зацікавлені сторони висловили сподівання щодо прискорення процедури реєстрації, підвищення її ефективності та прозорості.
396. На підставі результатів проведеного аналізу ми проаналізували хід процедури, яка представлена в розділі VІ.2 нижче.
397. Ми зробили декілька застережень з приводу можливих причин недостатньої ефективності та прозорості процесу. Наші спостереження з цього приводу представлені у розділах VІ.3, VI. 4 та VІ.5 нижче.
398. Крім того, у розділі VІ.6 нижче, наведено ряд рекомендацій, які можуть бути застосовані для вирішення деяких з вищезазначених проблем. У окремій секції представлені питання, пов’язані з організацією процесів, пов’язаних з державною реєстрацією лікарських засобів, у рамках Держлікслужби.
399. У зв’язку з тим, що ми не отримали приклади документів (реєстраційних справ) стосовно процедур перереєстрації та внесення змін, представлена схема процедури і наші висновки сфокусовані в основному саме на державній реєстрації, проте ми також додатково наводимо ряд рекомендацій та спостережень щодо процедури перереєстрації та внесення змін.
VI.2. СХЕМА ПРОЦЕДУРИ
400. Просимо звернути увагу, що представлена схема процедури була побудована частково на основі розгляду наданих реєстраційних справ за процедурами, що були розпочаті ще до набрання чинності останніх змін до Наказу МОЗ № 426, і за СОПами ДЕЦ, які, очевидно, ще не були оновлені з урахуванням останніх змін до Наказу МОЗ № 426. В результаті, опис деяких етапів може не повністю відображати поточну практику ДЕЦ. Відповідні застереження, де це необхідно, зроблені в тексті.
(а) Заявник подає заяву до МОЗ
401. Заява у вигляді короткої форми, що містить основні адміністративні дані про заявника, продукт і бажаний тип реєстрації (повне досьє, генеричний, біоеквівалентний т.д.)
Попередня процедура
Дії, перераховані в пунктах (b) – (е) більше не згадуються в поточній версії Наказу № 426. Тим не менш, вони все ще присутні у внутрішніх документах ДЕЦ, які ми розглянули під час зустрічі в ДЕЦ. Ми припускаємо, що у зв’язку з останніми змінами в Наказ МОЗ № 426 ці кроки вже не є релевантними.
(b) Міністерство охорони здоров’я направляє справу в ДЕЦ для первинної експертизи
Після отримання заяви Міністерство охорони здоров’я просить ДЕЦ виконати первинну експертизу заяви.
(с) Заявник подає реєстраційну форму з додатками для первинної перевірки
Після передачі справи МОЗ України в ДЕЦ, заявник зобов’язаний подати форму реєстрації безпосередньо в ДЕЦ.
Реєстраційна форма ґрунтується на основі формату ЗТД (ЗТД 1.2). Це більш детальний документ, ніж додаток, супроводжується декількома додатками, але ще не реєстраційним досьє.
(d) ДЕЦ виконує первинну експертизу заяви
Питання, які підлягають перевірці на рівні первинної експертизи включають в себе наступне:
- Чи була реєстраційна форма належним чином заповнена і чи були додані всі документи, необхідні на даному етапі;
- Чи не містять лікарські засоби заборонених речовин (аналіз зроблений на основі опису складу, не передбачено ніяких випробувань),
- чи відповідає продукт відповідному типу реєстрації (наприклад, загальному),
- чи є допустимою запропонована назва лікарського засобу.
ДЕЦ вже на даному етапі могла зв’язуватися безпосередньо з заявником, і попросити надати додаткові матеріали або пояснення.
Після попередньої експертизи ДЕЦ передає свій висновок (у формі «обґрунтованої думки щодо попередньої експертизи») в МОЗ.
(е) Кваліфікаційна комісія Міністерства охорони здоров’я надає свій висновок
Кваліфікаційна комісія, консультативний орган Міністерства охорони здоров’я, дає висновок про відповідність препарату до пропонованого типу заяви.(Ми не бачили якихось документів, що регламентують структуру та діяльність Кваліфікаційної комісії.)
Кваліфікаційна комісія також надає свою думку в разі відмови від розгляду заяви на початковому етапі експертизи.
(b) Міністерство охорони здоров’я передає справу в ДЕЦ для проведення експертизи
(с) Заявник підписує контракт з ДЕЦ, оплачує реєстраційний внесок та вартість експертизи і подає реєстраційне досьє
402. ДЕЦ готує контракт, що повинен бути підписаний заявником і рахунки за послуги, пов’язані з експертизою в розмірі, який встановлений для обраного типу заяви.
403. Заявник також зобов’язаний сплатити збір за державну реєстрацію.
404. Після оплати рахунку і збору заявник зобов’язаний надати ДЕЦ реєстраційні матеріали (реєстраційне досьє) на паперовому носії. За умови попередньої домовленості з ДЕЦ частину реєстраційних матеріалів можна надати також в електронному вигляді.
(d) Попередня експертиза
405. Метою попередньої експертизи є перевірка факту надання заявником усіх необхідних матеріалів для певного типу заяви (перевірка на повноту).
406. Попередня експертиза проводиться внутрішніми експертами ДЕЦ.
407. Попередня експертиза полягає в наданні експертом висновку щодо повноти документів. ДЕЦ інформує заявника про завершення і результати попередньої експертизи.
408. Якщо реєстраційні матеріали є неповними, ДЕЦ надсилає заявнику лист щодо необхідності подачі відсутніх матеріалів. Кожен запит про надання матеріалів повинен бути обґрунтованим з посиланням на відповідне положення процедури. ДЕЦ може надсилати тільки один запит щодо надання додаткових матеріалів.
409. Як правило, всі документи, передбачені Наказом МОЗ № 426 і відповідними додатками для даного типу заяви, повинні бути представлені на етапі попередньої експертизи. Як виняток, документ, що підтверджує відповідність умов виробництва вимогам GМР, що пов’язаний з лікарським засобом, може бути наданий на більш пізньому етапі процедури експертизи.
410. Попередня експертиза повинна бути виконана протягом 14 робочих днів з дня отримання реєстраційних матеріалів. Термін припиняється на час, необхідний для заявника, щоб подати додаткові реєстраційні матеріали.
411. Якщо результати експертизи є негативними на даному етапі, ДЕЦ відмовляє в передачі матеріалів для проведення спеціалізованої експертизи. Заявник може оскаржити таку відмову.
(е) Спеціалізована експертиза
412. Після позитивного результату попередньої експертизи, Директор ДЕЦ і начальник департаменту експертизи реєстраційних матеріалів надсилають супровідний лист разом з відповідними частинами досьє, експертам для проведення експертизи відповідних частин реєстраційного досьє та для складання висновків про безпеку, якість та ефективність лікарського засобу.
413. Реєстраційні матеріали можуть бути передані наступним підрозділам:
- КЕГ, орган, що складається з зовнішніх фахівців різних спеціальностей, відповідальних за розгляд доклінічної і клінічної документації і робить висновки про безпечність та ефективність;
- Управління експертизи інструкцій та номенклатури ДЕЦ;
- Департамент фармацевтичної діяльності ДЕЦ;
- Управління правового та інформаційно-аналітичного забезпечення ДЕЦ;
- Управління експертизи матеріалів з біоеквівалентності ДЕЦ.
414. Експерти не зв’язуються безпосередньо з заявником. Вони можуть задавати питання заявнику через ДЕЦ. Кожен експертний орган (комісія) має право двічі витребувати додаткову інформацію, що стосуються безпеки, ефективності або якості під час спеціалізованої експертизи. Експертам не дозволяється ставити запитання, що стосуються матеріалів, які вони вже переглянули.
415. Можна запросити представника заявника на нараду в ДЕЦ, щоб він міг відповісти на зауваження експертів або надати додаткові пояснення.
416. Якщо заявник не може надати запитувані документи або пояснення протягом встановленого періоду часу, ДЕЦ може рекомендувати МОЗ зняти заяву з розгляду на даному етапі. З цією метою справа має бути передана на засідання одного з консультативних органів – НЕР або НТР.
417. Після проведення експертизи кожна експертна комісія надає свій експертний висновок, що містить вмотивовані висновки про безпеку, ефективність та якість лікарського засобу.
(f) Лабораторні дослідження
418. На будь-якому етапі процедури державної реєстрації ДЕЦ може направити ЛЗ в лабораторію, акредитовану для досліджень.
419. Критерії для відправки продукту на лабораторні дослідження були встановлені в додатку 13 до Наказу МОЗ № 426.
420. Час очікування результатів від лабораторії не включається в загальну тривалість процедури.
(g) Підтвердження належних умов виробництва
421. Обов’язковим елементом кожної процедури державної реєстрації є підтвердження належних умов виробництва, пов’язаних із зазначеним лікарським засобом.
422. Для лікарських засобів, що виробляються в Україні достатньо мати діючу ліцензію на виробництво, видану Держлікслужбою. Для лікарських засобів, виготовлених за кордоном, відповідність умов виробництва з українським GMP повинна бути доведена. В останньому випадку Держлікслужба може покладатися на GMP сертифікати, видані членами PIC/S, проте може проводити інспектування виробничої дільниці з метою видачі GMP сертифікату.
(h) Рекомендація від консультативних органів, що функціонують в рамках ДЕЦ, до ДЕЦ
423. Результати експертизи окремих частин досьє повинні бути формалізовані і у подальшому направлятися для розгляду на засіданні відповідного консультативного органу (НЕР або НТР). У засіданні беруть участь керівники конкретної експертної ради.
424. Дорадчий орган приймає рекомендації для генерального директора ДЕЦ про державну реєстрацію, а також затвердження інструкції для медичного застосування, методи контролю якості та технології виробництва. (Насправді в Інструкції ДЕЦ НЕР/НТР зазначаються як орган, що приймає «рішення» щодо рекомендації ДЕЦ МОЗ).
425. НЕР скликається один раз на місяць. її роль полягає у вивченні та затвердженні результатів експертизи. НЕР дає рекомендації щодо:
- державної реєстрації лікарських засобів (або щодо відмови у державній реєстрації),
- затвердження змін, які потребують нової реєстрації лікарського засобу,
- припинення дії реєстраційного посвідчення.
426. НЕР може делегувати частину своїх функцій НТР.
427. НТР розглядає результати експертизи і висновки експертів для надання рекомендацій щодо:
- перереєстрації лікарських засобів;
- реєстрації (перереєстрації) АФІ;
- внесення змін до реєстраційних матеріалів.
428. НТР скликається двічі на місяць.
429. У процедурі внесення змін до реєстраційних матеріалів може взяти участь інший консультативний орган – ТЕК. TEK скликається щотижня. її обов’язок полягає у:
- розгляді скарг заявників щодо результатів експертизи;
- вивченні можливості відновлення експертизи відхиленої заяви після надання додаткових документів заявником;
- перевірці заяви на предмет незначних змін до реєстраційних матеріалів.
(і) Узгодження ДЕЦ проектів документів із Заявником
430. ДЕЦ повинен підготувати і передати заявнику пакет проектів документів для перегляду і редагування (за необхідності). Цей пакет включає в себе:
- проект реєстраційного посвідчення,
- проект інструкції для медичного застосування, МКЯ, маркування.
(j) Директор ДЕЦ підписує загальні висновки і рекомендації
431. Відповідно до рекомендацій консультативного органу, Директор ДЕЦ підписує загальний висновок щодо ефективності, безпеки та якості лікарського засобу, а також рекомендації МОЗ щодо реєстрації лікарського засобу.
(k) ДЕЦ складає перелік лікарських засобів, запропонованих на реєстрацію
432. Відповідно до висновків та рекомендацій ДЕЦ щодо реєстрації ЛЗ, ДЕЦ складає для МОЗ перелік лікарських засобів, запропонованих до реєстрації (що становлять частину наказу, який повинен бути складений МОЗ для реєстрації таких лікарських засобів).
433. Якщо ДЕЦ надасть негативну рекомендацію, інформація про це має бути вказана в окремому списку, що також додається до наказу МОЗ.
(l) ДЕЦ передає пакет документів в МОЗ
434. Протягом 5 робочих днів після узгодження документів із Заявником, ДЕЦ надає до МОЗ наступні документи:
- Перелік ЛЗ, рекомендованих до реєстрації і (за необхідності) рекомендованих до відмови в реєстрації;
- Обґрунтовані висновки щодо можливості реєстрації ЛЗ (або відмови в реєстрації);
- Проект реєстраційного посвідчення та екземпляри: інструкції для медичного застосування, SmPC (за необхідності), МКЯ, маркування, зовнішньої упаковки.
435. В той же час ДЕЦ інформує заявника про передачу документів до МОЗ.
(m) МОЗ складає проект наказу і передає його до ДЕЦ
436. Протягом 3 робочих днів МОЗ складає (затверджує) проект наказу і надає копію наказу в ДЕЦ протягом наступних 3 днів.
437. Дата підписання наказу є датою початку дії реєстраційного посвідчення.
438. Рішення про відмову в реєстрації лікарського засобу заявник може оскаржити в судовому порядку.
(n) ДЕЦ вносить інформацію про державну реєстрацію до ДРЛЗ (протягом 6 днів після отримання наказу від МОЗ).
(o) ДЕЦ повідомляє заявника про державну реєстрацію, готує для МОЗ проект реєстраційного посвідчення і передає його в МОЗ (протягом 3 робочих днів після отримання наказу від МОЗ).
(p) МОЗ підписує реєстраційне посвідчення і передає його назад до ДЕЦ (3 робочі дні)
(q) ДЕЦ готує пакет документів, які потрібно надати заявнику через «єдине вікно» і передає його до МОЗ
439. Документи для Заявника включають:
- Реєстраційне посвідчення,
- Затверджену інструкцію для медичного застосування, SmPC (за необхідності), МКЯ, маркування, макет упаковки.
(r) МОЗ передає документи заявнику через «єдине вікно»
440. Документи можна буди надані заявнику, лише після пред’явлення останнім:
- Довіреності на отримання документів,
- Заяви про відсутність порушення прав третіх осіб і Заяви про виконання умов затверджених документів,
- Підписаного Акту прийому-передачі, в якому заявник підтверджує належне виконання послуг з боку ДЕЦ відповідно до умов договору і відмовляється від претензій щодо роботи ДЕЦ.
VI.3. ВИЯВЛЕНІ ПРОБЛЕМИ ЩОДО ТРИВАЛОСТІ ПРОЦЕДУРИ
VI.3.1. Регламентування кінцевих строків (строків виконання)
441. Затримки під час процедури державної реєстрації становлять предмет занепокоєння для більшості представників індустрії, з якими були проведені співбесіди.
442. Відповідно до статистичних даних, у період з 1 січня 2013 р. до 29 жовтня 2015 р. (тобто, до внесення останніх змін до процедури):
- було подано 2727 заяв на реєстрацію, перереєстрацію та внесення змін;
- було відхилено 713 заяв,
- відповідно до наказів МОЗ було зареєстровано 1607 заяв,
- станом на 22 липня 2016 р. е 619 заяв досі перебувають на стадії розгляду (спеціалізована експертиза) (після 210 -ти робочих днів, що є максимальним строком проведення експертизи встановленим у Наказі МОЗ № 426).
443. З огляду на статистичні дані можна спостерігати критичну ситуацію з реєстрацією біосимілярів. Так, до ДЕЦ було подано 29 заяв, з них 12 заяв було відхилено, 10 і досі перебувають на етапі розгляду і лише 7 були успішно розглянуті ДЕЦ.
444. Більшість заяв було подано на реєстрацію генеричних лікарських засобів та АФІ.
445. У період з 30 жовтня 2015 р. (дата вступу в силу Наказу МОЗ № 460) до 22 липня 2016 р. 614 заяв щодо реєстрації генериків та АФІ було подано на реєстрацію, перереєстрацію та внесення змін.
- 39 заяв вже відхилено, 31 заява була успішно розглянута ДЕЦ.
- 544 заяв досі перебувають на етапі експертизи.
446. Аналіз законодавства та внутрішніх процедур показує, що багато уваги приділяється встановленню строків для конкретних процедур та певних дій в рамках процесу. Сам факт приділення значної уваги до встановлення граничних строків має позитивне значення, оскільки це слугує підтвердженням наміру відповідних регуляторних органів уникати затримок у процесі реєстрації. Однак, спосіб, яким визначаються такі строки, інколи є абсолютно неефективним.
447. По перше, строки часто визначаються у спосіб, що по своїй суті не є логічним та обґрунтованим.
448. У розділі VI.3, що присвячений спостереженням щодо строків процедури реєстрації, ми вже відзначали відсутність єдиного підходу до використання «робочих днів» або «календарних днів». Результати інколи дивують, наприклад, ДЕЦ має набагато більше часу для експертизи стандартної заяви на внесення змін до реєстраційного досьє (60 календарних днів), ніж для теоретично термінової процедури для внесення змін до реєстраційного досьє вакцин (45 робочих днів).
449. Іншим прикладом є непропорційні строки, що передбачені в СОПах ДЕЦ. Наприклад, Інструкцією ДЕЦ встановлено жорсткі часові обмеження у 30 хвилин для підготовки контракту для підписання, потім надається довший період в 4 дні для ДЕЦ для перевірки підписаного контракту і надання своїх коментарів до нього.
450. Кілька часових обмежень в СОПах встановлені без визначення початкового моменту їх відліку.
451. По-друге, відсутній загальний граничний строк процедури державної реєстрації. Ми вже описували наші спостереження з цього приводу у розділі ІІІ щодо відповідності українського законодавства до законодавства ЄС. Так, нами було зазначено, що строки, передбачені Директивою 2001/83 стосуються всієї процедури реєстрації, а не лише її окремої частини. Слід наголосити, що відсутність чітко визначеного загального строку процедури реєстрації є не тільки питанням відповідності законодавству ЄС, а й питанням ефективності моделі державної реєстрації лікарських засобів.
452. Наразі для кожного органу, що бере участь у процесі реєстрації, передбачені свої власні строки. МОЗ зобов’язаний прийняти рішення щодо державної реєстрації протягом одного місяця (нещодавно строк було скорочено до 10 днів), проте перед цим ДЕЦ має провести експертизу. ДЕЦ має провести експертизу в строки, які коливаються від 20 до 210 днів, при цьому ДЕЦ повністю незалежний від строків встановлених для МОЗ (експертиза не враховується у граничному при обрахуванні граничного строку для МОЗ). Крім того, якщо ДЕЦ надсилає зразок лікарського засобу для лабораторних випробувань, час на проведення таких випробувань не враховується у загальний строк проведення експертизи в ДЕЦ. Держлікслужба також має свої строки для проведення заходів для підтвердження відповідності.
454. По-третє, в українському законодавстві немає чіткої вказівки щодо того, який орган є відповідальним за дотримання загальних строків процедури. Усі задіяні у процедурі реєстрації регуляторні органи відповідають лише за дотриманням власних строків. Жоден з них не зацікавлений у дотриманні та не відповідає за загальний строк процедури реєстрації.
VІ.3.2. Відсутність ефективних механізмів для притягнення до відповідальності за затримки під час процедури реєстрації
454. Наш аналіз показує, що не зважаючи на загальну тенденцію щодо скорочення строків та встановлення окремих часових обмежень щодо кожного етапу/дії в рамках процедури реєстрації, не було вжито жодних заходів для забезпечення дотримання цих строків.
455. З точки зору заявника відсутні ефективні інструменти для уникнення затримок в процедурі. Заявник лише може подати запит з проханням проінформувати його про статус розгляду заяви у відповідний орган або перевірити статус в СВ.
VІ.3.3. Тривала процедура перереєстрації як загроза безперервності ведення бізнесу
456. Довготривала процедура є досить проблематичною, особливо для заявників, які мають на меті перереєструвати лікарський засіб. У випадку якщо процедура перереєстрації не завершена до закінчення строку дії реєстраційного посвідчення (5 років), заявник не може продовжувати виробництво лікарського засобу.
Рекомендація:
Запровадити норму (правило), що дозволить продовжувати виробництво лікарського засобу до завершення розгляду заяви про перереєстрацію.
Порівняльна оцінка (Польща)
Згідно з статтею 29.7 Польського фармацевтичного закону, якщо заява про поновлення, що була подана в межах встановлених строків (в Польщі мінімум за 9 місяців), не була розглянута вчасно, заявник може продовжувати виробництво та розміщення продукту на ринку після завершення дії реєстраційного посвідчення, до того моменту, поки не завершиться розгляд заяви про перереєстрацію.
VІ.4. СПОСТЕРЕЖЕННЯ У СФЕРІ ОРГАНІЗАЦІЇ ПРОЦЕСІВ
VІ.4.1. Заявник подає документи в три етапи
457. Заявник повинен подати документи в три етапи на різних стадіях процедури:
- заява, що містить базову інформацію про заявника та лікарський засіб (не документ ЗТД формату, 3-4 сторінки) – подається в МОЗ на початку процедури;
- реєстраційна форма (20+ сторінок, відповідає ЗТД формату) з 20+ додатками, що подаються в ДЕЦ після передачі справи з МОЗ в ДЕЦ (на практиці – декілька днів після подачі заяви);
- реєстраційне досьє в форматі ЗТД – подається в ДЕЦ, після підписання договору з ДЕЦ та сплати за послуги з експертизи.
458. Очевидно, що вимога подачі окремої заяви (яка є не супровідним листом, а скоріше скороченою версією реєстраційної форми) є зайвою.
459. Причина розриву в часі моменту подачі реєстраційної форми та частини документів, включаючи досьє, також є незрозумілою.
Попередня процедура
До останніх змін в Наказ № 426, були дві окремі стадії перевірки документації щодо їх повноти та формальної відповідності вимогам. На кожному з цих етапів заява могла бути відхилена. Перший етап був визначений у внутрішніх документах та в Наказі № 426 як «первинна експертиза».
Первинна експертиза проводилась експертом ДЕЦ, який мав надати «обґрунтовану думку щодо первинної експертизи». Остаточне рішення про допуск справи до подальшої стадії перевірки (тобто попередньої експертизи в ДЕЦ) приймалось шляхом рішення Кваліфікаційної Комісії МОЗ, яка була експертним консультативним органом.
Ми оцінюємо виключення цієї стадії з процедури реєстрації як позитивний приклад розвитку системи. Ми хотіли б, однак, відзначити, що первинна експертиза ще не була виключена з внутрішніх документів ДЕЦ, які ми розглянули.
VІ.4.2. Повноваження МОЗ і ДЕЦ, що перетинаються під час процедури реєстрації
460. Українська процедура державної реєстрації змішує обов’язки МОЗ та ДЕЦ. В результаті видається, що ці органи діють почергово і повинні багаторазово обмінюватися документами або листами в ході процедури реєстрації, принаймні у наступних випадках:
(a) МОЗ надсилає заяву до ДЕЦ,
(b) МОЗ направляє заяву на попередню та спеціалізовану експертизу в ДЕЦ,
(c) ДЕЦ надсилає МОЗ вмотивовані висновки щодо безпеки, ефективності та якості разом з рекомендацією щодо державної реєстрації та документів для затвердження (список лікарських засобів рекомендованих до реєстрації, перереєстрації, внесення змін або в відмови в реєстрації, перереєстрації внесення змін, інструкцію для медичного застосування, МКЯ, маркування),
(d) МОЗ надсилає наказ щодо державної реєстрації до ДЕЦ,
(е) ДЕЦ надсилає реєстраційне посвідчення МОЗ для підписання,
(f) МОЗ надсилає підписане реєстраційне посвідчення до ДЕЦ,
(g) ДЕЦ надсилає пакет затверджених документів у «Єдине вікно», що формально є частиною структури МОЗ.
461. Така значна кількість дій, пов’язаних з обміном документами, є причиною неефективної роботи МОЗ і ДЕЦ і може спричиняти затримки у процедурі.
VІ.4.3. Надмірне залучення колегіальних органів до процедури реєстрації
462. Виникають сумніви щодо надмірного залучення колегіальних органів (НЕР, НТР, ТЕК) під час експертизи в ДЕЦ.
463. ДЕЦ вже за визначенням є експертною установою. Особи, що проводять експертизу відповідних частин досьє є експертами у своїх сферах. Надсилаючи їх експертні висновки іншому органу кожного разу перед підписанням Директором ДЕЦ може розглядатися як відсутність впевненості у знаннях та здібностях конкретного експерта.
464. Залучення колегіальних органів також може призводити до затримок у процедурі, особливо у випадку залучення НЕР та НТР, які проводять свої засідання один раз на місяць (НЕР) та два рази на місяць (НТР).
465. Досить цікавим є той факт, що колегіальні органи ДЕЦ можуть вирішувати питання, що не мають жодного відношення до наукової оцінки. Наприклад, такі питання, як зняття заяви з розгляду через ненадання повного пакету документів, або внесення змін типу ІА.
Попередня процедура
До останніх змін до Наказу МОЗ № 426, інший колегіальний орган приймав активну участь в процесі реєстрації – Кваліфікаційна Комісія МОЗ. Її роль була надмірною по відношенню до попередньої експертизи, що пізніше проводилася експертами ДЕЦ. Виключення цього етапу процедури є позитивним кроком.
VІ.4.4 Зайва вимога щодо GMP сертифікації
466. З наших співбесід з представниками індустрії можна зробити висновок, що очікування сертифікату GMP чи підтвердження його відповідності є одним з ключових факторів, які затримують строки державної реєстрації.
467. Питання про відповідність виробника вимогам GMP не має відношення до державної реєстрації лікарського засобу. З точки зору Директиви 2001/83 досить того, щоб виробник мав діючу ліцензію на виробництво, і щоб постачальник АФІ пройшов GMP аудит та відповідав вимогам GMP.
VІ.4.5 Занадто ускладнена процедура після закінчення експертизи реєстраційних матеріалів
468. Беручи до уваги, що позитивна оцінка експерта рівносильна підтвердженню безпеки, якості та ефективності лікарського засобу, державна реєстрація повинна бути прямим наслідком такої позитивної оцінки. Проте, на практиці регуляторні органи та заявник до виведення ЛЗ на ринок мають вчинити ряд додаткових дій,. До таких дій відносяться:
- передача ДЕЦ рекомендацій в МОЗ разом з проектами документів для затвердження,
- підписання наказу в МОЗ,
- підготовка проекту реєстраційного посвідчення в ДЕЦ для підписання та направлення його в МОЗ,
- підписання реєстраційного посвідчення в МОЗ і направлення назад в ДЕЦ,
- надання заявником довіреності, додаткових документів та відмови від претензій до ДЕЦ,
- підготовка пакету документів для заявника в ДЕЦ та направлення в «Єдине вікно» МОЗ,
- отримання документів представником заявника,
- численні повідомлення ДЕЦ про статус процедури заявнику.
469. Ми схему взаємодії, що включає усі вищезазначені дій після надання позитивних висновків щодо можливості реєстрації лікарського засобу, як занадто складну.
VІ.4.6 Низький рівень якості СОП ДЕЦ
470. Після аналізу СОП, що розроблені в ДЕЦ, ми маємо сумніви щодо їх реального впливу на хід процедури реєстрації:
(a) Опис етапів процедури та строків не завжди викладений чітко та однозначно. Деякі строки не мають відправної точки.
(b) Порядок, в якому представлені конкретні етапи процесу дає помилкове враження, що всі етапи процесу виконуються в у прямій послідовності. Така форма не дозволяє визначити процеси та дії, які можуть виконуватися паралельно.
(c) Описи індивідуальних етапів процесу не містять посилання на відповідні законодавчі акти, стандарти та інші процедури. Застосовне законодавство представлено у формі списку в окремій главі кожної процедури та не пов’язане з певними етапами процесу. В результаті, є високий ризик того, що суб’єктивний підхід та індивідуальна інтерпретація експертів може мати вплив на трактування загального розуміння процесів.
471. Така структура СОП ускладнює організацію і контроль за виконанням процесів і може навіть заважати нормальній роботі експертів.
472. Крім того, ДЕЦ було затверджено Інструкцію, яка має аналогічні до СОП недоліки. Співвідношення між Інструкцією і СОП не визначено.
VІ.4.7 Неефективне використання «Єдиного вікна»
473. Ідея «Єдиного вікна», як правило, пропонується в якості інструменту для обмеження бюрократії, полегшення та збільшення ефективності контакту бізнесу з державними органами.
474. В реальності української системи реєстрації лікарських засобів, впровадження «Єдиного вікна» не полегшило процедуру. Заявник подає заяву і отримує реєстраційне посвідчення через те ж саме «Єдине вікно» в МОЗ. Проте, в той самий час, заява обробляється та оцінюється іншою установою – ДЕЦ, з якою заявник має вести комунікацію не регулярній основі (подавати реєстраційну форму, підписувати договір та робити оплати, подавати реєстраційне досьє, відповідати на запити з додатковими поясненнями, брати участь в зустрічах, узгоджувати формулювання документів, що оформляються ДЕЦ, надавати довіреність та додаткові документи, що потрібні для отримання реєстраційного посвідчення).
475. Враховуючи вищезазначене, в сфері державної реєстрації лікарських засобів «Єдине вікно» не працює на практиці. Воно радше є причиною додаткових проблем в організації процесу.
VІ.5. СПОСТЕРЕЖЕННЯ ЩОДО ПРИЙНЯТНОЇ МОДЕЛІ ПРОЦЕДУРИ
VІ.5.1. Нечіткий розподіл повноважень
476. Однією з визначальних особливостей української системи реєстрації лікарських засобів є відсутність чіткого розподілу обов’язків. Існує помітна тенденція розмивання відповідальності між різними елементами системи.
477. Яскравим прикладом цієї тенденції є те, яким чином законодавство описує компетенцію щодо прийняття рішень в області державної реєстрації:
(a) Закон про лікарські засоби встановлює, що МОЗ приймає рішення про реєстрацію або відмову в реєстрації лікарського засобу.
(b) Постанова КМУ № 376, будучи підзаконним актом, додає, що рішення має бути прийнято на підставі результатів експертизи ДЕЦ.
(c) Ще один підзаконний акт, Наказ МОЗ № 426 вводить правило, що експертиза повинна бути проведена кількома експертами і кожен експерт несе відповідальність за результати його чи її експертної оцінки. Крім того, результати експертизи мають бути розглянуті «розглянуті» на засіданні консультативно- дорадчого органу ДЕЦ, і вже потім буде надана рекомендація щодо реєстрації до МОЗ.
(d) Внутрішні документи ДЕЦ (Інструкції, СОПи) визначають три дорадчих органи для різних типів заяв та посилаються на них як на органи, що приймають рішення (наприклад, Інструкція ДЕЦ, п. 7.11: «Експерт ДЕРМ (Департамент експертизи реєстраційних матеріалів) готує повідомлення заявнику про рішення НЕР/НТР (форма 17) та передає його на підпис Директору ДЕЦ або особі, що його заміщує.»)
478. Як видно із вищезазначеного, чим нижчим є рівень підзаконного акту, тим складнішим є описання процесу прийняття рішень. Якщо судити тільки з формулювань, що містяться у Законі про лікарські засоби, можна припустити, що відповідальність за державну реєстрацію лікарського засобу покладається виключно на МОЗ. Після ознайомлення з підзаконними актами і внутрішніми процедурами, не можна чітко сказати, який орган є відповідальним за весь процес реєстрації.
479. Крім того, елементи інституційної відповідальності, що описані у законодавстві (наприклад, відповідальність МОЗ і ДЕЦ, обсяг якої передбачено Законом і Постановою КМУ № 376) переплітаються з елементами особистої відповідальності окремих експертів, які здійснюють експертизу, або з колективною відповідальністю колегіальних органів. В результаті не тільки заявник не може визначити, до кого він повинен звернутися у разі наявних претензій у випадку невдалої процедури реєстрації, але і самі учасники процедури реєстрації заохочуються не приймати такі рішення, які можуть слугувати підставою для притягнення їх до відповідальності.
480. Розмивання відповідальності є помітним не тільки на рівні прийняття рішень, але і на проміжних етапах процедури. Наприклад:
(a) Формально, рішення про направлення заяви до ДЕЦ на експертизу приймає МОЗ. На практиці до прийняття останніх змін до Наказу МОЗ № 426, МОЗ не приймав таких рішень самостійно, а залучав ДЕЦ до процесу первинної експертизи заяви.
(b) Рішення про позитивні результати експертизи заяви, як на рівні первинної експертизи (більше не діє), так і попередньої експертизи в ДЕЦ, не приймаються без попередніх експертних висновків, при цьому такі рішення могли б прийматися адміністративним персоналом без необхідності залучення експертів.
(c) Консультативні органи беруть участь на різних етапах процедури, в будь-який час приймається рішення, яке може привести установу до відповідальності. До нещодавніх змін в Наказ МОЗ № 426, рішення МОЗ, про те, чи може заява розглядатися за тим типом, що був обраний заявником (наприклад заява про державну реєстрацію генерика), чи ні, базувалося на рішенні Кваліфікаційної Комісії МОЗ. Будь-які рекомендації ДЕЦ для МОЗ завжди надаються на основі консультацій (або навіть «рішення», як це визначено внутрішніми правилами ДЕЦ) від консультативних органів ДЕЦ, в тому числі НЕР, НТР та ТЕК. Консультації необхідні як для позитивних так і негативних рекомендацій, а також не тільки для державної реєстрації, а й для перереєстрації та внесення змін.
481. Спосіб у який регулюються строки процедури реєстрації є ще одним інструментом розсіювання відповідальності. Немає чітких граничних строків, встановлених для всієї процедури (від моменту подання заяви до отримання реєстраційного посвідчення). Натомість є окремі строки, встановлені для МОЗ (для прийняття рішення про державну реєстрацію), а також для ДЕЦ (для проведення експертизи, щоб МОЗ могло прийняти рішення). Для деяких етапів процедури немає встановлених законодавством строків (строки можуть регулюватися тільки всередині установи).
482. МОЗ не має правових інструментів для втручання в процес реєстрації у разі затримки під час експертизи в ДЕЦ. МОЗ не може досліджувати причини затримок або випадки некоректності зі сторони експертів.
483. У цьому контексті, ми хотіли б звернути увагу на статтю 321 (2) Кримінального кодексу України. Відповідно до цього положення, порушення процедури державної реєстрації лікарських засобів підлягає покаранню у вигляді позбавлення волі.
484. Реєстрація лікарських засобів є одним з найскладніших питань, що вирішуються державними органами в кожній країні. У цьому контексті «порушення процедури» може відноситись до різних ситуацій, від реєстрації небезпечних продуктів до порушення строків.
485. Беручи до уваги вищезазначене, криміналізація дій і рішень, прийнятих під час процедури реєстрації лікарських засобів, а не захист належного ходу процедури реєстрації, швидше перешкоджає особам і установам, що беруть участь в процесі, приймати на себе відповідальність за перебіг і наслідки процедури.
486. Положення Кримінального Кодексу України, слугує причиною, яка пояснює надмірно складну модель прийняття рішень з розмитими обов’язками.
Рекомендація:
Ми вважаємо, що формулювання статті 321 (2) Кримінального Кодексу України має бути переглянутим, задля забезпечення гарантії належного ходу процедури реєстрації лікарських засобів, та, щоб в той же час уникнути непотрібного тиску на осіб, які беруть участь у державній реєстрації.
VІ.5.2. Залучення зовнішніх експертів
487. Під час проведення спеціалізованої експертизи, розгляд модулів 4 і 5 (доклінічні дослідження і документація щодо клінічних випробувань) здійснюється зовнішніми експертами.
488. Залучення зовнішніх експертів в процесі оцінки реєстраційного досьє не є незвичайним в практиці реєстраційних агентств в країнах ЄС. Однак прийняття такої моделі викликає певні труднощі для керівного персоналу органу реєстрації. Наприклад, існує менше інструментів для контролю щоденної роботи зовнішніх експертів, ніж для експертів, що працюють в агентстві. Це може привести до несвоєчасного виконання послуг зовнішніми експертами. Зовнішні експерти частіше можуть мати конфлікт інтересів.
489. Крім того, слід зазначити, що в ДЕЦ огляд документації клінічних випробувань (модуль 5) здійснюється виключно зовнішніми експертами. Це означає, що навіть у простих випадках ДЕЦ не в змозі завершити експертизу своїми власними зусиллями.
VІ.5.3. Неефективне залучення МОЗ до процедури реєстрації
490. МОЗ залучений в процедуру реєстрації на первинних та фінальних стадіях. МОЗ отримує заяву, яку раніше оцінював (наразі Наказом МОЗ № 426 цей етап виключено) та вирішував щодо направлення заяви в ДЕЦ на експертизу. Після отримання рекомендацій від ДЕЦ, МОЗ видає відповідні документи та надає їх заявнику.
491. На наш погляд, таке залучення МОЗ в актуальній моделі не є ефективним рішенням. МОЗ не має ресурсів, які б дозволили йому виконувати свої функції ефективно. Згідно з даними, які ми отримали від МОЗ, лише 3 особи в МОЗ займаються питаннями державної реєстрації лікарських засобів.
Попередня процедура.
До недавніх змін в Наказ № 426, залучення МОЗ в первинний розгляд заяви був зайвим, беручи до уваги, що ДЕЦ повторює формальну перевірку документів на рівні попередньої експертизи.
492. Крім того, необхідність в залученні МОЗ на завершальній стадії процедури є спірною. Відповідно до Закону, рішення про реєстрацію має бути прийнято на підставі результатів експертизи. Експертиза здійснюється ДЕЦ, а результати експертизи містяться у звітах експертів. Оскільки ресурси МОЗ у сфері реєстрації лікарських засобів є обмеженими, МОЗ фактично не може здійснювати повноцінний аналіз звітів, висновків та рекомендацій експертів. Як наслідок, рішення МОЗ щодо державної реєстрації (перереєстрації, внесення змін) базується виключно на довірі, яку Міністр має до точності та якості процедури, здійсненої ДЕЦ. В той самий час, МОЗ не має законних можливостей для перевірки рівня експертизи у конкретних випадках.
493. Слід також підкреслити, що наділення МОЗ адміністративними функціями є прикладом неправильного розподілу ресурсів. МОЗ слід зосередитись на розробці політики та нагляді, а не виконувати завдання, які є явно нижче рівня компетентності МОЗ в рамках системи.
VІ.5.4. Реєстрація шляхом прийняття наказу щодо декількох лікарських засобів
494. Процедура державної реєстрації завершується видачою наказу, який посилається на кілька лікарських засобів водночас. Наявність такого «колективного» елементу в процедурі державної реєстрації суперечить суті самої процедури, яка, як видається, від заяви та експертизи реєстраційних матеріалів до рекомендації ДЕЦ, має яскраво виражений «індивідуальний» характер і стосується конкретного лікарського засобу, а не їх групи.
495. Реєстрація через такий «колективний» наказ може бути однією з причин неефективності моделі державної реєстрації в Україні:
- Це може спричинити затримки в державній реєстрації. ДЕЦ може притримувати передачу рекомендації про реєстрацію певного лікарського засобу в МОЗ до тих пір, поки не завершить роботу над достатньою кількістю інших (паралельних) заяв. МОЗ може призупинити підписання наказу в разі виникнення сумнівів щодо хоча б одного з лікарських засобів, які охоплюються проектом наказу.
- Відсутній ефективний механізм адміністративного оскарження рішення, прийнятого у вигляді наказу МОЗ.
- Рішення, прийняте у формі наказу, не містить обґрунтування (переліку підстав, які зумовили його прийняття).
496. Крім негативних аспектів зберігання концепції «колективного» наказу, ми не виявили інших аргументів, які б виправдовували необхідність збереження такої форми прийняття рішень.
VІ.5.5. Квазі-комерційний статус ДЕЦ
497. Статус ДЕЦ в системі державної реєстрації є неоднозначним. З одного боку, ДЕЦ виконує функції, пов’язані винятково з виконанням державою своїх обов’язків у сфері охорони здоров’я (експертиза заяви, на підставі висновків якої МОЗ приймає рішення про реєстрацію). З іншого боку, ДЕЦ має правовий статус державного підприємства, що є особливим типом підприємства, яке має на меті отримання прибутку і надання послуг на комерційній основі, але знаходиться у власності держави через МОЗ. Така ситуація є нетиповою, і не є стандартом в ЄС.
498. Ми припускаємо, що цей статус має певні переваги (свій власний бюджет, більше гнучкості в кадрових питаннях). Нам відомо, що поточна модель (державне підприємство) є наслідком недоліків попередньої моделі(Державний Фармакологічний комітет- типова державна установа). У той же час поточна модель може спричинити наслідки, які будуть протирічити ролі ДЕЦ в системі.
499. Яскравим прикладом такої неоднозначності є відносини між ДЕЦ і заявником. Для того щоб ДЕЦ міг розпочати проведення експертизи, заявник повинен укласти договір з ДЕЦ. Договір нагадує стандартний договір про надання комерційних послуг, зокрема, в таких аспектах:
- Заявник називається «замовником».
- Експертиза визначається як «послуга», яка надається «на вимогу замовника».
- Плата за експертизу називається «винагородою». Договір дає замовнику право заперечувати проти суми рахунку-фактури/інвойсу, а також дозволяє платежі в іноземній валюті.
- Оплата визначається як «100% попередня оплата».
- Прийняття «послуг» «замовником» не вимагається. Якщо замовник не заперечує проти «Акту виконаних робіт», який видається ДЕЦ в чітко визначений строк, послуги вважаються прийнятими замовником.
- Встановлюється відповідальність обох сторін за можливе невиконання.
500. В країнах ЄС відносини між органом реєстрації лікарських засобів і заявниками є вертикальними за визначенням. Реєстраційні органи діють як регуляторні органи з повноваженнями приймати рішення щодо обігу лікарських засобів, а не в якості постачальників послуг для індустрії. Кваліфікація відносин між ДЕЦ і заявником як суто комерційних несе значні ризики як для обох сторін, так і для системи в цілому. Наприклад, логічним наслідком кваліфікації експертизи як «послуги» має бути право незадоволеного заявника отримати фінансову компенсацію за неналежне виконання договірних зобов’язань від «постачальника послуг» в суді. Ризик настання такої відповідальності може мати негативний вплив на незалежність експертів і може поставити під загрозу якість експертизи.
501. Крім того, якщо розглядати експертизу досьє ДЕЦ як суто надання комерційних послуг, може виникнути питання про можливість надання таких послуг іншими кваліфікованими підприємцями або експертами. З цієї точки зору діяльність ДЕЦ може розглядатися як монополія, і домінуюче становище ДЕЦ на відповідному ринку може підлягати аналізу Антимонопольним комітетом України.
VI.6. РЕКОМЕНДАЦІЇ
502. Перед тим, як розглянути питання щодо можливих змін в організації процесу реєстрації ЛЗ, ми хотіли б пояснити наш підхід до формування рекомендації в цій сфері.
503. Існуюча система, незважаючи на певні її недоліки, виконує свою основну функцію, забезпечуючи дієвий і відносно стабільний інституційний механізм допуску лікарських засобів до обігу на основі наукової оцінки їх якості, безпеки та ефективності. Система пройшла через декілька реформ в останні роки. Ефективність і якість системи в деяких аспектах все ще нижча за очікування фармацевтичної індустрії, однак реалізовані реформи демонструють справжню зацікавленість українських політиків у вдосконаленні процедур реєстрації лікарських засобів. Багато змін, реалізованих останнім часом, слід оцінювати як рух в позитивному напрямку (наприклад, скорочення строків виконання певних дій, обмеження чисельності запитів до заявників, усунення етапу первинної експертизи). Нам також повідомили, що всередині ДЕЦ були зроблені кроки для усунення затримок у процедурах (посилений внутрішній аудит, більш ретельний моніторинг затримок, змінені правила розподілу роботи серед експертів).
504. Зважаючи на вищевказане, ми хотіли б підкреслити, що наші рекомендації щодо можливих змін в системі повинні бути детально обговорені з представниками відповідних установ та інших зацікавлених сторін, щоб розробити такий спосіб їх реалізації, який би дозволив системі залишатися стабільною у перехідний період.
505. Ми хотіли б також зазначити, що, крім рекомендацій, представлених в цьому розділі, ми також пропонуємо додаткові рекомендації, які можуть бути корисними для вдосконалення процедури і які необхідні для того, щоб забезпечити дотримання законодавства ЄС з цих питань. Ці рекомендації були викладені окремо в розділі III вище, і їх слід розглядати в поєднанні з рекомендаціями, наведеними нижче.
506. З урахуванням зазначених вище зауважень, ми вважаємо, що слід розглянути можливість змін в системі в таких напрямках:
(a) Переведення всіх можливих завдань в сфері державної реєстрації до компетенції ДЕЦ
507. Повноваження МОЗ на початковій і кінцевій стадії процедури повинні бути або скасовані або зведені до мінімуму, в залежності від прийнятої моделі.
508. Заяви повинні подаватися напряму в ДЕЦ.
(b) [Опціонально] Переведення всіх можливих завдань у сфері державної реєстрації до компетенції новоствореного органу
509. [Опціонально] Для підвищення рівня прозорості процесів та загального рівня довіри до системи реєстрації як у заявників, так і у представників громадськості, основні функції у сфері державної реєстрації, що на сьогодні виконуються ДЕЦ, можуть бути передані до компетенції спеціалізованої новоствореної структури. При цьому, обов’язковими умовами для передачі відповідних функцій є:
- збереження новоствореною структурою статусу державного підприємства у сфері управління МОЗ (новостворена структура не повинна мати статусу органу державної влади);
- максимальна імплементація рекомендацій, що містяться у Звіті, при розробці (і) організаційної структури новоствореної структури та (іі) основних засад її діяльності.
(с) [Опціонально] Переведення компетенції приймати рішення про державну реєстрацію до ДЕЦ
510. В якості альтернативи теперішній моделі, необхідно розглянути можливість надати ДЕЦ/новоствореній структурі повноваження приймати рішення про державну реєстрацію, замість МОЗ. В такому разі МОЗ зберігатиме повноваження загального нагляду за роботою ДЕЦ/новоствореної структури і розгляду адміністративних скарг на рішення ДЕЦ/новоствореної структури.
511. Реалізація таких змін може вимагати внесення відповідних змін у правовий статус ДЕЦ і його працівників. За діючим законодавством прийняття рішення про реєстрацію від імені держави працівниками державного підприємства не є можливим.
(d) Індивідуальне рішення про державну реєстрацію
512. Поточну модель державної реєстрації через прийняття «колективного» наказу МОЗ слід замінити системою індивідуальних рішень про державну реєстрацію.
513. Реєстраційні посвідчення, видані на підставі «колективного» наказу, повинні бути перевидані в формі окремих наказів про державну реєстрацію.
514. Інститут наказу МОЗ про державну реєстрацію (перереєстрацію) лікарських засобів (медичних імунобіологічних препаратів) і внесення змін до реєстраційних матеріалів повинен бути скасований. Його інформативна функція (інформування пацієнтів, фахівців в області охорони здоров’я і ринку) може бути передана Державному реєстру лікарських засобів.
515. Необхідно запровадити інститут адміністративного оскарження рішення про державну реєстрацію (наприклад, до МОЗ, якщо рішення винесено ДЕЦ, а вже потім до суду).
(е) Консолідація процедури
516. Процедура державної реєстрації повинна бути спрощена і об’єднана, для того щоб обмежити кількість етапів і подачу/обмін документами.
517. Для ініціювання процедури державної реєстрації повинно бути достатнім подання лише однієї заяви (реєстраційної форми).
518. Всі документи на підтвердження заяви, в тому числі реєстраційне досьє, повинні бути додані до заяви на початку.
519. Заява разом з усіма додатками повинна бути перевірена на повноту і відповідність іншим формальним вимогам лише один раз, на початку процедури. У разі виявлення недоліків або помилок, заявник повинен бути повідомлений про них один раз для можливості їх виправлення; в іншому випадку заява буде відхилена.
520. [Опціонально] Вимога про підтвердження відповідності умов виробництва вимогам GMP як передумова державної реєстрації повинна бути скасована по відношенню до заявників, які мають дійсні GMP сертифікати видані в країнах-членах PIC/S (хоча б в країнах-членах ЄС) . Питання, що стосуються сертифікації GMP, повинні належати виключно до компетенції Держлікслужби. Держлікслужба повинна бути виключена з процедури державної реєстрації.
521. Час, необхідний для лабораторного тестування якості продукту і відтворюваності методів контролю, повинен враховуватися в загальний строк всієї процедури.
(f) Декомерціалізація процедури
522. Договір, як підстава для виконання ДЕЦ своїх обов’язків, повинен бути усунений.
523. У відповідних нормативно-правових актах повинно бути чітко зазначено, що здійснення експертизи ДЕЦ є виконанням ним обов’язків держави в сфері реєстрації лікарських засобів, а не наданням комерційних послуг.
524. Замість окремої оплати державного мита та вартості експертних послуг, необхідно впровадити єдиний реєстраційний внесок, який повинен сплачуватися авансом. Сума платежу не повинна базуватися на рішенні ДЕЦ та на рахунку-фактурі, а повинна бути врегульована на рівні наказу МОЗ.
(g) Перегляд повноважень консультативних органів
525. НЕР, НТР, ТЕК повинні бути замінені одним органом, що надаватиме консультації компетентному органу у сфері реєстрації лікарських засобів.
526. Новостворений орган повинен мати виключно консультативні функції. Він повинен надавати консультації лише з питань по суті, а не щодо процедури, і лише з питань, які є прецедентними або викликають сумніви з точки зору наукової оцінки. Рішення про передачу справи на розгляд консультативного органу повинно прийматися головою відповідного департаменту ДЕЦ або Директором ДЕЦ. В листі- направленні повинні вказуватися причини передачі справи в консультативний орган, а також точний обсяг питань, стосовно яких вимагається консультація.
(h) Відповідальність експертів виключно перед роботодавцем
527. Експерти, які здійснюють експертизу реєстраційних досьє, повинні бути звільнені від відповідальності по відношенню до заявників (за винятком випадків, коли експерт умисно порушив закон).
(і) Врегулювання відносин з зовнішніми експертами
528. Необхідно врегулювати у відповідному акті співпрацю ДЕЦ із зовнішніми експертами.
529. Залучення зовнішніх експертів повинно бути можливим у випадках, якщо виконання експертизи неможливе за рахунок власних ресурсів ДЕЦ.
530. Офіційний список зовнішніх експертів повинен бути доступний громадськості.
531. Зовнішні експерти повинні підписувати декларацію про відсутність конфлікту інтересів і підтверджувати відсутність такого конфлікту перед кожною експертизою. Неповідомлення про конфлікт інтересів повинне передбачати відповідні санкції, включаючи негайне виключення з переліку експертів.
(j) Введення в дію зрозумілих правил притягнення до відповідальності за затримку
532. Повинні бути введені загальні обмеження по строкам процедури державної реєстрації (перереєстрації, внесення зміни в реєстраційні матеріали), починаючи з дати подання повної заяви і до дати винесення рішення про державну реєстрацію (перереєстрацію, внесення змін до реєстраційних матеріалів).
533. Необхідно визначити єдиний компетентний орган в якості відповідального за здійснення державної реєстрації у встановлений законом строк. Цей орган повинен нести відповідальність за організацію процесу реєстрації у встановлені строки.
Приклад для порівняння (Польща)
Якщо справа не може бути вирішена протягом встановленого строку, компетентний орган повинен повідомити про це заявника, пояснити причини затримки і вказати нову дату розгляду справи.
Якщо справу заявника не було розглянуто протягом встановленого строку, або її розгляд затягується (наприклад, повторюються питання до документації), заявник може подати скаргу в МОЗ.
МОЗ аналізує причини затримки або тривалого розгляду справи, і, якщо скарга є обґрунтованою, то МОЗ:
534. Необхідно передбачити правовий механізм оскарження заявником затримок у процедурі реєстрації.
- встановлює новий строк розгляду справи;
- надає вказівку визначити причини затримки та осіб, відповідальних за затримку;
- надає вказівку вжити заходів для недопущення подібних затримок у майбутньому, якщо в цьому є необхідність,
- зазначає, чи була затримка грубим порушенням закону (що відкриває шлях для позовів про відшкодування збитків).
Працівник, який не розглянув справу протягом встановленого строку, підлягає дисциплінарній чи іншій відповідальності, передбаченій законом або внутрішніми правилами.
(k) Перегляд СОПів/підготовка нових СОПів
535. Стандартні операційні процедури в ДЕЦ, а також Інструкція з організації проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються з метою державної реєстрації, повинні бути переглянуті для того, щоб:
- виключити можливість їх різного трактування працівниками ДЕЦ,
- забезпечити виконання відповідних процесів без використання додаткових джерел інформації,
- встановлювати розумні й ефективні внутрішні строки виконання.
VІ.7. ОРГАНІЗАЦІЯ ПРОЦЕСІВ У ДЕРЖЛІКСЛУЖБІ
536. Держлікслужба бере участь в процесі реєстрації лікарських засобів в частині підтвердження відповідності умов виробництва лікарського засобу вимогам GМР. Закон про лікарські засоби вимагає, щоб заявники надавали сертифікат відповідності вимогам GМР до МОЗ щодо всіх лікарських засобів, заявлених до реєстрації.
537. Заявник подає в ДЕЦ або сертифікат відповідності умовам ОМР, виданий Держлікслужбою, або підтвердження Держлікслужбою ОМР-сертифіката, виданого уповноваженим органом країни-члена РІС/S, до кінця проведення спеціалізованої експертизи.
538. З точки зору організації процесу, робота Держлікслужби може бути вдосконалена, зокрема, в таких аспектах:
- паперовий документообіг повинен бути замінений на електронний;
- процедура первинної експертизи документів, поданих заявником для підтвердження GМР-сертифікатів, виданих уповноваженими органами країн- членів РІС/S, може бути скасована;
- залучення колегіального консультативного органу Держлікслужби (так званої Робочої групи) до процесу прийняття рішень має бути чітко врегульованим;
- випадки, за яких участь Робочої групи є необхідною, повинні бути чітко визначені.
VІ.7.1. Заміна процедури з використанням паперових носіїв електронною процедурою
539. Відповідно до Наказу МОЗ № 1130, всі документи подаються заявником до Держлікслужби в паперовій формі. Хоча деяка інформація повинна бути продубльована в електронному вигляді, процедура підтвердження умов виробництва вимогам GМР повністю здійснюється у паперовій формі.
540. Як і з іншими регуляторними процедурами у сфері реєстрації лікарських засобів, перехід від паперової до електронної процедури підтвердження умов виробництва вимогам GМР був би корисним для всіх сторін.
Рекомендації
Планувати перехід до здійснення процедури підтвердження відповідності вимогам GМР за допомогою електронних засобів, щоб це сприяло своєчасності, прозорості і більшій ефективності процедури.
Це б зробило необхідну інформацію легкодоступною для регуляторних органів, в тому числі і для МОЗ та ДЕЦ, а також зробило б можливим громадський контроль за процедурою.
VІ.7.2. Скасування етапу первинної експертизи під час процедури підтвердження GМР-сертифікатів, виданих уповноваженими органами країн-членів РІС/S
Пункт 2 Розділу I Наказу МОЗ № 1130
«первинна експертиза – перевірка комплектності поданих документів та відповідності таких документів законодавству, у тому числі вимогам цього Порядку, яка проводиться працівниками Держлікслужби України»
Пункт 5 Розділу II Наказу МОЗ 1130
«Держлікслужба України здійснює первинну експертизу поданих документів з метою перевірки їх комплектності та відповідності форми таких документів законодавству, у тому числі вимогам цього Порядку»
541. Відповідно до Наказу МОЗ 1130, Держлікслужба видає 2 види документів, що стосуються підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики:
- сертифікат відповідності умов виробництва лікарських засобів вимогам належної виробничої практики, якщо заявник не має відповідного сертифіката, виданого уповноваженим органом країни-члена PIC/S;
- висновок про підтвердження відповідності умов виробництва лікарських засобів вимогам GMP (підтвердження відповідного сертифікату, виданого уповноваженим органом країни-члена PIC/S).
542. Обидві процедури включають первинну та спеціалізовану експертизу поданих документів. Первинна експертиза має на меті перевірку комплектності та відповідності поданих документів формальним вимогам, той час як спеціалізована експертиза включає в себе аналіз змісту і достовірності поданих документів.
543. Незважаючи на той факт, що експертиза при підтвердженні відповідності повинна займати 2-5 і 3-15 робочих днів (в залежності від типу процедури), на практиці навіть просте підтвердження GMP-сертифікатів, виданих FDAабо однією з європейських інспекцій, може зайняти 3-6 місяців і більше. Такі затримки можуть поставити під загрозу саму процедуру реєстрації. Цьому сприяють різні фактори, зокрема:
- Використання повноважень щодо призупинення перебігу строку посадовими особами Держлікслужби на будь-якому етапі первинної чи спеціалізованої експертизи (це не суперечить чинному законодавству, але може спричинити значну затримку в ході процедури);
- Недостатня кількість експертів в Держлікслужбі;
- Затримки з боку заявників.
544. Заявники, що подають документи на підтвердження GMPсертифікатів, часто скаржаться на те, що немає необхідності проводити первинну оцінку наданих документів, так як цей етап просто перевантажує та ускладнює процедуру, і зумовлює необхідність збільшення часу на її проведення. Всі необхідні питання можуть бути підняті на етапі спеціалізованої експертизи через призупинення перебігу її строку відповідним регуляторним органом.
Рекомендації
Етап первинної експертизи під час процедури підтвердження GMP- сертифікатів, виданих уповноваженими органами країн-членів PIC/Sмає бути скасовано. Слід забезпечити на рівні нормативно-правового акту, що повноваження щодо призупинення перебігу строку регуляторним органом можуть бути використані ним тільки один раз в тих випадках, коли посадові особи Держлікслужби запитують додаткові документи (за умови, що заявник відповідає на запит і надає всі запитувані дані).
VI.7.3. Функціонування Робочої групи
545. Робоча група є консультативним органом в межах ДЕЦ, в завдання якої входить надання остаточних рекомендації щодо відповідності вимогам GMPв тих випадках, коли Держлікслужба проводить інспектування виробництва.
546. На практиці, в разі інспектування виробництва заявника на місці, наявність позитивних рекомендацій Робочої групи є обов’язковою, і без них Держлікслужба не видає сертифікат відповідності вимогам GMP.
547. Повноваження Робочої групи, що функціонує в рамках Держлікслужби, не врегульовані чинним законодавством, незважаючи на її важливу роль у процедурі підтвердження відповідності. Єдине положення в відповідних наказах МОЗ, що стосується Робочої групи, викладено нижче:
Пункт 11 Розділу III Наказу МОЗ 1130
«Розгляд Держлікслужбою України на робочому засіданні звіту, складеного за результатами інспектування, з метою його оцінки та обґрунтованості викладених у ньому порушень здійснюється не пізніше 10робочих днів від дати його складання.»
548. Це робить процес прийняття рішень Робочою групою непередбачуваним і непрозорим. Відповідальність за прийняття рішень є розмитою.
549. Діяльність Робочої групи регулюється певними внутрішніми документами Держлікслужби. Проте вони лише встановлюють вимоги до її складу, і не встановлюють вимоги до її діяльності та до процесу прийняття нею рішень.
Рекомендації
Скасувати вимогу обов’язкового схвалення Робочою групою результатів експертизи під час процедури підтвердження GМР-сертифікатів, або чітко регламентувати її роль, повноваження і процес прийняття рішень на рівні Наказу МОЗ № 1130.
VII. СПОСТЕРЕЖЕННЯ У СФЕРІ КОМУНІКАЦІЇ ТА ПРОЗОРОСТІ
VII.1. ПРОГАЛИНИ В КОМУНІКАЦІЇ З БОКУ РЕГУЛЯТОРНИХ ОРГАНІВ
550. Дуже важливим інструментом удосконалення процедури реєстрації лікарських засобів є постійна комунікація між усіма її учасниками. Відсутність порозуміння між учасниками процедури реєстрації та широкої громадськості має своїм результатом багато негативних висловлювань і необгрунтованих взаємних звинувачень.
551. Доведення до відома громадськості і заявників всіх внутрішніх процедур в регуляторних органах дозволить зменшити кількість скарг до функціонування системи реєстрації.
VII.1.1. Комунікація з громадськістю
552. Посилення взаємодії між регуляторними органами, які беруть участь в процесі реєстрації лікарських засобів, та громадськістю надзвичайно важливо для розвитку української системи охорони здоров’я в цілому.
553. Важко переоцінити роль процедури реєстрації лікарських засобів в складі системи охорони здоров’я в Україні. Тому надзвичайно важливо забезпечити, щоб вся необхідна інформація про державну реєстрацію лікарських засобів належним чином доводилася до відома громадськості.
554. Основні проблеми з точки зору комунікації з громадськістю полягають в:
- обмежених функціональних можливостях офіційних веб-сайтів МОЗ, ДЕЦ і Держлікслужби;
- відсутності загальнодоступної інформації про поточні процедури в Держлікслужбі і МОЗ, дуже обмежена інформація про поточні процедури в ДЕЦ;
- відсутності загальнодоступної інформації щодо організації процесу експертизи ДЕЦ.
VII.1.2. Обмежені функціональні можливості офіційних веб-сайтів
555. В сучасному світі більша частина громадськості, як правило, отримує інформацію за допомогою цифрових каналів, зокрема через мережу Інтернет. Це посилює роль вебсайту в якості каналу зв’язку між громадськістю та регуляторними органами.
556. Офіційні сайти Держлікслужби, ДЕЦ і МОЗ можуть бути вдосконалені в багатьох аспектах, зокрема, в питаннях змісту та форми надання інформації.
557. Найбільш поширеними проблемами офіційних веб-сайтів є недосконала система навігації, незручний інтерфейс, а також те, що англійські версії веб-сайтів не оновлюються на постійній основі, а більша частина веб-сайтів не перекладається англійською взагалі.
558. Використання застарілої інформації в англійській версії сайтів є спільною проблемою ДЕЦ, МОЗ та Держлікслужби. Це має своїм наслідком ситуацію, коли англомовні користувачі веб-сайтів не можуть ознайомитись з останніми важливими змінами в процедурі реєстрації лікарських засобів або навіть деякими загальними законодавчими положеннями.
Приклад
На офіційному сайті Держлікслужби в англійській версії, в підрозділі «Контроль якості лікарських засобів – Загальна інформація» все дані щодо загальної процедури контролю якості та ролі Держлікслужби в ній востаннє оновлювалися в 2011 році.
Українська версія цього підрозділу востаннє оновлювалася в 2016році. Вона додатково містить важливу інформацію про діюче українське законодавство у сфері охорони здоров’я з посиланнями на законодавчі акти, а також контакти відповідальних осіб Держлікслужби.
Рекомендації:
Оновити офіційні веб-сайти МОЗ, ДЕЦ та Держлікслужби, щоб зробити їх зручними та доступними для користувачів, регулярно оновлювати в тому числі і англійські версії веб-сайтів.
VII.1.3 Відсутність загальнодоступної інформації про поточні процедури, статистичні дані, результати щорічних внутрішніх і зовнішніх аудитів, звіти про результати оцінки
569. Відповідно до нещодавніх змін до Закону України «Про лікарські засоби», інформація про подану заяву про державну реєстрацію, її статус і прийняті на основі її розгляду рішення оприлюднюється безкоштовно на веб-сайті установи, що здійснює експертизу реєстраційних матеріалів (ДЕЦ). Проте, для реалізації цього положення необхідно прийняти відповідні зміни до Наказу МОЗ № 426.
560. На даний час інформація про поточні процедури, звіти про результати оцінки лікарського засобу, а також статистичні дані, які б демонстрували результати роботи і містили стисле викладення змісту внутрішніх і зовнішніх аудитів ДЕЦ і МОЗ, не є загальнодоступною.
Інформація про поточні процедури:
561. На сьогодні офіційний сайт ДЕЦ містить певну інформацію щодо поточних процедур в ДЕЦ, але ця інформація обмежується списками лікарських засобів, поданих на державну реєстрацію, та списками лікарських засобів, рекомендованих/не рекомендованих ДЕЦ для реєстрації. Хоча ця інформація публікується регулярно (як правило, щотижня), вона завантажується на веб-сайт у вигляді консолідованого документу в форматі MSWord.
562. Веб-сайт ДЕЦ не містить єдиного загального інструменту пошуку, доступного заявникам та широкому загалу.
563. Для довідки, на веб-сайті Європейського агентства з лікарських засобів (ЕМА) можна легко знайти перелік заяв про реєстрацію лікарських засобів (marketingauthorization), які знаходяться на етапі розгляду11, та перелік заяв, які були відкликані12. Крім того, на веб-сайті EMA кожен може переглянути інформацію про лікарські засоби, щодо яких була здійснена експертиза Комітетом EMA з лікарських препаратів для людини (CHMP) і щодо яких в даний час очікується рішення Європейської комісії.
Рекомендації:
Привести Наказ МОЗ №»426 у відповідність до останніх змін в Законі України «Про лікарські засоби».
Публікувати статус щодо кожної поточної процедури реєстрації.
Статистичні дані, результати внутрішніх та зовнішніх аудитів:
564. Свіжі статистичні дані та результати внутрішніх і зовнішніх аудитів є показником того, чи виконують регуляторні органи свої функції, чи ні. Ця інформація дає можливість громадськості контролювати строки здійснення процедур та показує загальний обсяг робіт, що виконують регуляторні органи.
Рекомендації:
Розміщувати у відкритому доступі статистику та підсумки внутрішніх та зовнішніх аудитів.
Звіти за результатами оцінки:
565. Ні заявники, ні пацієнти/широка громадськість не мають доступу до звітів за результатами оцінки лікарських засобів, поданих на реєстрацію.
566. В ЄС звіти за результатами оцінки лікарських засобів належать до загальнодоступною інформації. Будь-хто може переглянути ці звіти на сайті ЕМА13. Сайт EMA містить детальну інформацію про історію реєстрації лікарського засобу, що дає можливість громадськості перевірити цю інформацію. Такий підхід забезпечує більшу прозорість та відповідальність регуляторних органів за прийняття відповідних рішень.
Рекомендації:
Звіти за результатами оцінки лікарських засобів повинні розміщуватися у відкритому доступі на сайті ДЕЦ.
Беручи за зразок EMA, кожен лікарський засіб, який був зареєстрований, повинен мати власну сторінку з історією реєстрації засобу, перереєстрації та всіх внесених змін.
_______________________
11Перелік заяв про реєстрацію лікарського засобу, які знаходяться на етапі розгляду, можна знайти за цим посиланням:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/document_listing/document_listing_000349.jsp&mid=WC0b01ac05805083eb
12Перелік заяв, які були відкликані, можна знайти за цим посиланням:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/wapp_search.jsp&mid=WC0b01ac058001d128.
http://www.ema.europa.eu/ema/index.isp?curl=pages/medicines/landing/wapp search.isp&mid=WC0b01ac058001d128.
13Європейські звіти за результатами публічної оцінки (EPAR) лікарських засобів для використання людиною, що публікуються EMA, можна знайти за наступним посиланням:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124.
VII.2. КОМУНІКАЦІЯ МІЖ ДЕЦ ТА ЗАЯВНИКАМИ
567. Постійний зворотний зв’язок від заявників щодо можливих недоліків у процедурі реєстрації є необхідним для того, щоб вдосконалити саму процедуру і зробити її більш зручною для користувачів. Зв’язок між ДЕЦ в якості органу, який здійснює експертизу, з одного боку, і заявниками, з іншого боку, відіграє важливу роль в розвитку системи реєстрації лікарських засобів.
568. Чим більш прозорим і інформативним є рівень спілкування, тим менше буде виникати питань у заявників щодо ефективності системи і її професійного рівня. Це також дасть заявникам краще розуміння всіх особливостей процесу реєстрації, а для ДЕЦ – інформацію про оцінку процедури реєстрації заявниками.
569. Основні фактори, які вливають на неналежний рівень комунікації між заявниками та ДЕЦ, полягають в наступному:
- обмежений обсяг інформації (рекомендацій), доступний заявникам;
- право на отримання консультації перед поданням документів не працює на практиці, платна наукова консультація не є доступною;
- неналежний рівень комунікації між ДЕЦ і заявниками.
VII. 2.1. Обмежений обсяг інформації (рекомендацій), доступний заявникам, обмежена кількість документів-зразків
570. Офіційний сайт ДЕЦ містить дуже обмежену інформацію щодо особливостей подачі документів в ДЕЦ та інших важливих аспектів процедури реєстрації. В мережі Інтернет доступні тільки кілька шаблонів документів, необхідних для комунікації з ДЕЦ.
Рекомендації:
Додати на офіційний веб-сайт ДЕЦ рекомендації щодо здійснення кожної процедури, а також секцію FAQ та відповідні зразки всіх документів, які використовуються заявниками у межах процедури.
VII.2.2. Право на отримання консультації перед поданням документів не працює на практиці, платна наукова консультація не є доступною
571. Процедура реєстрації лікарських засобів включає право заявників на отримання консультації від експертів ДЕЦ до подачі документів на експертизу.
Пункт 1 Розділу IV Наказу МОЗ №426:
Проведенню експертизи за бажанням заявника передує надання [ДЕЦ] безкоштовних консультацій з питань державної реєстрації лікарських засобів.
572. Право на отримання безкоштовної консультації експерта перед поданням документів не працює на практиці, що було підтверджено і заявниками, і представниками ДЕЦ.
573. Крім того, в Україні немає усталеної практики надання платних наукових консультацій або будь-якої іншої аналогічної процедури, що дозволила б заявнику отримати детальну консультацію від експертів ДЕЦ до подачі документів на експертизу.
Рекомендації:
Застосовувати на практиці положення Наказу МОЗ №426 стосовно права отримання консультації від експертів ДЕЦ перед подачею документів на експертизу.
Ввести можливість платної наукової консультації для заявників.
Приклад для порівняння (Німеччина)
Відповідно до німецького законодавства, наукова консультація має на меті сприяти більш ефективній розробці лікарського засобу і надати можливість заявнику пройти майбутню процедуру реєстрації лікарського засобу в прискореному порядку.
Наукова консультація надається Федеральним інститутом лікарських засобів та виробів медичного призначення (Bundesinstitut für Arzneimittel und Medizinprodukte, BfArM).
Наукові консультації можуть надаватися в будь-який час до або після початкової реєстрації лікарського засобу, і може охоплювати питання, пов’язані з:
- фармацевтичною якістю (включаючи біологічні та біотехнологічні аспекти),
- оцінкою виробів медичного призначення,
- розробкою і проведенням неклінічних досліджень та клінічних випробувань лікарських засобів та виробів медичного призначення (включаючи біостатистику),
- фармаконаглядом та планом управління ризиками.
Обсяг консультації визначається переліком питань, який надається заявником щодо програми розробки лікарського засобу або виробу медичного призначення.
Питання заявника повинні обґрунтовуватися тим, що вони або не врегульовані чинним законодавством, або що конкретні положення законодавства вимагають інтерпретації.
На додаток до переліку питань заявник повинен надати актуальну супутню документацію для обґрунтування своєї позиції.
Запит на надання консультації повинен подаватися в письмовій формі з використанням аплікаційної форми, доступної на веб-сайті BfArM.
Документи, які необхідно подати:
- супровідний лист:повинен узагальнювати основну інформацію щодозапитуваної консультації, включаючи ім’я та адресу заявника та опис продукту;
- аплікаційна форма: повинна включати розширену інформацію про заявника, порядок розгляду заяви, якому заявник віддає перевагу (письмово чи з проведенням слухань), інформацію про продукцію, обсяг запитуваної наукової консультації;
- перелік питань;
- інформаційна записка;
- список представників заявника, що будуть присутні на слуханні – група від 4 до 7 чоловік максимум. Рекомендується включати до цього списку саме тих внутрішніх експертів та зовнішніх консультантів (якщо такі є), які ознайомлені з відповідним проектом і які можуть брати участь в обговоренні.
BfArM може надавати консультації або шляхом безпосереднього спілкування з заявником (у формі консультативних слухань) або в письмовій формі.
Консультативне слухання триває не більше двох годин. Спочатку заявник виступає з короткою презентацією суті проблеми. Потім обговорюється список питань заявника.
BfArM надає консультацію в письмовій формі в тих випадках, коли питання заявника стосуються компетенції конкретного наукового відділу BfArM. Заявник може очікувати відповідь на свій запит протягом чотирьох-шести тижнів з дня отримання BfArM відповідних документів, в залежності від складності запиту.
Вартість консультації залежить від фактичного використання ресурсів BfArM та може становити від 1020 до 4600 євро за консультацію.
VII.2.3. Неналежний рівень комунікації між ДЕЦ та заявниками
574. Наказ МОЗ № 426 містить декілька положень, спрямованих на покращення рівня взаємодії між заявниками та регуляторними органами.
Пункт 5 Розділу IV Наказу МОЗ №426
Під час проведення спеціалізованої експертизи матеріалів реєстраційного досьє … кожна з експертних комісій Центру може дворазово запитати у заявника необхідні матеріали, що доповнюють дані з ефективності, безпеки та якості лікарського засобу… , не допускається нових запитів щодо матеріалів, які вже розглядалися експертом, за винятком випадків, коли додаткові матеріали надані не в повному обсязі.
575. Інколи саме заявники не дають належних відповідей на запити ДЕЦ, або їх представляють особи, які не мають професійного досвіду в сфері реєстрації лікарських засобів, і т.д.
576. Щоб усунути ці проблеми, необхідні нові методи комунікації між заявниками та ДЕЦ, наприклад, спеціалізовані воркшопи для спеціалістів з питань реєстрації від заявників. Це дозволить вдосконалити розуміння заявниками процесу реєстрації лікарських засобів та посилить їх впевненість в роботі відповідних регуляторних органів.
Рекомендації:
Розробити і впровадити для заявників і їх фахівців тренінги/освітні курси ДЕЦ з фінальним екзаменом, що дозволить учасникам отримати сертифікат по завершенню (в результаті, сертифікат менеджера з регуляторних питань має розглядатися працедавцями як перевага).
VII.3. ВІДСУТНІСТЬ ПРОЗОРОСТІ В РОБОТІ РЕГУЛЯТОРНИХ ОРГАНІВ
577. У зв’язку з непрозорою роботою МОЗ, ДЕЦ та Держлікслужби, у заявників та громадськості виникає багато запитань щодо процедури реєстрації.
578. На прозорість роботи органів системи державної реєстрації лікарських засобів вливають наступні фактори:
- необгрунтовані обмеження доступу до інформації;
- незрозумілий розподіл повноважень всередині ДЕЦ;
- відсутність загальнодоступної інформації про експертів.
VII.3.1. Необґрунтовані обмеження доступу до інформації
579. Під час підготовки цієї доповіді Експерти отримали сотні сторінок інформації про внутрішні процедури реєстрації, доступ до якої обмежений МОЗ та ДЕЦ.
580. Не зовсім зрозуміло, чому інформація про внутрішні процедури реєстрації, перереєстрації та внесення змін до реєстраційних матеріалів є конфіденційною.
581. Оскільки ця інформація є конфіденційною, заявники не мають чіткого розуміння процедури реєстрації. Це в багатьох випадках призводить до непорозумінь в комунікації між заявниками та ДЕЦ.
582. Внутрішні документи ДЕЦ описують відповідні процедури більш детально, аніж Наказ МОЗ №426, доступ до якого є відкритим. Ці внутрішні документи також визначають підрозділи ДЕЦ, відповідальні за конкретний етап процедури реєстрації.
583. Наприклад, Наказ МОЗ №426 не передбачає існування Науково-експертної ради та Науково-технічної ради ДЕЦ, які на практиці приймають основні рішення під час процедури реєстрації.
Пункт 7 Розділу IV Наказу МОЗ №426:
Результати проведених експертиз розглядаються під час засідань відповідного дорадчого органу [ДЕЦ] за участі голів та заступників голів робочих експертних груп відповідно до напрямів.
За результатами засідань надаються рекомендації щодо складання [ДЕЦ] для МОЗ рекомендацій про можливість державної реєстрації лікарських засобів.
584. ДЕЦ має детальні положення про Науково-експертну раду та Науково-технічну раду, які встановлюють їх склад, повноваження та процедури прийняття рішень.
Рекомендації:
Доступ до документів, які визначають внутрішні процедури ДЕЦ, Держлікслужби, МОЗ, не повинен бути обмеженим.
VII.3.2. Нечіткий розподіл повноважень
585. Оскільки внутрішні процедури ДЕЦ є конфіденційними, відсутня загальнодоступна інформація про розподіл повноважень між підрозділами ДЕЦ.
586. Основне питання, яка виникає, полягає в тому, хто приймає остаточне рішення рекомендувати лікарський засіб до реєстрації, перереєстрації, чи внесення змін до реєстраційних матеріалів.
587. Після завершення експертизи експерти готують проект рішення рекомендувати лікарський засіб до реєстрації. Проекти таких рішень і результати експертизи перевіряються дорадчим органом ДЕЦ. Дорадчий орган схвалює або не схвалює позицію експертів.
588. В такий спосіб розмивається відповідальність за рішення рекомендувати чи не рекомендувати лікарський засіб до реєстрації, перереєстрації, внесення змін до реєстраційних матеріалів.
VII.3.3. Відсутність інформації про експертів
589. Відсутня загальнодоступна інформація про внутрішніх і зовнішніх експертів, які проводять експертизу реєстраційних матеріалів. Заявники не володіють інформацією про освіту чи досвід роботи експерта, призначеного здійснювати експертизу досьє їх лікарського засобу.
590. Реєстрація лікарських засобів відіграє дуже важливу роль для держави. У зв’язку з цим, список експертів, інформація про їх освіту та досвід роботи повинні бути загальнодоступними на офіційному веб-сайті ДЕЦ. Така інформація буде корисною для заявників та громадськості, та може використовуватися для цілей громадського контролю.
591. Експерти, які здійснюють експертизу реєстраційних матеріалів, повинні бути незалежними та не мати конфлікту інтересів. Загальнодоступна інформація про експертів, викладена в зрозумілій формі, допоможе заявникам та громадськості слідкувати за виконанням цих вимог.
592. На веб-сайті EMA кожен може переглянути інформацію про всіх експертів14. На веб- сайті зазначається контактна інформація про експерта, його резюме (curriculum vitae), публічна заява про відсутність зацікавленості та зобов’язання зберігати конфіденційність.
___________________
14Список діючих членів Комітету з лікарських засобів для використання людиною (CHMP) та їх заступників можна переглянути за цим посиланням:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/contacts/2010/02/people_listing_000002.jsp&mid=WC0b01ac0580028c7c
Рекомендації:
Публікувати на веб-сайті ДЕЦ довідкову інформацію про внутрішніх та зовнішніх експертів, які проводять експертизу реєстраційних матеріалів, беручи за приклад EMA.
VIII. СПОСТЕРЕЖЕННЯ У СФЕРІ ФІНАНСУВАННЯ
VIII.1. ВІДНОСНО НИЗЬКА ВАРТІСТЬ ДЕРЖАВНОЇ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ
593. Загальна вартість державної реєстрації лікарських засобів в Україні включає два елементи:
- збір за державну реєстрацію лікарського засобу, що сплачується в державний бюджет;
- вартість послуг з проведення різних видів експертизи, що сплачується ДЕЦ напряму на підставі договору з заявником.
594. Розмір реєстраційного збору встановлюється Постановою КМУ №376:
- за державну реєстрацію лікарських засобів, у тому числі медичних імунобіологічних препаратів, крім радіоактивних лікарських засобів, діагностичних засобів, простих або складних (галенових) препаратів з рослинної лікарської сировини, – у сумі, еквівалентній 100 євро за кожну лікарську форму, 10 євро за кожну наступну дозу, 10 євро за кожну наступну упаковку лікарського засобу;
- за державну реєстрацію радіоактивних лікарських засобів, діагностичних засобів, простих або складних (галенових) препаратів з рослинної лікарської сировини, препаратів з донорської крові або плазми – у сумі, еквівалентній 25 євро за одне найменування, 5 євро за кожну наступну дозу, 5 євро за кожну наступну упаковку лікарського засобу.
595. Вартість послуг з проведення експертизи реєстраційних досьє встановлюється у Наказі ДЕЦ №139 та становить від 500 грн. за зміну типу ІА до 80 880 грн. за спеціалізовану експертизу матеріалів клінічних випробувань.
596. У порівнянні з європейськими країнами, вартість державної реєстрації лікарських засобів в Україні є відносно низькою.
597. Для прикладу, нижче можна побачити вартість державної реєстрації генеричного лікарського засобу в різних європейських країнах (за національною процедурою) в порівнянні з Україною15:
| Країна | Вартість, в євро |
| Польща | 5.234,5 |
| Литва | 3.199 |
| Латвія | 4.268,62 |
| Україна | 1.962,72 + 120 за 1 дозу та 1 тип упаковки |
______________
15Інформація стосовно дійсної вартості державної реєстрації лікарського засобу взята з офіційних джерел:
Польща – http://www.urpl.gov.pl/sites/default/files/zalaczniki/ENG table fees.pdf.
Литва – https://www.e-tar.lt/portal/lt/legalAct/bd022be03f5e11e58568ed613eb39a73.
Латвія – https://www.zva.gov.lv//doc upl/SAM-pricelist-20160108.pdf.
Україна – http://www.dec.gov.ua/site/files/vutratu/dvp.pdf
598. Для збільшення фінансування ДЕЦ необхідно спланувати та запровадити поступове та обґрунтоване підвищення вартості експертизи.
599. Відповідно до відгуків заявників, збільшення вартості експертизи є для них прийнятним, якщо кошти, отримані в результаті такого збільшення, будуть спрямовані на покращення функціонування ДЕЦ та підвищення рівня якості послуг зі здійснення експертизи.
Рекомендації:
Спланувати поступове та обґрунтоване підвищення вартості експертизи.
Ввести преференційні умови (зменшену вартість експертизи) задля промоції певних видів лікарського засобу і/або типу заявника, наприклад:
(a) для малих та середніх підприємств,
(b) для препаратів-сиріт і лікарських засобів з педіатричними показаннями.
VІІІ.2. ВАРТІСТЬ ЕКСПЕРТИЗИ НЕ РЕГУЛЮЄТЬСЯ ЗАКОНОМ
600. Відповідно до чинного законодавства ДЕЦ не є державним органом, а має статус державного підприємства під управлінням МОЗ.
601. Факт того, що вартість експертизи не регулюється на рівні закону або нормативного акту, а встановлюється внутрішнім рішенням ДЕЦ, не суперечить українському законодавству. Нам також відомо, що історично регулювання вартості в наказах МОЗ було відмінено через невідповідність вимогам Податкового кодексу.
602. Для забезпечення прозорості, зрозумілості і передбачуваності тарифів ДЕЦ, рекомендується встановити хоча б/по меншій мірі максимальні ставки для всіх видів експертизи на рівні закону або підзаконного нормативно-правового акту. Це може потребувати відповідних змін в податкове законодавство.
Рекомендації:
Встановити максимальні ставки вартості всіх видів експертизи на рівні закону або підзаконного нормативно-правового акту
VIII.3. НЕЗНАЧНИЙ ОБСЯГ ІНВЕСТИЦІЙ В РОЗВИТОК СИСТЕМИ
603. Відповідно до інформації з відкритих джерел, діяльність ДЕЦ була прибутковою останні кілька років16. Не зважаючи на це, з різних причин не робилися значні інвестиції в розвиток процедури реєстрації лікарських засобів.
604. Для пришвидшення розвитку реєстраційної системи України необхідно покращити систему фінансового менеджменту, бюджету і планування, а також здійснити значні інвестиції в розвиток системи електронного урядування та електронного документообігу, інфраструктури, прозорості та комунікації, мотивації та розвитку персоналу.
605. Одним зі способів збільшити фінансування ДЕЦ є підвищення вартості державної реєстрації лікарських засобів. Інші опції також необхідно брати до уваги.
Рекомендації:
Знайти альтернативні джерела фінансування, окрім підвищення вартості експертизи.
Однією з таких альтернатив є надання платних наукових консультацій (див. пункт 571 вище)
Наукова консультація має на меті сприяти більш ефективній розробці лікарських засобів і пришвидшенню процедури їх реєстрації у майбутньому.
__________________________________________
16https://youcontrol.com.ua/contractor/?id=8983101#tab2
IX. СПОСТЕРЕЖЕННЯ У СФЕРІ ЛЮДСЬКИХ РЕСУРСІВ В РАМКАХ ДЕЦ
IX.1. РЕЗЮМЕ
606. У штаті ДЕЦ зараз числяться 482 штатні одиниці, з них 105 експертів (21% від загального числа співробітників). Станом на сьогодні, ДЕЦ додатково залучив 88 позаштатних експертів і приблизно 50 позаштатних співробітників у регіональних центрах фармаконагляду ДЕЦ.
607. Із 482 штатних одиниць Центру, 50 – це вакантні, в тому числі декретні позиції. За словами Тетяни Думенко, Директора Центру, ці вакансії стосуються передусім допоміжних функцій і не перешкоджають діяльності Центру.
608. Ми вважаємо, що ДЕЦ має достатньо людських ресурсів у допоміжних підрозділах, таких як адміністративний та фінансовий відділи. Разом з тим, Центр стикається з проблемами у рекрутингу і утримані висококваліфікованих спеціалістів, і в першу чергу експертів Центру, головним чином через низький рівень оплати праці.
609. Крім того, нещодавні зміни в законодавстві, зокрема запровадження спрощеного порядку реєстрації ЛЗ, створюють додаткові виклики для Центру з точки зору навантаження на його експертів. Для підвищення ефективності роботи експертів керівництвом Центру вже реалізовано ряд змін, зокрема змінено розподіл завдань між експертами відповідно до їх досвіду, поточного навантаження, а також обсягу матеріалів досьє.
610. Широке залучення Центром позаштатних експертів та колегіальних органів у процес реєстрації ЛЗ знижує брак досвіду в його штатних працівників. Проте, щоб уникнути можливого розмивання відповідальності, ДЕЦ слід обмежити використання Науково- експертної ради та Науково-технічної ради у процесі реєстрації ЛЗ до розгляду лише нестандартних та суперечливих випадків, коли штатним експертам може дійсно бракувати досвіду або знань.
611. Як і в багатьох інших державних підприємствах, структура ДЕЦ сукупно з низькими рівнями зарплат стимулює створення численних проміжних рівнів управління і позицій на кшталт “заступник керівника” і концентрації відповідальності на рівні Директора та Заступників Директора Центру. Ми позитивно оцінюємо зміни в організаційній структурі, запропоновані діючим керівництвом ДЕЦ, так як вони покликані перерозподілити відповідальність на керівників середньої ланки та спростити ланцюжок прийняття рішень.
IX.1. ПОТОЧНА ОРГАНІЗАЦІЙНА ТА HRСТРУКТУРА ДЕЦ
612. Структура Центру багаторазово змінювалася, у відповідь на зміни його керівництва, яке відбувається, як правило, разом зі змінами у Міністерстві. Поточний дизайн організаційної структури веде до концентрації контролю та відповідальності на верхніх рівнях управління Центру, зокрема. Ми вважаємо, що така структура може бути ефективним інструментом реалізації організаційних змін, або у кризових ситуаціях, проте не може бути ефективною в довгостроковій перспективі.
IX.1.1 Поточна організаційна структура ДЕЦ
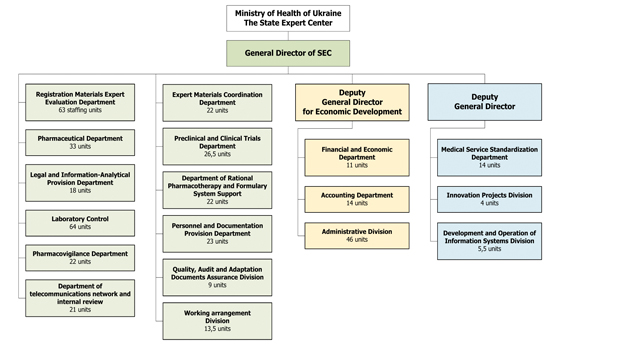
IX.1.2. Запропоновані зміни в організаційній структурі ДЕЦ (пропозиція ДЕЦ)
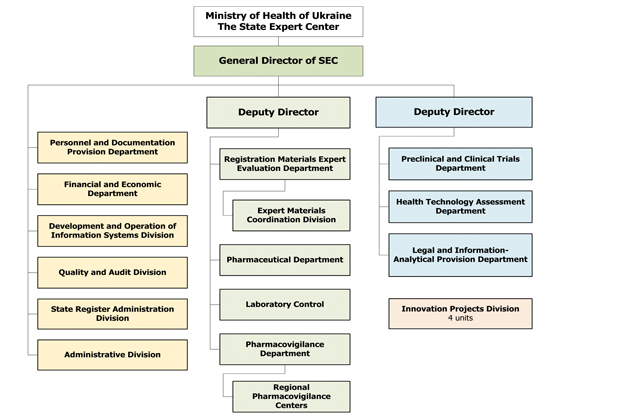
IX.1.3. Оцінка пропонованих змін
613. Запропоновані керівництвом зміни в організаційній структурі Центру, спрямовані на перерозподіл функцій і відповідальності від Директора ДЕЦ до середнього рівня управління – Заступників Директора і керівників департаментів та відділів ДЕЦ.
614. Крім того запропонована структура передбачає консолідацію основних функцій Центру в кількох підрозділах під управлінням функціональних заступників Директора Центру (один з них відповідає за усі функції, пов’язані з реєстрацією ЛЗ, інший – за напрям клінічних випробувань та пов’язані з ним функції), тоді як всі допоміжні підрозділи будуть підпорядковані безпосередньо Директору Центру.
615. Також запропонована структура передбачає виокремлення Відділу інноваційних проектів і передачу його в підпорядкування МОЗ, оскільки він виконує функції, не властиві ДЕЦ (розробка медичних стандартів, форм та іншої технічної документації для Міністерства).
616. Крім того ми звернули увагу на кілька випадків дублювання функцій, наприклад , між Департаментом оцінки технологій охорони здоров’я та Фармакологічним відділом. Узгодженнях таких випадків дублювання може підвищити ефективність процесів у Центрі, а також оптимізувати допоміжний персонал, що дозволить керівництву перерозподілити штатні одиниці і фонд оплати праці на ключових працівників і в першу чергу експертів Центру.
617. Детальна оцінка ефективності запропонованих змін в організаційній структурі ДЕЦ, а також виявлення дублюючих функцій вимагають подальшого вивчення внутрішніх процесів.
ІХ.2. РЕКРУТИНГ, ПРОФЕСІЙНА ПІДГОТОВКА І АТЕСТАЦІЯ ЕКСПЕРТІВ ЦЕНТРУ
618. За нашими оцінками рекрутингова діяльність Центру не є достатньо ефективною. Відділ кадрів заповнює значну частину вакансій за рахунок випускників, яких рекомендують позаштатні експерти (багато з них – вчені і викладачі у провідних медичних університетах). У рідкісних випадках, на вакансії ДЕЦ відгукуються кандидати з досвідом.
619. В Україні не існує університетських або поза-університетських навчальних програм, спрямованих на підготовку фахівців для роботи у сфері реєстрації ЛЗ. Винятком є спеціалізовані курси та інші навчальні заходи, які організовує ВООЗ та інші відповідні організації. Тому кандидати на позиції початкового рівня часто потребують базової підготовки та наставництва. У той же час, багато експертів Центру є досвідченими фармакологами і лікарями.
620. Крім того, у минулому менеджмент ДЕЦ не брав до уваги обсяг кожного конкретного досьє при розподіленні навантаження між експертами. Зараз, в рамках ДЕЦ запроваджено підхід, відповідно до якого більш досвідченіші експерти отримують більші за обсягом досьє.
ІХ.2.1. Використання позаштатних експертів
621. Для того, щоб розширити медичну експертизу Центр залучає на субпідрядної основі зовнішніх консультантів (переважно відомих лікарів з практичним досвідом). Вартість послуг позаштатних експертів для ДЕЦ є набагато нижчою у порівнянні зі штатними експертами Центру. В свою чергу, позаштатні експерти розглядають ці ролі більш як елемент престижу і визнання серед колег.
622. В цілому, в ДЕЦ працюють 88 позаштатних експертів, які об’єднані у 18 Консультаційно-експертних груп (кожна з них відповідає за окремий напрям медицини). Крім того, вони беруть участь у засіданнях двох дорадчих органів Центру (див. нижче).
ІХ.2.2. Використання колегіальних органів
623. ДЕЦ створив кілька консультативних органів, як засіб залучення різносторонньої експертизи від лікарів різного профілю щодо практичних аспектів застосування лікарських засобів. Оскільки питання, які піднімаються на засіданнях цих Рад, можуть вплинути на рішення експертів, ми розглядаємо їх як можливий фактор розмивання відповідальності щодо остаточного рішення рекомендувати конкретний ЛЗ для реєстрації Міністерством.
624. У процесі реєстрації ЛЗ задіяні наступні колегіальні органи:
- Техніко-експертна Комісія – проводить первісну оцінку для визначення переліку процедур, необхідних для експертизи матеріалів досьє кожного ЛЗ, який подається у ДЕЦ. Дана Комісія складається лише з працівників ДЕЦ.
- Науково-експертна Рада і Науково-технічна Рада проводять заключні слухання за результатами оцінки матеріалів досьє і висновків експертів щодо особливостей та наслідків використання ЛЗ.
625. Науково-експертна Рада приймає остаточні рішення щодо наступних рекомендацій для Міністерства охорони здоров’я:
- Реєстрація ЛЗ,
- Відмова в реєстрації ЛЗ
- Повна або тимчасова заборона на використання ЛЗ через припинення дії його реєстрації,
- Внесення змін до інструкції для медичного застосування ЛЗ,
- Затвердження початку клінічних досліджень ЛЗ та змін у матеріалах клінічного випробування,
- Інші пов’язані питання.
626. Науково-технічна Рада приймає остаточні рішення щодо наступних рекомендацій для Міністерства охорони здоров’я:
- Перереєстрації ЛЗ,
- Реєстрації (перереєстрації) субстанцій (активних фармацевтичних інгредієнтів),
- Внесення змін у матеріали, які не вимагають реєстрації ЛЗ,
- Відмови в реєстрації ЛЗ,
- Затвердження початку клінічних досліджень ЛЗ та правок у матеріалах клінічного випробування,
- Інші пов’язані питання.
627. Крім того, обидві Ради розглядають питання, пов’язані з припиненням експертизи матеріалів досьє ЛЗ і вирішують спірні випадки, що виникають в ході експертизи. Представники заявника можуть бути присутніми на слуханнях, щоб адресувати будь- які аргументи не рекомендувати МОЗ реєструвати ЛЗ.
628. Консультативно-Експертна Група – є головним консультативним органом, що бере участь в процесі оцінки матеріалів досьє ЛЗ. У ДЕЦ створено 18 таких груп. Вони охоплюють різні напрямки медицини, і складаються з відповідних експертів, в першу чергу позаштатних експертів, досвідчених практикуючих фармакологів і медичних працівників, які надають свої зауваження щодо особливостей і сторонніх ефектів застосування ЛЗ, якщо такі є. ДЕЦ надає копію досьє ЛЗ відповідній групі, щоб вона підготувала свої висновки паралельно з висновками штатних експертів ДЕЦ.
629. Представники КЕГ беруть участь в засіданнях Науково-Експертної Ради та Науково- Технічної Ради, і надають спеціалізовану консультаційну допомогу та експертні висновки при оцінці матеріалів досьє ЛЗ.
ІХ.3. ВИНАГОРОДИ ТА ІНШІ ІНСТРУМЕНТИ МОТИВАЦІЇ В ДЕЦ
630. Середній дохід експертів ДЕЦ коливається від 10 до 11,5 тис. грн. до вирахування податків, що, з врахуванням недавньої девальвації гривні становить від 345 до 397 євро17. Це як мінімум в двічі менше ринкового рівня і втричі менше, ніж 3 роки тому. Варто також відзначити, що керівництво ДЕЦ не переглядало рівні винагороди протягом останніх п’яти років.
631. Недавнє зниження заробітної плати у валютному виразі є основною причиною зростання плинності кадрів. Це створює найбільші проблеми для фармацевтичного Департаменту ДЕЦ, де співробітники можуть порівняно легко знайти роботу провізорів/ технологів у місцевих та іноземних фармацевтичних компаніях. Серед іншого це також є причиною того, чому більшість експертів Центру є лікарями, порівняно з фармакологами.
632. Разом з тим, базовий оклад працівників ДЕЦ є одним із найвищих у державному секторі системи охорони здоров’я України. За словами Тетяни Думенко, вони можуть підвищити зарплати до 30% за умови збереження прибутковості Центри. Міністерство має затвердити відповідні зміни в бюджеті і зарплатній відомості ДЕЦ.
633. Змінна частина винагороди експертів (виплачується щомісяця і щокварталу) становить до 100% від базової винагороди. Із загального бонусу 60% – це щомісячна премія за виконання обов’язків експерта та належного проходження процесу реєстрації. Керівник кожного відділу розподіляє премію серед співробітників на основі оцінки ним їх відносного робочого навантаження і вкладу в загальні результати за період. Ще 40% змінної винагороди – це надбавки, які залежать від ряду факторів, таких як стаж роботи.
_________________
17Ми зібрали інформацію про рівні заробітних плат та кадрову політику ДЕЦ станом на 31 серпня2016 року. Ми зібрали фінансові дані в українській гривні і конвертували їх у євро заміжбанківським курсом станом на 31 серпня 2016 року
634. Оскільки бонуси, які виплачуються у ДЕЦ, не прив’язані до об’єктивних показників ефективності і вони рідко не виплачуються, лише у разі суттєвого порушення експертом своїх обов’язків, у більшості випадків люди розглядають їх як обов’язкову частину заробітної плати. Тому, вони поки що не є повноцінним інструментом управління ефективністю, і ми розглядаємо його як частину базової винагороди.
635. Робота експертів Центру поки що не прив’язана до набору ключових показників ефективності. В ДЕЦ відсутня комплексна система управління ефективністю. Строки проведення процедур оцінки матеріалів досьє відстежуються вручну керівниками структурних підрозділів ДЕЦ. Можливе відставання у реагуванні керівництва Центра на затримки в процесі реєстрації ЛЗ, оскільки вони обговорюються на щотижневій нараді керівництва Центру у понеділок.
636. Керівництво ДЕЦ рідко застосовує негативні стимули. В ДЕЦ відсутні штрафи за помилки – у разі порушення працівником процедур, він отримає менший бонус і догану. Суворі дисциплінарні заходи у Центрі практично не застосовуються через низький рівень зарплат, адже керівництво прагне в першу чергу зберегти колектив.
637. Законодавство також не передбачає адміністративну відповідальність за порушення процедури реєстрації ЛЗ. Кримінальна відповідальність виникає лише у разі судових позовів проти ДЕЦ.
ІХ.4. ІНШІ КАДРОВІ ТА ОРГАНІЗАЦІЙНІ ПИТАННЯ
638. Державний експертний центр генерує достатньо коштів для фінансування своєї діяльності. Однак, за давно сформованою практикою, ДЕЦ зобов’язаний покривати різні витрати Міністерства охорони здоров’я, зокрема фінансувати: повсякденні офісні витрати Міністерства, організацію заходів та поїздок персоналу. Деякі із працівників ДЕЦ, що входять до штатного розпису та організаційної структури ДЕЦ (Адміністративний відділ), виконують роботу/ надають послуги МОЗ. Ми розглядаємо це як перешкоду на шляху до незалежності Центру, прозорості його взаємодії з Міністерством та іншими органами в українській системі охорони здоров’я, а також якості управлінських рішень, що підтримують процес реєстрації ЛЗ.
639. Ми пропонуємо перемістити відповідний персонал до відокремленого підрозділу для подальшої передачі його у Міністерство. Підтримка існуючої системи буде утримувати Центр від підвищення власної ефективності.
640. Керівництво ДЕЦ позитивно сприймає ідею створення органу громадського нагляду (Громадської ради) для підвищення прозорості його діяльності та суспільного іміджу. Вони визнають вимогу громадськості до розкриття Центром своєї неконфіденційної інформації (наприклад, публікації т. зв. «помаранчевої книги»). Тим не менш, підготовка відповідних матеріалів повинна бути ретельно спланована, щоб відповідати поточним можливостям Центру і узгоджуватися з його зобов’язаннями про нерозголошення конфіденційної інформації.
X. ВИКОРИСТАННЯ ІНФОРМАЦІЙНИХ СИСТЕМ У ПРОЦЕСІ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ В УКРАЇНІ.
641. Мета цього розділу полягає в визначенні рівня супроводу інформаційних систем в рамках процесу реєстрації лікарських засобів в Україні, враховуючи ефективність, обсяг функціональності, поточні ризики, недоліки та можливості до вдосконалення.
642. Цей розділ Звіту було підготовлено на основі попереднього аналізу документації, доступної консультантам, та співбесід, проведених у період з 14.06.2016 по 16.06.2016 та з 05.07.2016 по 07.07.2016 зі співробітниками ДЕЦ, Держлікслужби і Асоціації представників міжнародних фармацевтичних виробників України (AIPM). Ця доповідь зосереджена, в основному, на функціонуванні інформаційних систем, що підтримуються та використовуються ДЕЦ.
X.1. ВСТУПНІ ПРИМІТКИ
X. 1.1. Застереження
643. Доповідь обмежується інформацією, яка була доступна для консультантів на час підготовки цього документа. Процеси, функціональність інформаційних систем, документи та інші дані, які збирали та вивчали консультанти, відбиралися методом вибірки з урахуванням розуміння важливості відповідних процесів. Огляд функціональності інформаційної системи ДЕЦ щодо експертного супроводу лікарських засобів (MES) було проведено в тестовому середовищі. Консультаційний проект не включав в себе технічне тестування інформаційних систем, тому спостереження і висновки, що стосуються технічних можливостей систем, зроблено відповідно до інформації, отриманої в ході інтерв’ю.
644. У доповіді пропонується ряд удосконалень процесу реєстрації лікарських засобів з використанням сучасних інформаційних технологій та заміною існуючих інформаційних систем. Ці вдосконалення можуть бути обмежені наявною законодавчою базою і організаційною структурою процесу. Для підготовки ефективного плану реалізації рекомендацій першочергово необхідно узгодити внесення змін до діючої нормативно-правової бази.
645. Деякі рекомендації підготовлені з урахуванням майбутніх потреб та статусу процесу реєстрації лікарських засобів у рамках загальної структури інформаційних систем в області охорони здоров’ я. Подальша розробка таких рекомендацій не входить до предмету цього Звіту/консультаційного проекту.
X.2. ПОПЕРЕДНІ ВИСНОВКИ
646. Більшість заявників у ході опитування, проведеного консультантами, визнають переваги впровадження електронного обміну даними з ДЕЦ і, можливо, – системи електронного подання реєстраційних досьє. Перехід від паперового процесу реєстрації до процесів, що використовують електронні документи та інформаційні системи, дозволить підвищити продуктивність, ефективність та прозорість процедури реєстрації. Процеси, повністю засновані на використанні інформаційних систем, дозволять встановлювати та контролювати відповідні ключові показники ефективності, що в результаті дозволить краще оцінювати ефективність та приймати відповідні управлінські рішення задля підвищення якості обслуговування клієнтів. Забезпечення двосторонньої електронної комунікації між ДЕЦ і заявниками за допомогою вдосконаленої системи візуалізації інформації (або нового порталу ДЕЦ) також поліпшить досвід заявників і сприятиме покращенню обслуговування.
647. Необхідна належна правова база для використання електронного обміну даними, що може включати використання вдосконалених цифрових підписів або двосторонніх угод між ДЕЦ та заявниками щодо автентифікації та авторизації електронного подання документів. Перехід до електронної обробки даних буде не тільки ІТ- проектом, а й потребуватиме відповідних комунікацій з усіма зацікавленими сторонами, підготовки кадрів та зв’язків з громадськістю. Використання технологій також визначатиме нові вимоги до надійності та доступності інфраструктури. У такому випадку, вирішальне значення має забезпечення резервних та безвідмовних засобів обробки інформації для ключових інформаційних систем, які будуть розташовуватися в різних локаціях.
648. Однією з ключових характеристик вдосконалених інформаційних систем є забезпечення автоматизованої інтеграції, яка дозволить усунути необхідність ручного введення даних, скоротити витрати та покращити доступність та надійність інформації. Інформація щодо реєстрації лікарських засобів є одним з ключових об’єктів, які використовуватимуться в інших інформаційних системах охорони здоров’я, включаючи статистичні дані, відшкодування витрат, видачу електронних рецептів, інформаційні системи лікарень, тощо. Для досягнення цих цілей важливо забезпечити доступні та клієнто-орієнтовані інтерфейси даних. Для цього необхідно розробити процеси кодування даних та управління класифікаторами.
649. Організації, які беруть участь у впровадженні нових інформаційних систем, повинні розробити та дотримуватися загальної ІТ-стратегії, яка забезпечить основу для внутрішнього планування та координації діяльності між різними органами. Очевидно, що власником та основною рушійною силою цієї стратегії має бути МОЗ. З огляду на обсяг і масштаби запропонованих змін, важливо забезпечити наявність політичної волі та взяття на себе відповідних зобов’язань вищим керівництвом та політиками щодо забезпечення наявності ресурсів та підтримки у прийнятті рішень для успішної реалізації проекту.
Х.3. ВИКОРИСТАННЯ ІНФОРМАЦІЙНИХ СИСТЕМ У ПРОЦЕДУРИ РЕЄСТРАЦІЇ ЛІКАРСЬКИХ ЗАСОБІВ В УКРАЇНІ. СПОСТЕРЕЖЕННЯ, ВИСНОВКИ ТА РЕКОМЕНДАЦІЇ
650. У стратегічних документах в сфері охорони здоров’я визначені кілька цілей, які стосуються розвитку ІТ систем, зокрема:
Програма економічних реформ в Україні на 2010-2014 рр. передбачає:
- Розробку та впровадження електронних реєстрів пацієнтів,
- Впровадження електронного документообігу в установах охорони здоров’я,
- Оптимізацію статистичної звітності в установах охорони здоров’я.
Національна програма охорони здоров’я передбачає:
- Розробку інтегрованих баз даних щодо надання послуг у сфері охорони здоров’я та стану здоров’я населення;
- Розробку та впровадження електронних реєстрів пацієнтів, електронного документообігу в установах охорони здоров’я, системи електронних рецептів, електронних листів очікування пацієнтів, реєстрів установ охорони здоров’я та спеціалістів сфери охорони здоров’я.
651. Однак, практичне впровадження інформаційних систем залишається повільним переважно через юридичні питання та відсутність співпраці між відповідними органами. При плануванні ІТ-проектів надзвичайно важливо пам’ятати про необхідність технічної та семантичної інтеграції з метою забезпечення взаємодії цих систем та повторного використання даних. Наприклад, дані з реєстрації лікарських засобів повинні у подальшому використовуватися для видачі електронного рецепта, статистичних цілей, відшкодування витрат та ін. Крім того, існує потреба в створенні реєстрів аптек, фармацевтів, фахівців первинної медичної допомоги, інших закладів охорони здоров’я, тощо. Тому вкрай важливо визначити ці питання під час планування загальної електронної структури в області охорони здоров’я та забезпечення інтеграції між різними проектами та програмами.
652. На схемі нижче (Рисунок 1) представлено загальний потік даних за нинішнього процесу реєстрації лікарських засобів:
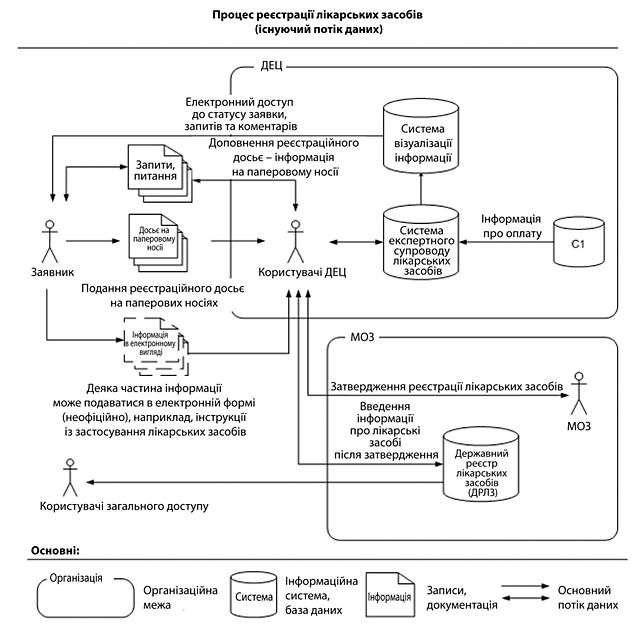
Рисунок 1. Процес реєстрації лікарських засобів (існуюча структура)18
653. У наступних розділах цієї доповіді коротко представлено результати, висновки та рекомендації стосовно інформаційних систем, що використовуються в межах цих процесів.
Х.3.1. Структура процесів реєстрації лікарських засобів. СОПи
(а) Спостереження
654. Процеси реєстрації лікарських засобів визначаються в так званих «стандартних операційних процедурах» – СОПах. Процедури задокументовані та затверджені на відповідному рівні.
__________________
18 Примітка: Організаційні межі, представлені на малюнку, є логічними. Власником ДРЛЗ є МОЗ, але система використовується в приміщеннях ДЕЦ. Детальний опис див. у розділі X.
655. Кожна СОП містить докладний опис етапів процесу, представлений у вигляді таблиці, відповідальних осіб та строків виконання для кожного етапу процесу. Додаткова інформація – форми, контрольні переліки – наведена в додатках до процедур.
656. В описі процесу в СОП немає чітко прописаних підстав для переривання/ відновлення процесів та алгоритму дій для виняткових ситуацій. Інформація, подана у формі таблиці, створює враження, що всі етапи процесу виконуються в лінійному порядку. Дана форма не допускає зазначення розгалуження процесів та етапів, які можуть виконуватися паралельно. Така інформація також не включена в коментарях.
657. Описи окремих етапів процесу не містять посилань на конкретні законодавчі акти, стандарти та інші процедури. Перелік актів застосовного законодавства наведено в окремих розділах процедури, але не зазначено зв’язку між окремими вимогами законодавства та етапами процесу (однак, певний зв’язок передбачено в контрольних переліках, що додаються до СОП). Такий підхід містить високий ризик того, що трактування законодавства буде залежати від індивідуальних знань, навичок та підходу окремого експерта. СОПи самі по собі без доступу до законодавчої бази не можуть використовуватися в якості єдиного інструменту для організації та контролю роботи експертів, і таким чином, не тільки ускладнюють організацію та нагляд, а і не полегшують роботу експертів та вимагають додаткового часу для роботи з ними.
658. Форми, шаблони і контрольні переліки, зазначені в додатках до СОП, розроблені для документообігу на паперових носіях. Інформація кожного попереднього етапу процесу переходить до наступного етапу за допомогою паперової документації. Інформаційна система використовується для відстеження процесу, але не для керування ним.
(b) Висновки, вплив та рекомендації
659. Кращі практики процесів документообігу полягають в використанні як текстових, так і графічних форм. Графічне подання інформації у вигляді блок-схеми є найкращим варіантом для демонстрації послідовності та розгалуженості етапів процесу, тоді як текст може використовуватися для надання додаткової інформації, необхідної для трактування та точного визначення умов обробки.
660. Блок-схеми вперше були введені Франком Гілбертом у 1921 році і спочатку називалися «технологічними схемами». Існують різні методи та варіанти графічного відображення блок-схем. Найбільш поширеними19 є, зокрема, такі:
«Основна схема послідовності операцій», що базується на стандарті GС20-8152-1 – «Методика обробки даних ІБМ: методика створення блок-схем». З деякими поправками цей стандарт на практиці досі використовується найчастіше для аналізу та документування процесів;
BPMN- система умовних позначень (нотацій) для моделювання бізнес-процесів;
IDEF0 – методологія функціонального моделювання та графічна нотація, призначена для формалізації та опису бізнес-процесів.
______________
19Цей перелік не є вичерпним.
661. СОПи повинні бути досить детальними для можливості виконання процесу без використання додаткових джерел даних. Це означає необхідність включення законодавчих положень до технологічної документації на такому рівні, щоб особа, яка виконує процес, могла використовувати СОП без пошуку законодавчих положень. Структура СОПів також має враховувати можливе використання документації процесу для вибору, розробки та адаптації програмного забезпечення в майбутньому.
662. Слід встановити вимоги до СОПів з розробки інших СОПів.
663. На відміну від процесу, заснованого на паперових носіях, майбутні процеси необхідно розробляти з використанням концепції процесу, що керується за допомогою системи. Завдяки цій концепції процес зберігає всю інформацію в базі даних. Одразу після введення та перевірки цієї інформації вона має бути доступною для всіх інших користувачів у рамках процесу. Завдяки використанню концепції процесу, що керується за допомогою системи, не повинно виникати вираженої необхідності друкувати та підтримувати паралельний потік паперових документів за межами інформаційної системи.
X.3.2. Система експертного супроводу лікарських засобів (MES)
(a) Спостереження
Огляд функціональності:
664. Система MES використовується для управління роботою експертів у межах ДЕЦ. Існує близько 200 користувачів системи MES (персонал ДЕЦ).
665. Система підтримує такі ключові процеси/ функції:
- відстеження замовлень (заявок на реєстрацію),
- розподілення та визначення завдань для експертів,
- визначення та контроль дотримання строків виконання,
- збереження звітів та проміжної інформації, наданої експертами
- додавання файлів звітів,
- збереження питань до заявників,
- збереження коментарів,
- збереження документів для цілей офіційного листування між ДЕЦ та заявниками під час процесу реєстрації,
- створення підсумкового звіту.
666. Система MES не обробляє дані реєстраційного досьє в електронному вигляді. МОЗ подає реєстраційні досьє в ДЕЦ на паперових носіях. Відповідні частини досьє копіюються та надсилаються експертам для виконання їх обов’язків в ДЕЦ на паперових носіях. Однак, деяка інформація також подається ДЕЦ в електронному форматі в якості додаткової робочої версії (зокрема, модулі 3, 4 та 5 для генеричних лікарських засобів). Заявник гарантує, що інформація, представлена в електронному вигляді, є достовірною та відповідає інформації на паперовому носії.
667. MES має функціональні можливості для відстеження строків виконання експертизи відповідно до визначеного графіку. Завдання для експертів призначаються напівавтоматично. Це означає, що головний відповідальний експерт розділяє роботу з проведення експертизи на частини. Кожна частина розподіляється на відповідну групу експертів – зазвичай підрозділам ДЕЦ. Потім керівник відповідного підрозділу розподіляє завдання на конкретного фахівця. Експерти можуть додавати коментарі та запити для отримання додаткової інформації стосовно предмету завдання. Система здатна генерувати офіційні листи заявникам та відстежувати вхідну/вихідну кореспонденцію. Додавання запиту про надання інформації до експертизи призупиняє перебіг процесу до отримання відповіді на запит. Характер призупинення залежить від ступеня важливості питання/коментарів. Якщо процес експертизи розгалужений, то перебіг строків призупиняється тільки для визначеної частини експертизи. Статус перебігу експертизи та питання, що виникли, відображаються заявникам через систему візуалізації інформації (СВ, див. опис нижче). Експерти надають свої звіти у вигляді файлів формату Microsoft Word або PDF, що прикріплюються до реєстраційного досьє. Остаточний звіт формує головний експерт, який є відповідальним протягом всього процесу реєстрації.
668. Система не має функціональних можливостей для реєстрації часу, витраченого експертами, або іншої інформації, яка могла б сприяти ефективності використання часу та ресурсів під час експертизи. Проте, можливо створити звіт з зазначенням експертів, які працювали над конкретною експертизою, та звіт з зазначенням кількості експертиз, проведених кожним експертом.
669. В системі частково вбудований функціонал електронного підпису, але він не використовується. Згідно з інформацією, наданою персоналом ДЕЦ під час інтерв’ю, ця функція потребує доповнення для її повноцінного використання.
Технології, що використовуються, та розробка системи:
670. Систему MES було розроблено з використанням мови C# та структури додатків .Net4 зовнішньою компанією «E-life» і введено в 2012 році на заміну старого додатка [Visual] FoxPro. Компанія «E-life» є власником прав інтелектуальної власності (ПІВ) на програмне забезпечення і тому має обов’язково виконувати роботи з будь-яких модифікацій та інші роботи, що виникають з коду системи. Роботи з технічного супроводу системи MES виконуються персоналом ДЕЦ (7 посад, 2 вакантні) та обмежуються вилученням інформації і підготовкою звітів без самостійного внесення змін до коду системи MES та зміни структури MES даних. Наявна технічна специфікація (документ SRS рівня) системи, яка спочатку виступала в якості бази для договору з компанією «E-life», а потім була доповнена в ході розробки проекту.
Обладнання засоби та приміщення:
671. Система використовується в центрі даних ДЕЦ (в комп’ютерному залі) співробітниками ДЕЦ. Існують резервні технічні ресурси (надкомпактні сервери). Таке рішення пропонує лише обмежену ступінь безперервності бізнес-процесу, оскільки всі сервери розташовані в одному приміщенні. В даний час відсутнє місце для резервного копіювання системи на випадок, якщо первинні приміщення стануть непридатними для експлуатації (тобто у випадку пожежі, повені або в іншиханалогічних випадках). Наявне окреме середовище для тестування, яке використовується для перевірки змін у системі перед їх впровадженням. Технічна платформа (надкомпактні сервери) не оновлювалася з 2008 року, тобто з дня введення в експлуатацію. Згідно з інформацією наданою представниками ДЕЦ, підтримка ОЕМ технічної платформи буде припинена найближчим часом.
Обмін даними з іншими інформаційними системами:
672. Система MES має односпрямований інтерфейс з системою візуалізації інформації ДЕЦ (СВІ). СВ використовується для відображення статусу процесу реєстрації лікарського засобу для зовнішніх користувачів (детальний опис СВ див. далі). Інтерфейс реалізується за допомогою тригерів SQL.
673. Існує односпрямований інтерфейс з бухгалтерською інформаційною системою C1. Інтерфейс використовується для передачі інформації про оплату з програми C1 до системи MES. Інтерфейс реалізується за допомогою обміну даними у форматі XML.
674. Обмін даними між системою MES та Державним реєстром лікарських засобів України виконується вручну.
675. Система не має інтерфейсу з Реєстром юридичних осіб або іншими державними інформаційними системами для перевірки особи та статусу заявників. Тому, будь-яка необхідна інформація має бути включена до заявки у формі коментарів.
(b) Висновки, вплив та рекомендації
676. Організація в повній мірі не використовує можливості обробки електронних документів. Подання досьє в електронному вигляді (принаймні частково) може бути більш зручним для заявників. Електронна інформація краще підходить для обробки (можливості пошуку, доступність, одночасна доступність багатьом користувачам без копіювання, онлайн коментування, тощо) та повторного використання. Велика частина інформації, яка включена в реєстраційні досьє, попередньо існує в компанії- заявнику/компанії-виробнику в електронній формі та готується з використанням комп’ютеризованих систем. Вимога друкувати цю інформацію та надавати її в паперовому вигляді вимагає непотрібної ручної роботи та займає додатковий час. Згідно з результатами опитування, проведеного консультаційним консорціумом, тільки друк та перевірка друкованих матеріалів займає від 24 до 128 людино-годин з боку заявника. Доставка паперових документів вимагає додаткових 1-5 днів.
677. Більшість респондентів позитивно сприймають використання системи подання електронних документів до всіх або, принаймні, до деяких частин досьє (наприклад, до модулів 2-5 досьє). Це дозволить забезпечити більш ефективний зворотній зв’язок та обмін інформацією по відношенню до додаткових запитів і додаткової інформації (ця функція пов’язана з СВ).
678. При здійсненні електронного обміну даними між ДЕЦ та заявниками необхідно враховувати правові та технічні аспекти автентифікації та захисту електронної документації. Відповідно до частини 3 XVII поправки до Угоди про асоціацію між Європейським Союзом та його державами-членами, з одного боку, і Україною, з іншого боку, встановленим терміном для прийняття Регламенту №910/2014 Європейського парламенту та Ради від 23 липня 2014 року щодо електронноїідентифікації та трастових послуг для електронних трансакцій на внутрішньому ринку та скасування Директиви 1999/93/ЄС є 17.17.201720. Іншими варіантами реалізації обміну електронною інформацією є прийняття відповідних положень та укладення двосторонніх угод між ДЕЦ та заявниками.
679. Важливо зазначити, що обробка інформації повністю в електронному вигляді потребує більш високої ефективності та потужності IT-інфраструктури. Впровадження технічних засобів, ліцензій та інших ресурсів, необхідних для обробки електронної документації, повинно ретельно плануватися. Для забезпечення високої якості обслуговування необхідно постійно контролювати наявність та використання ресурсів інфраструктури під час впровадження змін та експлуатації нових систем.
680. Система не має функціональної можливості реєстрації часу, затраченого експертами, чи іншої інформації, яка б сприяла підвищенню ефективності процесів. Додання такої функціональності має вирішальне значення для можливості встановлення відповідних ключових показників ефективності та для вдосконалення процесів. Вбудована функція реєстрації часу є бажаною, оскільки вона забезпечить точний зв’язок між завданнями та кількістю зареєстрованих годин.
681. Подальші зміни та вдосконалення системи MES може здійснювати тільки одна компанія – «E-life», яка є власником відповідних ПІВ. Ця опція звужує конкуренцію в разі необхідності впровадження таких змін, та може привести до збільшення витрат на їх впровадження та до ризику одного постачальника. Організація повинна приділити належну увагу питанню здійснення модернізації одним постачальником або забезпечення можливості відкритої конкуренції в разі необхідності внесення суттєвих змін до системи MES. Слід також зазначити, що для деяких варіантів фінансування чітко вимагається, щоб організація-одержувач була власником відповідних ПІВ.
682. Організація спирається на єдиний центр обробки даних, розташований в приміщенні ДЕЦ. Це робить систему надзвичайно уразливою в разі знищення або виходу з експлуатації центру обробки даних з інших причин. Рівень поточного ризику не настільки високий, оскільки основна документація (досьє, комунікація з заявниками) зберігається на паперових носіях. Однак, з огляду на очікування повністю електронізованого процесу у майбутньому, рекомендується мати резервні засоби обробки даних в іншому місці. Технічна платформа повинна бути модернізована, щоб забезпечити подальшу підтримку OEM.
683. Обмін даними між системою MES та іншими системами забезпечується вручну. Це уповільнює процеси, підвищує вірогідність виникнення проблем, пов’язаних з ручним введенням даних (помилки, упущення), та вимагає постійної роботи працівників. Вимога до заявників надавати всю необхідну інформацію для перевірки заявок у досьє переносить витрати на отримання та підготовку необхідної інформації на заявників. Заявники виступають в якості посередників між ДЕЦ та іншими установами (наприклад, з Реєстром юридичних осіб). Інформація, отримана таким чином, можебути неточною через затримку у часі. Автоматизовані інтерфейси даних можуть покращити загальну ефективність процесу та знизити вищезазначені ризики.
__________________
20http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv:OJ.L_.2014.161.01.0003.01.ENG&toc=OJ:L:2014:161:TOC
684. Можлива структура інтеграції між оновленими системами MES, ДРЛЗ, СВ та іншими інформаційними системами в області охорони здоров’я наведена в Додатку B цієї доповіді.
X.3.3. Система візуалізації інформації ДЕЦ (СВ)
(a) Спостереження
Огляд функціональності системи:
685. СВ забезпечує односторонній зворотній зв’язок із заявниками стосовно:
- статусу процесу реєстрації лікарських засобів;
- запитів про надання додаткової інформації;
- коментарів;
- надання шаблонів документів для заявників.
686. Для заявників відсутні можливості зворотнього зв’язку щодо цих запитів, надання відповідей на запити та комунікації з експертами.
687. Система не відправляє повідомлення користувачам у разі появи нової інформації або зміни статусу заяви.
688. На даний час існує близько 200 активних користувачів системи. Всі користувачі реєструються на підставі письмового запиту. Форма реєстрації доступна на веб-сайті СВІ. Для реєстрації користувача компанія повинна завантажити шаблон, заповнити його та відправити до ДЕЦ у друкованому вигляді разом з довіреністю і супровідним листом. Реєстраційна інформація включає адресу електронної пошти та номер мобільного телефону користувача. Після отримання форми вона затверджується, і адміністратор ДЕЦ створює користувача. Користувачеві відправляється початковий пароль. Зв’язок між ДЕЦ і користувачами захищений за допомогою шифрування SSL.
689. Відповідно до інтерв’ю з представниками АІРМ, інформація, представлена в СВІ, не завжди є точною і своєчасною. Тому вона не вважається корисною. Це шкодить задоволеності користувачів якістю сервісу.
690. Подальша робота над цим аспектом включає 2 етапи:
- ми можемо почати з дослідження можливості системи (наприклад, статуси є дуже стислими та не надають заявнику достатньої інформації);
- далі можна досліджувати якість та своєчасність введення інформації експертами ДЕЦ.
Технології, що використовуються, та розробка системи:
691. Систему розроблено сторонньою компанією та впроваджено у 2014 році. На відміну від Система Експертизи, ПІВ на код СВ належить ДЕЦ.
692. Інтерфейс користувача розроблено мовою С#.
Технічні засоби та приміщення:
693. СВ має ті ж технічні ресурси, що і Система Експертизи (див. п. X.3.3).
Обмін даними з іншими інформаційними системами:
694. Інтерфейс між системою Система Експертизи та СВ реалізується за допомогою тригерів SQL (безпосередній доступ до таблиць MSSQLбази даних Системи Експертизи).
(b) Висновки, вплив та рекомендації
695. Забезпечити функцію електронного повідомлення користувачів. Ця опція здатна забезпечити негайні результати з точки зору вдосконалення зручності та простоти використання системи. ДЕЦ уже збирає контактну інформацію про користувачів системи. Надсилання електронних повідомлень про зміни статусу, запити та коментарі на електронну пошту та/або SMS дозволить скоротити часову затримку між датою публікації інформації та датою отримання цієї інформації користувачами. Ця опція є бажаною для користувачів системи відповідно до електронного опитування, проведеного консультантами. Опція також розглядалася в ДЕЦ, але її не було реалізовано через відсутність належної політичної волі для прийняття відповідних рішень.
696. Забезпечити самостійну реєстрацію користувачів або дворівневе адміністрування користувачів для заявників. Процес реєстрації користувача СВ за допомогою паперових носіїв забирає багато часу та ресурсів. Консультанти радять запровадити дворівневе адміністрування користувачів або процедуру самостійної реєстрації.
697. Функція дворівневого адміністрування означатиме, що ДЕЦ буде створювати та керувати тільки адміністраторами верхнього рівня від організацій-заявників. Потім адміністраторам організацій будуть надані права управління користувачами в своїх організаціях. Ця функція дозволяє знизити навантаження на адміністраторів ДЕЦ, скоротити час процедури реєстрації та скасувати реєстрацію користувача. Впровадження такої функції потребує укладання двосторонніх угод між ДЕЦ та організаціями-заявниками, в яких буде передбачатися відповідальність організацій щодо управління правами користувача.
698. Процедура самостійної реєстрації користувача буде також сприяти підвищенню ефективності процесу. Однак, в будь-якому випадку необхідна процедура забезпечення доступу до системи тільки авторизованих користувачів. Ця опція уже розглядалася в ДЕЦ, але її не було реалізовано через відсутність належної політичної волі для прийняття відповідних рішень.
699. Реалізувати двосторонній зв’язок із заявниками в електронній формі. Ця опція вже обговорювалася в розділі про можливі вдосконалення системи MES (див п. X.3.2 Звіту).
X.3.4. Державний реєстр лікарських засобів (ДРЛЗ)
(a) Спостереження
Огляд функціональності системи:
700. Система містить офіційну інформацію про зареєстровані лікарські засоби.
701. Власником ДРЛЗ є МОЗ. ДЕЦ, що діє в якості адміністратора інформації, відповідає за внесення даних та ведення реєстру. Інформація про реєстрацію (перереєстрацію та внесення змін до реєстраційних матеріалів) лікарських засобів вноситься після відповідного затвердження в МОЗ.
702. Права користувачів та рівні доступу:
- На даний час існує 4 користувача системи, які можуть вносити/змінювати інформацію (відділ адміністрування лікарських засобів). Для забезпечення достовірності інформації дотримується принцип «чотирьох очей»: введення даних завжди перевіряється та затверджується будь-яким іншим співробітником, аніж той, що ввів інформацію.
- Інші відділи ДЕЦ мають доступ до бази даних тільки для читання.
- Зовнішні користувачі мають обмежений доступ тільки для читання. Відкритий інтерфейс Реєстру доступний тут: http://www.drlz.com.ua/. Починаючи з 2010 року, було зроблено близько 8 мільйонів запитів на лікарські засоби через загальний веб- інтерфейс.
703. Існує намір надання особливого доступу до ДРЛЗ для митниці. Реалізацію цього наміру було призупинено через реорганізацію та зміни в управлінні ДЕЦ.
704. Реєстраційний номер лікарських засобів (номер реєстраційного посвідчення) формується з таких частин: групи лікарських засобів/форми/дозування. Код не містить інформації про упаковку (наприклад, 10 або 20 таблеток в упаковці).
705. Інформація про дозування та концентрацію надається в довільній формі. Одна і та ж інформація може вводитися по-різному та може бути непридатною для подальшої автоматизованої обробки (наприклад, «50 µg» – грецькою «мікро» або «50 мкг» – кирилицею; «50,00 г» – континентальний стиль десяткового числа + кирилиця або «50.00 §» – США/ англійський стиль десяткового числа)21.
706. Класифікатори системи (тобто МНН, АТХ) утримуються спеціалістами МОЗ.
707. ДРЛЗ не містить інформації про ціни та статистики використання лікарських засобів. Існує окремий реєстр цін на лікарські засоби, який ведуть спеціалісти МОЗ.
708. Офіційний веб-сайт ДРЛЗ також має функцію звітності про побічні ефекти/ відсутність ефективності лікарського засобу при його медичному застосуванні (форма № 137/о). Ця опція доступна для будь-якого (неавторизованого) користувача. Інформація, що повідомляється, далі обробляється в ДЕЦ (Департаментом післяреєстраційного моніторингу), а потім експертами Держлікслужби. Система незабезпечує зворотного зв’язку з особою, що надіслала повідомлення, стосовно результатів дослідження, проведеного експертами.
_______________
21У ході огляду системи ми знайшли тільки узгоджені між собою записи. Як видається, ДЕЦ має необхідні ручні процедури, дотримання яких дозволяє уникнути таких відхилень. Проте, наскільки нам відомо, деякі установи стикаються з проблемами якості відповідних даних. Потім такі дані не можуть використовуватися для автоматизованої обробки та звітності без внесення виправлень вручну. Елементи управління, вбудовані в систему, завжди забезпечують кращу якість даних, аніж процедури, що виконуються вручну.
709. Система не має функції реєстрації дозволів на проведення клінічних досліджень. Такі дозволи видаються МОЗ. Процес здійснюється за допомогою паперових носіїв.
Технології, що використовуються, та розробка системи:
710. Для системи використовується платформа IBM Domino Lotus Notes. Програмне забезпечення клієнта використовує систему WindowsXP. Вперше систему було введено в експлуатацію в 1996 році.
711. Розробка та супровід системи забезпечується двома аутсорсинговими програмістами. Протягом останніх двох років система не змінювалася і не вдосконалювалась.
Технічні засоби та приміщення:
712. Система використовується в приміщеннях ДЕЦ, тим самим маючи одну точку відмови з системою MES.
Обмін даними з іншими інформаційними системами:
713. Система не має інтерфейсу автоматичного обміну даними ані з системою MES, ані з іншими інформаційними системами.
(b) Висновки, вплив та рекомендації
714. Забезпечити вдосконалене кодування та опис інформації про лікарські засоби. Сучасний рівень кодування інформації (інформації про упаковку, дозування, концентрацію) обмежує використання ДРЛЗ для аналізу ринку, контролю цін, статистичних даних та інших способів використання цієї інформації.
715. Ми рекомендуємо:
- включити інформацію про упаковку (наприклад, 10 або 20 таблеток) до форми, що обробляється комп’ютером;
- включити інформацію про дозування та концентрацію до форми, що обробляється комп’ютером. Встановити відповідні правила та класифікатори для написання одиниць вимірювання та їх відповідного перекладу (наприклад, 5 міліграм = 5000 мікрограм).
716. Застарілі платформи розробки додатків. Платформа розробки додатків Lotus Domino колись була популярною. Але, за деякими джерелами, ця платформа втратила свою могутність, і відсоток її використання знизився з 50% в 1998 р. до 15% в 2014 р. Операційна система Windows XP, яка використовується для клієнтського програмного забезпечення, більше не підтримується корпорацією Майкрософт (з 8 квітня 2014 року). З огляду на зміни та нові розробки, на додаток до існуючих опцій системи необхідно дослідити та проаналізувати зміни платформи розробки додатків. Звичайними ризиками, пов’язаними з використанням застарілих платформ, є:
- можливість потрапити до технологічного тупику та в будь-якому випадку бути вимушеним списати інвестиції та почати нові розробки;
- наявність і вартість ресурсів розробки, оскільки вони можуть стати рідкістю на ринку;
- відсутність підтримки постачальника, включаючи відсутність оновлень безпеки.
717. Системи СВ та MES не мають автоматичного інтерфейсу. Це питання вже розглядалося вище та викладено в розділі VI.1 Звіту.
718. Інформація про реєстрацію лікарських засобів в значній мірі використовується в інших процесах в сфері охорони здоров’я (наприклад, в процесі виписування рецепту на лікарський засіб та подальшої реімбурсації його вартості). Тому наявність інформації про реєстрацію лікарських засобів має вирішальне значення для реалізації програми електронної системи охорони здоров’я. Створення відкритого інтерфейсу та послуг з передачі даних для інших інформаційних систем і організацій повинні бути частиною загальної стратегії інформаційної системи. Важлива частина цієї стратегії полягає в узгодженні належного кодування даних і використання класифікаторів, оскільки вони є основними компонентами автоматизованої обробки та аналізу.
719. Наявність відповідної інформації у митних та контролюючих органів може покращити ефективності їх роботи, що було визнано в ході інтерв’ю та електронного опитування.
X.3.5. Управління розвитку інформаційних систем
(a) Спостереження
720. Згідно з інформацією, отриманою в ході первинних інтерв’ю, видається, що кожна організація прагне розробляти свої власні інформаційні системи. Інтеграція та загальне використання інформаційних систем розвинуті недостатньо. Розвиток інформаційних систем фінансується на локальному рівні за рахунок бюджету відповідної установи. Бюджети складаються щорічно. Процес формування бюджету виглядає більше як збір потреб різних відомств та мало стосується належного планування, аналізу можливостей для вдосконалення та оцінки вартості майбутніх проектів. Чіткі правила прийому та реалізації ІТ-проектів відсутні. Наприклад, повідомлялося, що ІТ-відділ ДЕЦ вже розглянув можливість додавання процесу самостійної реєстрації користувача та повідомлення до СВІ, проте така можливість не була реалізована.
(b) Висновки, вплив та рекомендації
721. На цьому етапі ми б хотіли наголосити на дотриманні таких принципів та настанов:
722. Інформація про реєстрацію лікарських засобів має бути одним з наріжних каменів загальної структури електронної системи охорони здоров’я. Ця інформація є абсолютно необхідною для інформаційної системи з надання електронних рецептів, контролю застосування лікарських засобів, розробки системи закупівель лікарських засобів для установ у сфері охорони здоров’я, що фінансуються з коштів державного бюджету, реімбурсації вартості лікарських засобів, статистичних цілей, та ін.
723. Щоб ця інформація була корисною, вона має бути:
- надійною;
- своєчасно доступною, включаючи між комп’ютерний інтерфейс даних;
- стандартизованою (з точки зору структури, кодування, мови).
724. Інформаційні послуги з реєстрації лікарських засобів повинні реалізовуватися у відповідності з іншими електронними інформаційними системами в області охорони здоров’я.
725. Необхідно впровадити процес загального планування та прийняття рішень щодо інформаційних систем як між установами, так і всередині установ. Для впровадження такого процесу має бути чітке визначення понять «повноваження» та «прийняття рішень», які мають бути зафіксовані в єдиному документі планування (наприклад, «Стратегія розвитку інформаційних систем в галузі охорони здоров’я»).
726. У багатьох випадках користувачі інформації можуть знаходитися за межами організації, в той час як витрати на підготовку цієї інформації, вартість розробки, обслуговування та експлуатації інформаційних систем здійснюються всередині організації. Тобто ДЕЦ може витратити деяку суму на розробку процесу самостійної реєстрації користувача та системи повідомлень, але перевагу від цих змін будуть отримувати організації-заявники. Оскільки відносини між заявником та ДЕЦ не є повністю ринковими, то, швидше за все, таке вдосконалення не забезпечить збільшення доходу ДЕЦ, проте забезпечить покращення комунікації з заявниками.
727. Такі проблеми повинні враховуватися при виділенні бюджетних коштів та прийнятті рішень про пріоритети розвитку. Для доведення необхідності удосконалень можуть бути розроблені і використані різні ключові показники ефективності, зокрема, рівень задоволеності користувачів або час виходу на ринок.
Х.4. ЗАПРОПОНОВАНІ ЗМІНИ ТА ДІЇ
Х.4.1. Структура майбутніх інформаційних систем
728. На схемі нижче (Рисунок 2) представлено загальний потік даних запропонованої структури майбутніх інформаційних систем для процесу реєстрації лікарських засобів:
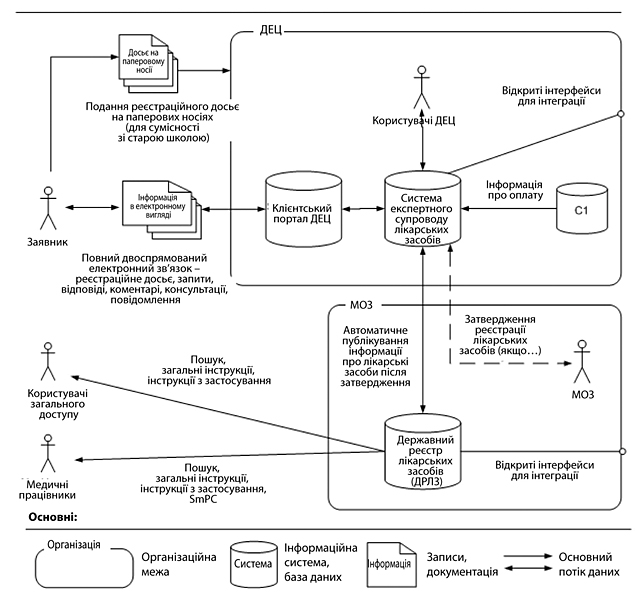
Рисунок 2. Процес реєстрації лікарських засобів (структура майбутніх інформаційних систем).
729. Необхідно впровадити новий портал ДЕЦ для заявників. Портал повинен забезпечити всі необхідні функції для комунікації між заявниками та ДЕЦ:
- подачу реєстраційних досьє в електронній формі,
- можливості для надання запитів, відповідей, коментарів та надсилання повідомлень,
- електронні засоби зв’язку повинні переважно замінити існуючий документообіг на паперових носіях.
730. Заявники можуть подавати паперові досьє протягом перехідного періоду, а також для забезпечення сумісності з попередніми версіями і безперервності бізнесу.
731. Схвалення реєстрації лікарських засобів повинно вводитися в систему посадовими особами МОЗ (якщо буде прийнято рішення залишити цей етап процесу). Післяавторизації система повинна автоматично публікувати інформацію про лікарські засоби в ДРЛЗ. Необхідно створити новий інтерфейс для обміну даними між MES та ДРЛЗ.
732. MES та ДРЛЗ повинні бути доповнені відкритими інтерфейсами, що дозволяють інтегрувати інші інформаційні системи (третіх сторін), тобто системи лікарень, аптек, електронних рецептів та статистики. Електронна інформація може використовуватися іншими установами відповідно до їх рівня авторизації, наприклад, митницею, Держлікслужбою, тощо.
733. Система MES потребує впровадження інших вдосконалень відповідно до рекомендацій, описаних у попередніх розділах.
X.4.2. Переваги
734. Посадових осіб ДЕЦ буде звільнено від необхідності переписування поданих заявок у систему, оскільки основне введення даних в систему будуть здійснювати заявники.
735. Імовірність помилкового введення даних через переписування буде зменшено.
736. Людські ресурси ДЕЦ будуть використовуватися більш ефективно, оскільки працівники зможуть зосередитися на своїй основній функції – здійснення експертизи.
737. Заявники будуть звільнені від необхідності друкувати та доставляти на паперових носіях інформацію, що попередньо існує в електронному вигляді в організаціях- заявниках. Ресурси заявників будуть використовуватися більш ефективно.
738. Інформація буде доставлятися швидше, а факт доставки може негайно бути відслідкований в системі.
739. Система забезпечить кращу прозорість та контроль процесу. Всі етапи та дії в рамках процесів можуть реєструватися та перевірятися.
740. Інформація, що створюються в рамках процесу реєстрації лікарських засобів, буде готовою до використання іншими інформаційними системами та процесами, які її потребують.
X.4.3. Ризики та питання, на які слід звернути увагу під час впровадження структури майбутніх інформаційних систем
741. Законодавчі ризики. Пропоновані зміни до інформаційної системи залежать від відповідних змін законодавчої бази, запропонованих групою з надання консультаційних послуг у рамках даного проекту. Затримка прийняття цих змін буде стримувати розробку нового ІТ. Якщо розробка нового ІТ почнеться без відповідної законодавчої бази, існує ризик того, що остаточний проект не буде відповідати законодавству і підлягатиме зміні.
742. Фінансові ризики. Не всі переваги системи можуть виражатися в суто фінансових цілях. Низька заробітна плата працівників ДЕЦ та інших посадових осіб може не виправдати покращення внаслідок скорочення робочого навантаження. Це може перешкодити прийняттю рішень, необхідних для затвердження та призначення належних ресурсів.
743. Знання та досвід співробітників ДЕЦ та інших державних організацій. Наявні працівники можуть мати недостатній досвід і кваліфікацію для виконання масштабних проектів між організаціями. Це може зумовити необхідність використання досвіду зовнішніх організацій (постачальника) в цих питаннях.
744. Ризики, пов’язані з впровадженням інформаційних систем. Перевитрати бюджету та часу.
745. Високі очікування. Зміни можуть бути успішно реалізовані тільки за умови їх поступової реалізації. Бажання змінити все та в короткі строки може призвести до того, що більшість результатів проекту не будуть використовуватися належним чином.
746. Опір впровадженню змін. Опір впровадженню змін може виходити в особливості від тих відділів і працівників, які підлягають скороченню в результаті впровадження нових інформаційних систем та функцій. У деяких випадках експерти можуть розуміти, що нова система буде забезпечувати посилений контроль за їх роботою. Такі ситуації повинні постійно контролюватися в ході процесу впровадження та негайно вирішуватися керівництвом у рамках загального процесу управління змінами.
Х.4.4. Резюме результатів та рекомендацій
| № | Сфера | Питання | Вплив | Коментарі/ Рекомендації |
| 1. | MES | Права інтелектуальної власності на MES належать приватній компанії – “E-life” | Подальші зміни та вдосконалення системи MES можуть здійснюватися тільки однією компанією – власником відповідних прав інтелектуальної власності. | Права інтелектуальної власності на програмне забезпечення повинні належати державі, в разі якщо розробка програмного забезпечення фінансується з державного бюджету або інших ресурсів, що належать державі. Такий підхід повинен реалізовуватися на основі загального законодавства про державні закупівлі та має застосовуватися до програмного забезпечення, розробленого на замовлення.Щодо прав інтелектуальної власності на існуюче програмне забезпечення, необхідно зрозуміти, чи є доцільним продовження відносин власності у старому форматі, чи необхідно організувати переведення цих прав на державну установу, або розробити новий формат та відмовитися від старої системи. |
| 2. | MES | Паперова документація вважається первинною. | Перешкоджання реалізації електронного документообігу. Процеси на основі паперової документації є менш ефективними. Ненадійна інформація про статус процесу реєстрації.
Менша прозорість процесу. |
Процеси та інформаційні системи повинні бути відповідним чином змінені для функціонування на базі процесів, що керуються системою.Необхідно розглянути доцільність введення правових та технічних заходів автентифікації і захисту електронної документації.Подання і обробка інформації повністю в електронному вигляді буде потребувати більш потужної ІТ- інфраструктури. |
| 3. | MES | Завдання для експертів призначаються через інформаційні системи лише частково. Система не має функціональної можливості реєстрації часу, витраченого експертами, або іншої інформації, збереження якої сприяло б кращій ефективності експертизи з точки зору часу і ресурсів. | Фактичне навантаження експертів не може контролюватися. | Забезпечити функціональні можливості для реєстрації часу в рамках MES.Розробити відповідні ключові показники ефективності для експертів.Розробити і утримувати систему управління, яка сприятиме більш продуктивній роботі експертів. |
| 4. | MES/СВ | Система MES має односпрямований інтерфейс з Системою візуалізації інформації (СВ).СВ не містить можливостей для заявників. | Перешкоджання комунікації між заявниками та ДЕЦ. | Необхідно впровадити новий портал ДЕЦ для заявників. Портал повинен забезпечити всі необхідні функції для комунікації між заявниками та ДЕЦ:- подачу реєстраційних досьє в електронній формі,- можливості для надання запитів, відповідей, коментарів та надсилання повідомлень,- електронні засоби зв’язку повинні переважно замінити існуючий документообіг на паперових носіях..За допомогою двонаправленого інтерфейсу, ця інформація буде автоматично з’являтися на робочому місці експерта. |
| 5. | MES/ ДРЛЗ | Обмін даними між MES та ДРЛЗ проводиться вручну. | Ризики, пов’язані з ручним введенням даних.
Надмірний обсяг роботи. |
Забезпечити автоматизований інтерфейс передачі даних між MES та ДРЛЗ.Схвалення реєстрації лікарських засобів повинно вводитися в систему посадовими особами МОЗ (якщо буде прийнято рішення залишити цей етап процесу). Після авторизації система повинна автоматично публікувати відповідну інформацію в ДРЛЗ. |
| 6. | MES/ ДРЛЗ | Неповний процес обміну даними з іншими інформаційними системами. MES не пов’язана ні з Єдиним державним реєстром юридичних осіб, фізичних осіб-підприємців та громадських формувань, ні з іншими державними інформаційними системами для перевірки особи та статусу заявника. | Заявники виступають в якості посередників між ДЕЦ та іншими установами.
Інформація, отримана в автономному процесі, може бути неточною через затримку у часі. Уповільнення процесу у зв’язку з ручним введенням даних (помилки, упущення), і необхідністю в постійній ручній роботі. |
Створити інтерфейси для обміну даними з іншими інформаційними системами.MES та ДРЛЗ повинні бути доповнені відкритими інтерфейсами, що дозволяють інтегрувати інші (третіх сторін) інформаційні системи, тобто системи лікарень, аптек, електронних рецептів та статистики. Електронна інформація може використовуватися іншими установами відповідно до їх рівня авторизації, наприклад, митницею, Держлікслужбою.Первинна інформація, яка має бути доступною автоматично в зазначених інтерфейсах, – це класифікатори та інформація щодо реєстрації лікарських засобів.MES повинна мати можливість автоматично отримувати інформацію, яка зберігається в інших державних інформаційних системах та є необхідною для її процесів, наприклад, дані з Реєстр юридичних осіб для перевірки правового статусу заявника. |
| 7. | СВ | Функція відправки повідомлень заявникам не доступна в СВ. | Поганий досвід користувачів. Користувачі повинні постійно перевіряти систему на наявність нових змін. | Забезпечити функцію електронного повідомлення для користувачів. Ця опція здатна забезпечити негайні результати з точки зору вдосконалення зручності та простоти використання системи. |
| 8. | СВ | Всі користувачі реєструються співробітниками ДЕЦ. | Повільний процес реєстрації.
Повільний та ненадійний процес скасування реєстрації користувача. Надмірний обсяг роботи адміністраторів ДЕЦ. |
Забезпечити самостійну реєстрацію користувачів або дворівневе адміністрування користувачів для заявників. |
| 9. | ДРЛЗ | Недоліки в кодуванні та описі інформації про лікарські засоби. | Нинішній рівень кодування інформації (інформації про упаковку, дозування, концентрація) обмежує використання ДРЛЗ для аналізу ринку, контролю цін, статистичних даних та інших способів використання цієї інформації.Реєстраційний номер лікарських засобів не дозволяє ідентифікувати конкретний продукт (включаючи упаковку). | Забезпечити вдосконалені кодування та опис інформації про лікарські засоби.Додати необхідну інформацію до реєстраційного номеру лікарського засобу. Розробити необхідні процедури для застосування нового кодування, які повинні застосовуватися до всіх нових реєстрацій.Необхідно визначити період передачі, протягом якого будуть оновлені старі записи.
У разі необхідності власники реєстраційних посвідчень повинні надати додаткову інформацію для поновлення реєстраційних записів. Забезпечення процедур та правової бази для використання реєстраційного номеру лікарських засобів в якості первинного ключа для ідентифікації лікарського засобу в рамках інтеграції ДРЛЗ до іншої інформаційної системи. |
| 10. | ДРЛЗ | Інформація про дозування та концентрацію надається в довільній формі. | Якість введених даних залежить від співробітників, які дотримуються певних правил їх введення. Цей контроль вважається слабким. Відхилення в даних, в разі його наявності, зробить цю інформацію непридатною для автоматизованої обробки без попереднього внесення необхідних змін вручну (наприклад, для звітності). | Забезпечити контроль введення даних в інформаційну систему. |
| 11. | ДРЛЗ | Застаріла платформа розробки ДРЛЗ | Ризик технологічного тупика.Ризик відсутності ресурсів розробників.
Відсутність підтримки постачальника, включаючи відсутність оновлень безпеки. |
Інші опції зміни технології повинні досліджуватися та оцінюватися. |
| 12. | ДРЛЗ | ДРЛЗ не містить інформації щодо цін і статистики використання лікарських засобів. Існує окремий реєстр цін на лікарські засоби, який утримується МОЗ. | Відсутність контролю за ціноутворенням на лікарські засоби. Можливі цінові відмінності можуть вплинути на доступність лікарських засобів. | Перевірити та визначити вимоги до управління ціноутворенням на лікарські засоби. Ціни можуть бути визначені в процесі реєстрації лікарських засобів. Тому інформацію стосовно ціноутворення необхідно включити в MES. |
| 13. | MES/ДРЛЗ/ СВ | Відсутня функція резервного копіювання інформації. | Ризик втрати даних в разі проблем з мережею, у випадку пожежі, стихійного лиха, та ін.
Обробка інформації повністю в електронному вигляді потребує більш високої ефективності та потужності ІТ інфраструктури. |
Забезпечити резервні засоби обробки даних. |
| 14. | Стратегія | Відсутня ефективна стратегія розвитку інформаційних систем у сфері охорониздоров’я . МОЗ не забезпечує дотримання такої стратегії. | Кожна установа дотримується своїх власних планів. Процес планування є досить ситуативним та фокусується на короткострокових питанняхтехнічного обслуговування та експлуатації.Відсутність інтеграції між організаціями, системами та проектами. Відсутність бачення та труднощі у формулюванні довгострокових цілей.Інтеграція та загальне використання інформаційних систем є недостатньо розвиненими. Немає чітких правил прийому та реалізації ІТ-проектів. | Необхідно впровадити процес загального планування та прийняття рішень щодо інформаційних систем як між організаціями, так і всередині організацій. Для впровадження такого процесу має бути чітке визначенняпонять «повноваження» та «прийняття рішень», які мають бути зафіксовані в єдиному документі планування («Стратегічний ІТ план інформаційних систем в області охорони здоров’я»). |
Х.4.5. Операційна сумісність інформаційних систем
747. На блок-схемі нижче представлені можливі варіанти інтеграції між оновленими системами MES, ДРЛЗ, СВ та іншими інформаційними системами та/або процесами в сфері охорони здоров’я.
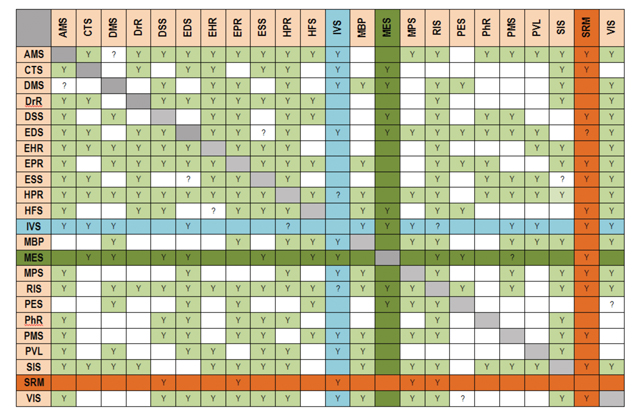
Рисунок 3. Операційна сумісність інформаційних систем
Додаток 1. РЕЗУЛЬТАТИ ОПИТУВАННЯ
У цьому додатку представлені консолідовані результати електронного опитування, що проводився Консорціумом серед представників фармацевтичних компаній в Україні.
Просимо зауважити що представлені результати опитування, включаючи коментарі, були попередньо відібрані, оброблені і відредаговані (без зміни їх змісту) для задоволення потреб проекту.
ЧАСТИНА 1: Аналіз процесу реєстрації лікарських засобів ХІМІЧНОГО ПОХОЖДЕННЯ
1. Скільки часу (місяці) Ваша Компанія витрачає на реєстрацію лікарського засобу за стандартною процедурою?
8 -18 місяців
2. Скільки часу (місяці) Ваша Компанія витрачала на реєстрацію лікарського засобу за стандартною процедурою до 30 жовтня 2015 року? (Дата набуття чинності Наказу МОЗ № 460)
8 місяців – 3 роки
3. Скільки часу (місяці) Ваша Компанія витрачає на реєстрацію лікарського засобу за спрощеною процедурою (вкажіть за якою саме)?
1-8 місяців
4. Скільки часу (місяці) Ваша Компанія витрачала на реєстрацію лікарського засобу за спрощеною процедурою (вкажіть якою саме) до 30 жовтня 2015 року?
6 – 18 місяців без лабораторних досліджень, продукти EMA 6-7 місяців
5. Скільки часу (місяці) Ваша Компанія витрачала на перереєстрацію лікарського засобу до 30 жовтня 2015 року?
7- 18 місяців
6. Скільки часу (місяці) Ваша Компанія витрачає на внесення змін до реєстраційного досьє?
3- 10 місяців, адміністративні – 4 місяці, зміни в реєстраційне – 6-9 місяців, зміни в Інструкцію – 6-8 місяців
7. Скільки часу (місяці) Ваша Компанія витрачає на внесення змін до реєстраційного досьє до 30 жовтня 2015 року?
3 -14 місяців
8. Скільки запитів та якого змісту Ви зазвичай отримуєте від МОЗ та ДЕЦ під час стандартної процедури реєстрації?
До 18 запитів, як правило 3-4
Вкажіть які:
- Запити щодо:
- заяви
- інструкції
- методів контролю якості
- фармакобезпеки
- матеріалів про методи контролю якості
- зауважень юридичного департаменту
9. Скільки запитів та якого змісту Ви зазвичай отримуєте від МОЗ та ДЕЦ під час спрощеної процедури реєстрації?
до 5
Вкажіть які:
Запити щодо:
- централізованої процедури ЄМА (згідно з Наказом МОЗ № 460 заявнику не надаються зауваження)
- технічні, якщо продукт схвалений FDA(за скороченою процедурою 45 днів) то неможливо йти по скороченій процедурі, оскільки потрібні вмотивовані висновки, які FDA не видає
- зауважень юридичного відділу, департаменту післяреєстраційного нагляду, управління номенклатури та інструкцій
10. Чи мають представники Вашої Компанії (незалежні експерти) можливість обговорювати результати експертної оцінки ДЕЦ (наприклад, брати участь у внутрішніх нарадах ДЕЦ, засіданнях науково-експертної ради) згідно з п.16 розділу IV Наказу МОЗ № 460?
 11. У який спосіб Ваша Компанія підтримує зв’язок з експертами ДЕЦ та посадовими особами МОЗ в ході реєстрації лікарських засобів?
11. У який спосіб Ваша Компанія підтримує зв’язок з експертами ДЕЦ та посадовими особами МОЗ в ході реєстрації лікарських засобів?
12. Чи легко Вам зв’язатися з посадовими особами МОЗ?
Коментарі:
- МОЗ не має достатньо ресурсів, для того щоб контролювати процес реєстрації
- Більше ніж 1 місяць потрібно, щоб записатися до вченого секретаря ДЕЦ
- Важко по телефону, деколи Вам потрібно стояти в черзі, щоб зустрітись з потрібним експертом
- При необхідності вдається запланувати робочу зустріч
13. Просимо Вас оцінити рівень знань фахівців ДЕЦ (від 1 до 5, де 1 – незадовільно, 5 – не маю жодних нарікань)
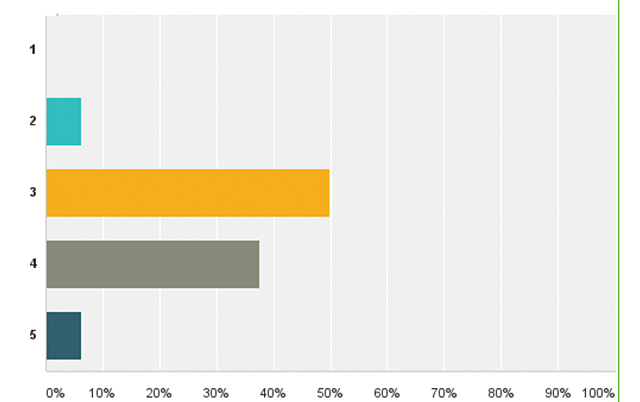
14. Чи існують які-небудь галузі, в яких експертам ДЕЦ не вистачає знань? (наприклад, щодо конкретних видів лікарських засобів)
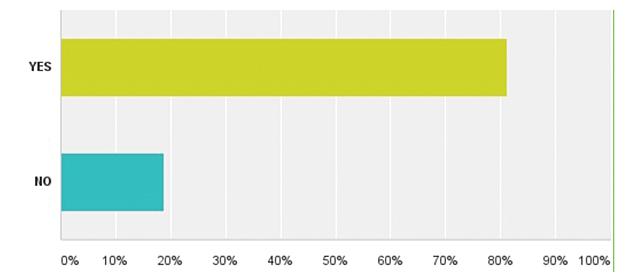
Коментарі:
- Фармацевтична частина документації
- Показники специфікації та методи контролю якості оригінальних ЛЗ, вимоги фармаконагляду
- Біосиміляри
- Експерти з реєстрації не володіють знаннями щодо GМР
- Продукти генної інженерії. інноваційні продукти із методами контролю якості що потребують не стандартних методик
- У ДЕЦ дуже висока рухомість кадрів (звільнення працівників за власним бажанням) як і структурах МОЗ
- Щодо типу заяви лікарського засобу
- Проблема з розумінням централізованої процедури ЄМА
- Проблеми з розумінням положень щодо Плану управління ризиками
- Європейське та міжнародне регуляторне законодавство загалом, а також для імунобіологічних препаратів та препаратів крові
- На даний момент, якщо продукт схвалений вже ЕМА або FDA, експертиза виглядає як освітній проект, в якому заявник ознайомлює експертів з правилами ЄС/ICHкерівними принципами. В такому випадку ДЕЦ повинна розширити свої знання в сфері експертизи генериків та біосимілярів
- Не вистачає знання європейського законодавства, структури Модуля 3 та комунікаційних навичок.
15. Чи виникали у Вас проблеми комунікації з представниками ДЕЦ або інших державних установ, що беруть участь в процесі реєстрації??
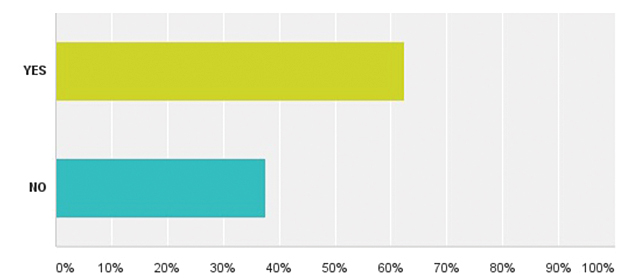
Коментарі:
- Листи в МОЗ часто залишаються без відповіді або ми отримуємо відповідь з затримкою
- Держлікслужба – затримки видачі висновків, що унеможливлює реєстраційні процеси; уповноважені лабораторії – застаріле оснащення і як результат неможливість відтворення методик
- Як правило, комунікація була ефективною
- Не можливо записатися до експертів для отримання інформації на найближчий час. Зустріч із внутрішніми експертами можлива через 3 -4 тижні після звернення. Затримки у внутрішніх процедурах ДЕЦ із введенням реєстраційних форм у внутрішню базу даних та отриманням рахунків на оплату
- Іноді бувають непорозуміння завдяки постійним змінам у законодавстві та постійним звільненням досвідчених кадрів ДЕЦ
- Юридичний департамент вимагає документи, які не передбачені Наказом
- Є позитивний досвід у комунікації з управлінням Фармацевтичної діяльності та якості фармацевтичної продукції МОЗ України
- Погана комунікація між ДЕЦ та Держлікслужбою та всередині установ між департаментами.
16. Чи помітили Ви позитивні зміни в роботі ДЕЦ та МОЗ протягом останніх 12 місяців?
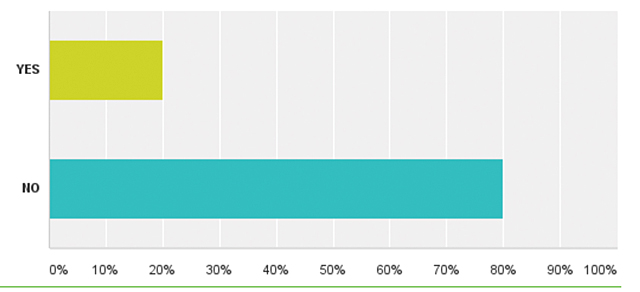
Коментарі:
- У зв’язку із великою плинністю персоналу ДЕЦ (велике навантаження, неконкурентний рівень зарплатні тощо), експертиза РД проводиться із затримкою на 2-3 місяці
- Є позитивні та негативні зміни. ДЕЦ не впливає на регуляторну політику. Інформованість в ДЕЦ о регуляторних змінах дуже низька. Завантаженість експертів дуже зросла. Кількість експертів (особливо професіональних) зменшується, що відображається на роботі установи. Терміново необхідно надати допомогу збільшивши штат професіоналами .
- Позитивно: покращилась система Візуалізації.
- Реформування законодавства та інших документів не означає реформування стилю відносин та автоматичного вирішення проблем, більшість яких не мають до законодавства ніякого відношення
- Постійна зміна експертів, завантаженість експертів
- ДЕЦ потрібно тримати персонал (експертів), тому що будь-яка зміна персоналу призводить до поганого рівня процесу реєстрації, оскільки нові кадри не мають достатнього досвіду.
17. Які аспекти процесу реєстрації на Вашу думку потребують покращення?
- Недостатньо ресурсів, Строки
- Внутрішній документообіг ДЕЦ (листи передаються з кабінету в кабінет тижнями), кількість експертів незадовільна (неможливість здати своєчасно досьє, реєстраційну форму, додаткові матеріали і т.д.)
- Робота відділу змін та можливість запису на зустріч
- Прозорість процедур реєстрації, підвищення кваліфікації експертів, орієнтація на інтереси Заявників при спрощенні процедур тощо
- Терміни, кратність перегляду досьє і кваліфіковане формування зауважень
- Спрощення бюрократичних процедур. Введення можливості листування електронною поштою представникам Заявника із експертами
- Стабільна та прозора робота державних структур. Оцінка людей за досвідом та професіоналізмом
- Комплектація досьє у відповідності до діючих вимог ЕС
- Недотримання термінів експертизи через велике навантаження на експертів (до 400 препаратів на одного експерта)
- Накази та слідування їм
- Розширення штату експертів.
18. Якщо Ви зіштовхувались з випадками неефективної або непрозорої процедури реєстрації в 2013-2016 роках і готові дати експертам Консорціуму матеріали справи для аналізу, просимо Вас вказати назву лікарського засобу та номер заяви про державну реєстрацію
- Критичних випадків з вини працівників ДЕЦ не було, переважно технічні моменти та непорозуміння які можна було вирішити
*Є інші відповіді. Вони містять конфіденційну інформацію.
19. Чи інформує Вас ДЕЦ про винесення позитивного рішення за результатами експертизи до офіційного схвалення МОЗ?
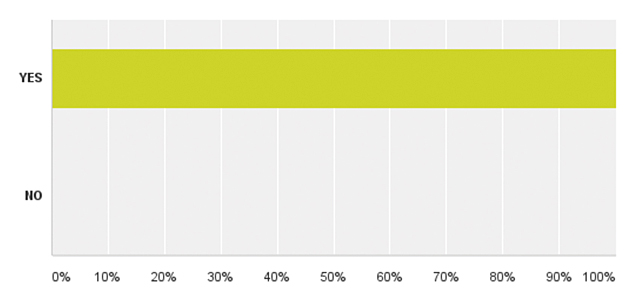
Коментарі:
- За два місяці
- За 2 місяці до схвалення МОЗ 2
- Є спеціальна форма, в якій ДЕЦ вказує про проходження тієї чи іншої наукової ради
- 45-60 днів
- 1.5-2 місяці
- Отримуємо листа щодо рекомендації до затвердження на засіданні НТР/НЕР.
- 30-45 днів.
20. Чи існують на Вашу думку етапи процедури реєстрації в МОЗ або ДЕЦ, які є зайвими або надлишковими??
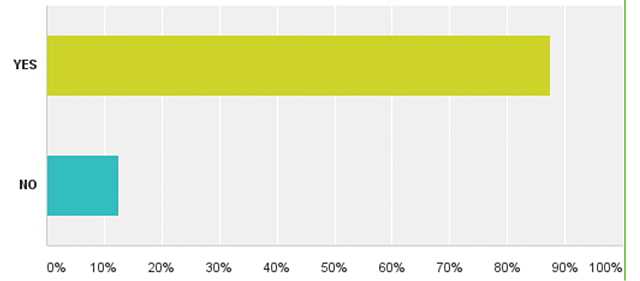
Коментарі:
- Офіційне схвалення МОЗ є несуттєвим, оскільки ДЕЦ є представником МОЗ, і етап схвалення МОЗ є суто бюрократичною процедурою
- Бюрократична процедура підписання висновків
- Наразі заява та реєстраційна форма містять ту саму інформацію
- Подання заяв до МОЗ через «єдине вікно», перевірка (аудит) висновків ДЕЦ у МОЗ, підготовка МОЗ наказів для реєстрації
- Передача справ до МОЗ з підписаним наказом в «єдине вікно», деякі аспекти фармаконагляду, які відрізняються від європейських стандартів
- 2 типи заяв. Подача документів у кілька етапів, необхідність попереднього запису для зустрічі з експертами. Крім попереднього запису, ще треба стояти в черзі протягом кількох годин. Неможливість призначення зустрічі протягом тижня у випадку необхідності
- Постійні зміни до законів та внутрішніх процедур ДЕЦ не вирішують проблеми, а створюють їх, збільшуючи кількість документів для подачі замість їхнього зменшення і спрощення процедури
- Подача заяв до «єдиного вікна» відповідно до Наказу №220
- Правова експертиза не виключена Наказом МОЗ № 460. Передача документів для перереєстрації та внесення змін з безпеки до Консультативно-експертної групи. Лабораторне тестування. Схвалення методів контролю якості
- Подача заяви через «єдине вікно»
- Схвалення позитивних висновків експертів на зустрічі Науково-експертної ради особами, які не ознайомлені з матеріалами Реєстраційного досьє
- Спеціалізована експертиза лікарських засобів схвалена за централізованою процедурою EMA, FDA
21. Просимо Вас дати оцінку рівня плати за експертизу матеріалів:
Too high-зависока
Acceptable-допустима
Too low–низька
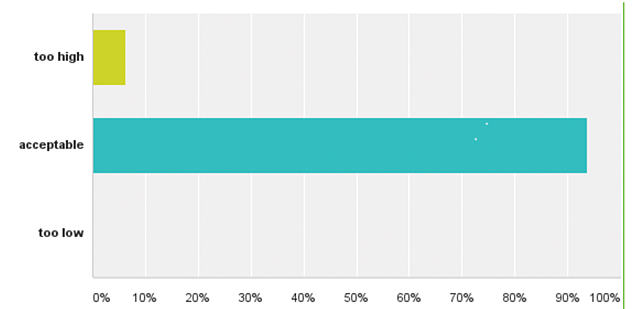
22. Яке підвищення вартості проведення експертизи (у відсотках) Ви вважатимете прийнятним задля покращення процесу реєстрації??
- Якщо це посприяє мотивації експертів та збільшить їхню кількість – я б запропонував 50%
- 20 %
- Прийнятним можна бути вважати рівень вартості експертизи, обґрунтований у калькуляції витрат ДЕЦ. В цих калькуляціях повинна бути вказана частина витрат на з/п (яка повинна бути на рівні з/п фахівців фарм. компаній). Кількість співробітників ДЕЦ повинна бути адекватною рівню навантаження
- зазвичай допустимо 20-30% з поступовим ще підвищенням, проте питання термінів і якості роботи відповідно повинні бути відображені через КРІ
- Необхідно враховувати, що компанії мають різні доходи в цілому та різну прибутковість від реалізації різних продуктів. та різні доходи самих компаній, цей фактор необхідно враховувати. Тому сума може бути як занадто високою, так і прийнятною залежно від ситуації
- 10 %
- 10-15 %
- ціни підвищують постійно, але на покращення процесу це не впливає
- Питання за межами моєї компетентності
- це залежить від того, яким буде механізм взаємозв’язку між вартістю експертизи і покращенням процесу експертизи
- не більше 10%, і у разі підвищення вартості якість експертизи також має підвищуватись.
23. Просимо підтвердити або спростувати припущення: більша частина інформації, яка включена в реєстраційне досьє, створена та зберігається в електронному вигляді.
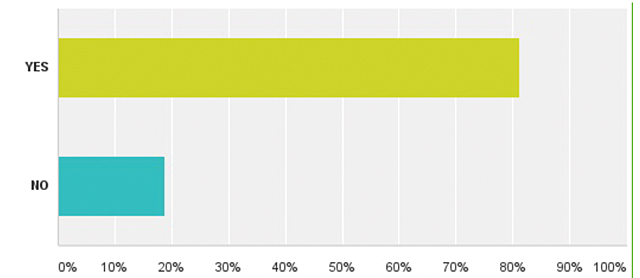
24. Скільки часу (днів) у Вас займає підготовка досьє для подання??
А: Друк файлів
Від 1 дня до 3 місяців.
В: Підготовка досьє, перевірка документів у паперовому вигляді
Від 8 годин до 10 днів.
С: Подання досьє
Від 1 години до 5 днів.
25. Визначте середню кількість робочих годин, витрачених на дії, зазначені в пунктах А та В.
Від 12 до 200 годин.
26. Визначте строки доставки (дні) та витрати (грн) на кур’єра, поштові послуги, тощо за дії, зазначені в пункті С.
А: Строки (дні)
0,1- 5 робочих днів
В: Витрати (гривня)
600 -10000 гривень
27. Які сфери реєстрації лікарських засобів потребують впровадження інформаційних технологій чи покращення їх рівня?
- Впровадження електронної комунікації як офіційної комунікації під час подачі досьє
- Електронний документооборот; візуалізація – етапи проходження експертизи; обіг документів в МОЗ після завершення експертизи в ДЕЦ; єдина база ЛЗ, якою можуть користуватися всі державні органи (залучені до обороту ЛЗ), включаючи митницю
- Впровадження електронного подання
- Процедура подачі заявок
- Подання заяви в електронній формі, модулі досьє 2-5 – на електронному носії (наприклад, СО)
- Візуалізація.
28. Загальні коментарі до Частини 1
ЧАСТИНА 2: Аналіз процесу реєстрації БІОЛОГІЧНИХ лікарських засобів
29. Скільки часу (місяці) Ваша Компанія витрачає на реєстрацію лікарського засобуза стандартною процедурою?
18 місяців
30. Скільки часу (місяці) Ваша Компанія витрачала на реєстрацію лікарського засобу за стандартною процедурою до 30 жовтня 2015 року? (Дата набуття чинності Наказу МОЗ №460)?
24 місяці
31. Скільки часу (місяці) Ваша Компанія витрачає на реєстрацію лікарського засобу за спрощеною процедурою (вкажіть за якою саме)?
6 місяців
32. Скільки часу (місяці) Ваша Компанія витрачала на реєстрацію лікарського засобу за спрощеною процедурою (вкажіть за якою саме) до 30 жовтня 2015 року??
–
33. Скільки часу (місяці) Ваша Компанія витрачала на перереєстрацію лікарського засобу до 30 жовтня 2015 року??
12 місяців
34. Скільки часу (місяці) Ваша Компанія витрачає на внесення змін до реєстраційного досьє??
6-8 місяців
35. Скільки часу (місяці) Ваша Компанія витрачає на внесення змін до реєстраційного досьє до 30 жовтня 2015 року??
3-9 місяців
36. Скільки запитів та якого змісту Ви зазвичай отримуєте від МОЗ та ДЕЦ під час стандартної процедури реєстрації??
До 10
37. Скільки запитів та якого змісту Ви зазвичай отримуєте від МОЗ та ДЕЦ під час спрощеної процедури реєстрації??
–
38. Просимо Вас оцінити рівень знань фахівців ДЕЦ (від 1 до 5, де 1 – незадовільно, 5 – не маю жодних нарікань)
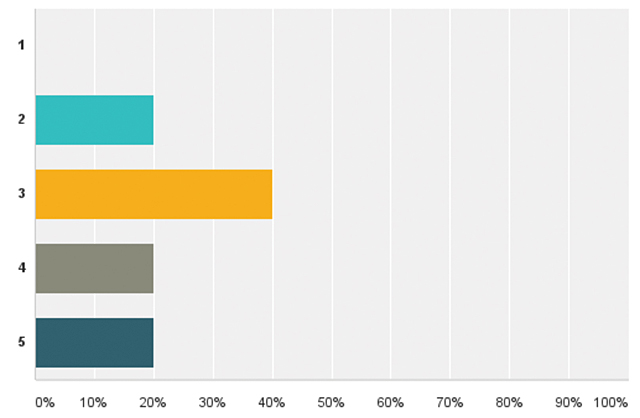
39. Якщо Ви зіштовхувались з випадками неефективної або непрозорої процедури реєстрації в 2013-2016 роках і готові дати експертам Консорціуму матеріали справи для аналізу, просимо Вас вказати назву лікарського засобу та номер заяви про державну реєстрацію.
–
40. Чи інформує Вас ДЕЦ про винесення позитивного рішення за результатами експертизи до офіційного схвалення МОЗ?
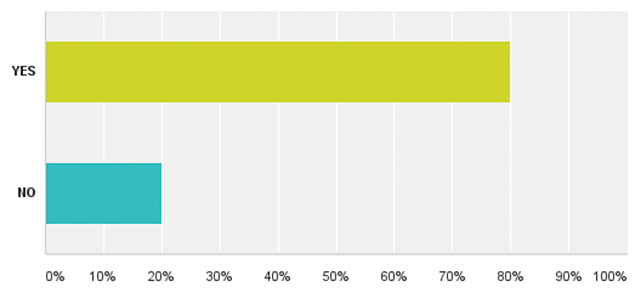
41. Скільки часу (дні) Ви витрачаєте на підготовку досьє для подання?
Див. пит. 24
42. Визначте середню кількість робочих годин, витрачених на дії, зазначені в пунктах А та В.
Див. пит. 25
43. Визначте строки доставки (дні) та витрати (грн) на кур’єра, поштові послуги, тощо за дії, зазначені в пункті С.
44. Загальні коментарі до Частини 2?
–
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим