Державне підприємство
Державний експертний центр Міністерства охорони здоров’я
України»
ПОСІБНИК
ЗАГАЛЬНІ ПРИНЦИПИ ОРГАНІЗАЦІЇ ДІЯЛЬНОСТІ КОМІСІЙ З ПИТАНЬ ЕТИКИ ПРИ ЛІКУВАЛЬНО-ПРОФІЛАКТИЧНИХ ЗАКЛАДАХ, В ЯКИХ ПРОВОДЯТЬСЯ КЛІНІЧНІ ВИПРОБУВАННЯ ЛІКАРСЬКИХ ЗАСОБІВ
(РЕКОМЕНДАЦІЇ ДЛЯ ЕКСПЕРТІВ)
Матеріали роботи науково-практичної конференції з міжнародною участю:
25 років діяльності
Державного експертного центру МОЗ України»
УСТАНОВА РОЗРОБНИК:
ДП «Державний експертний центр Міністерства охорони здоров’я України»
АВТОРИ УКЛАДАЧІ:
к.м.н. Л. Ковтун, Л. Янкова, С. Распутняк, к.м.н. О. Сілантьєва, Я. Мальцева, к.б.н. Л. Попова, М. Калашнікова, Т. Федорчук
В умовах постійно зростаючої кількості клінічних випробувань та участі в них досліджуваних, постає необхідність забезпечення належного рівня етичної експертизи клінічних випробувань. Метою такої експертизи є попередження можливого ризику для досліджуваних та моніторингу дотримання етичних та морально-правових принципів під час проведення клінічних випробувань. Ефективність роботи етичних комісій залежить від рівня компетентності членів комісій в питаннях етичної експертизи, що значною мірою обумовлюється їх навчанням та методичним забезпеченням.
Достатньо гостро в Світі стоїть проблема підготовки спеціалістів для проведення етичної експертизи. Останнім часом особлива увага приділяється створенню системи етичної експертизи: її складовим, основним принципів діяльності, критеріям якісного функціонування комісій з питань етики, а процес етичної експертизи потребує постійного вдосконалення та чіткого методологічного підходу.
Даний посібник розроблений для членів комісій з питань етики при лікувально-профілактичних закладах, лікарів-дослідників, спонсорів, заявників та інших зацікавлених осіб, які приймають участь у проведенні експертизи матеріалів клінічних випробувань.
ЗМІСТ
| Вступ | 5 |
| 1. Загальні положення щодо захисту прав людини | 8 |
| 2. Нормативно-правова база України щодо проведення клінічних випробувань лікарських засобів | 9 |
| 3. Склад та діяльність ЛЕК | 13 |
| 4. Алгоритм діяльності комісії з питань етики при ЛПЗ | 15 |
| 5. Інформована згода | 18 |
| 5.1. Вимоги до оформлення та змісту інформації для пацієнта та форми згоди | 19 |
| 5.2. Умови отримання інформованої згоди від пацієнтів та здорових добровольців | 21 |
| 6. Етичні питання клінічних випробувань за участю малолітніх та неповнолітніх дітей | 23 |
| 7. Клінічні випробування за участю недієздатних осіб (в тому числі, осіб, які перебувають в критичних та невідкладних станах) | 28 |
| 8. Клінічні ситуації, що вимагають невідкладної медичної допомоги | 31 |
| 9. Етичні питання використання плацебо | 34 |
| 10. Моніторинг щодо забезпечення захисту прав, безпеки, благополуччя досліджуваних, етичних та морально-правових аспектів у процесі проведення клінічного випробування | 35 |
| 10.1. Аналіз наявної в ЛЕК інформації щодо КВ | 36 |
| 10.2. Проведення моніторингу (перевірки) КВ в місці його проведення, яку проводить ЛЕК | 36 |
| 10.3. Планування проведення моніторингу (перевірки) | 36 |
| 10.4. Підготовка до моніторингу (перевірки) | 36 |
| 10.5. Інформування дослідника про моніторинг (перевірку) | 37 |
| 10.6. Безпосереднє проведення моніторингу (перевірки) | 37 |
| 10.7. Представлення результатів моніторингу (перевірки) | 39 |
| 10.8. Тимчасове або повне зупинення клінічного випробування | 39 |
| 11. Засоби самооцінки Комісії з питань етики при ЛПЗ | 40 |
| 12. СПИСОК ВИКОРИСТАНИХ ДЖЕРЕЛ | 41 |
ВСТУП
Клінічним випробуванням (КВ) відводиться важливе місце при розробці нових лікарських засобів, тому що рішення про медичне застосування лікарського засобу (ЛЗ) може бути прийняте тільки після систематичного вивчення його на людині та на підставі даних доведеної ефективності та безпечності.
Загальноприйнятим міжнародним стандартом, планування, організації та проведення КВ за участі людини, а також оформлення та представлення їх результатів є Належна клінічна практика (GCP), розроблений в рамках міжнародної конференції по гармонізації технічних вимог до реєстрації ЛЗ для людини (ICH GCP (Guideline for good clinical practice E6(R2)), останні доповнення до якого вступили в силу в 2016 р. Основні принципи Належної клінічної практики (ICH GCP) базуються саме на етичних засадах проведення клінічних випробувань.
Загальні принципи проведення КВ зазначені в ICH GCP:
1. Клінічні випробування слід проводити згідно з етичними принципами Ґельсінська декларації, правилами належної клінічної практики і чинними регуляторними вимогами.
2. До початку клінічного випробування має бути проведена оцінка співвідношення ризику, що передбачається, і незручностей з очікуваною користю для суб’єкта випробування та суспільства. Клінічне випробування може бути почате й продовжене лише в тому разі, якщо очікувана користь виправдовує ризик.
3. Права, безпека і благополуччя суб’єктів випробування важливіші від інтересів науки і суспільства.
4. Наявних даних доклінічного і клінічного вивчення досліджуваного препарату має бути достатньо для обґрунтування клінічного випробування, що планується.
5. Клінічне випробування має бути науково обґрунтоване, докладно і чітко описане у протоколі.
6. Випробування слід проводити відповідно до протоколу, щодо якого заздалегідь отримане схвалення/позитивне рішення експертної ради медичного закладу/ незалежного етичного комітету.
7. Лише кваліфікований лікар або стоматолог може взяти на себе відповідальність за надання суб’єктам випробування медичної допомоги і прийняття рішень медичного характеру.
8. Усі особи, які беруть участь у проведенні клінічного випробування, повинні мати освіту, професійну підготовку й досвід, що відповідають виконуваним функціям.
9. До включення суб’єкта у клінічне випробування необхідно отримати його добровільну інформовану згоду.
10. Реєструвати, обробляти і зберігати всю інформацію, що отримують в ході клінічного випробування, слід таким чином, щоб забезпечити коректне подання, інтерпретацію та верифікацію даних.
Цей принцип поширюється на всі записи, зазначені у цьому керівництві, незалежно від типу використовуваного носія.
11. Необхідно забезпечити конфіденційність документів, що дозволяють установити особистість суб’єкта, при дотриманні прав на недоторканність приватного життя та конфіденційність згідно з відповідними регуляторними вимогами.
12. Виробництво й зберігання досліджуваних препаратів, а також поводження з ними слід здійснювати відповідно до правил належної виробничої практики (GMP). Препарати необхідно застосовувати відповідно до затвердженого протоколу.
13. Слід використовувати систему процедур, що забезпечують якість клінічного випробування у всіх його аспектах.
В центрі уваги таких систем мають бути аспекти клінічного випробування, що необхідні для забезпечення захисту суб’єктів дослідження та надійності (достовірності) результатів випробування.
Етичні принципи, які мають значення при проведенні клінічних випробувань це перш за все-повага до людини, яка має право на захист; милосердя-це мінімізація ризиків, оцінка переваг та справедливість. Більш детальне тлумачення цих принципів зазначено в інших міжнародних документах, а саме:
- • Гельсінська декларація Всесвітньої медичної асоціації «Етичні принципи медичних досліджень за участю людини у якості об’єкта дослідження» 1964–2013 рр. (http://uacm.kharkov.ua/download/2014_11/22.pdf);
- • Міжнародне керівництва по етиці для біомедичних досліджень за участі людини в якості досліджуваних Ради міжнародних організацій медичних наук (CIOMS) (http://cioms.ch/ethical-guidelines-2016/WEB-aOMS-EthicalGuidelines.pdf.);
- • Бельмонтський звіт (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (1978). The Belmont Report. Ethical Principles and Guidelines for the Protection of Human Subjects of Research. http://www.hhs.gov/ohrp/humansubjects/guidance/belmont.html);
- • Нюрнберський кодекс (The Nuremberg Code.(1947).
- • (https://history.nih.gov/research/downloads/nuremberg.pdf.).
У вищенаведених міжнародних документах визначено, що інтереси науки та суспільства не повинні мати переваги над інтересами та правами кожної окремої людини, в тому числі право на життя, здоров’я та безпеку. Повага до людини та захист її прав обумовлена необхідністю всебічно ознайомити досліджуваного щодо умов клінічного випробування та отримання добровільної інформованої згоди на участь у такому випробуванні.
Існуюча на даний час в Україні нормативно-правова база щодо проведення КВ лікарських засобів, відповідає міжнародним стандартам щодо проведення КВ та Директивам ЄС.
В той же час, за наявності чіткого порядку проведення та експертизи матеріалів таких випробувань в Україні, залишаються поза увагою деякі організаційні питання діяльності Комісій з питань етики при лікувально-профілактичних закладах (ЛПЗ). Тому існує потреба у створенні посібника, який спрямований надати допомогу комісіям з питань етики (ЛЕК) при лікувально-профілактичних закладах щодо етичної оцінки матеріалів клінічних досліджень з метою надання незалежного рішення про можливість проведення клінічного дослідження в Україні.
1. ЗАГАЛЬНІ ПОЛОЖЕННЯ ЩОДО ЗАХИСТУ ПРАВ ЛЮДИНИ
Проведення клінічних випробувань лікарських засобів можливе лише за умови дотримання особливих положень щодо захисту прав людини. Ці положення забезпечують захист учасників або населення в цілому від необґрунтованих ризиків при проведенні досліджень.
Всі випробування за участю людини як суб’єкта дослідження повинні проводитися відповідно до загальновизнаних етичних принципів, а саме:
- принцип автономії (визнання спроможності людини робити свій вибір). Принцип автономії реалізується, зокрема, шляхом процедури добровільної інформо- ванної згоди, яка може бути відкликана в будь-який час і без будь-яких наслідків для людини;
- принцип користі і непричинення шкоди (користь і непричинення шкоди-моральне зобов’язання забезпечувати максимум потенційної користі і мінімізації можливої шкоди);
- принцип справедливості (справедливість-неупередженість і рівність). Справедливість має велике значення особливо для відбору учасників дослідження. Критерії відбору повинні бути пов’язані з метою дослідження, а не мати за основу лише, наприклад, легкість, з якою могла бути отримана інформована згода.
Ці принципи відображені в різних керівництвах з біомедичної етики, а також в документах, які мають обов’язкову юридичну силу, що стосуються захисту учасників біомедичних досліджень, наприклад, в Конвенції про права людини та біомедицину. Дані принципи взаємопов’язані, і при їх застосуванні цей взаємозв’язок необхідно враховувати. У цьому керівництві висвітлюється застосування зазначених основоположних принципів та норм на практиці.
Основою базових принципів є необхідність поваги і захисту людської гідності та, відповідно принцип пріоритету людини, її життя і здоров’я, честі і гідності, недоторканості і безпеки, захисту прав і свобод. Принцип пріоритету людини має безпосереднє відношення до галузі біомедичних досліджень. Відповідно до цього принципу інтереси і благополуччя людини, що бере участь в дослідженні, завжди повинні переважати над інтересами науки і суспільства.
Незалежна оцінка наукової користі проекту (протоколу КВ) дослідження і його експертиза з точки зору етичної прийнятності гарантує дотримання етичних та морально-правових принципів проведення біомедичних досліджень. Комісії з питань етики (ЛЕК) відповідають за забезпечення прав, безпеки, благополуччя досліджуваних та за надання суспільству відповідних гарантій.
Діяльність ЛЕК базується на дотриманні та забезпеченні основних етичних принципів.
Терміни, які використовуються у даному посібнику зазначені у нормативних документах, які визначають проведення КВ в Україні.
2. НОРМАТИВНО-ПРАВОВА БАЗА УКРАЇНИ ЩОДО ПРОВЕДЕННЯ КЛІНІЧНИХ ВИПРОБУВАНЬ ЛІКАРСЬКИХ ЗАСОБІВ
На теперішній час в Україні існує нормативно-правова база, яка забезпечує проведення клінічних випробувань відповідно до міжнародних стандартів. Зокрема, це:
- Конституція України 1996 р.;
- Сімейний Кодекс України, 2002;
- Закон України «Основи законодавства України про охорону здоров’я» від 19.11.1992 № 2801-ХІІ;
- Закон України «Про лікарські засоби»,1996 р.;
- Закон України «Про захист персональних даних» від 01.06.2010 № 2297-VI;
- «Настанова. Лікарські засоби. Належна клінічна практика. СТ-Н МОЗУ 42- 7.0:2008», затверджена наказом МОЗ України № 95 від 16.02.2009, зі змінами, внесеними наказом МОЗ України від 26.09.2017 №1169
- Порядок проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань і Типове положення про комісії з питань етики, затверджені наказом МОЗ України № 690 від 23.09.2009 зі змінами;
- «Настанова Лікарські засоби. Дослідження біоеквівалентності» СТ-Н МОЗУ 427.1:2016, що затверджена наказом МОЗ України № 22 від 12.01.2017р.;
- «Настанова. Лікарські засоби. Подібні біологічні лікарські препарати, що містять моноклональні антитіла-неклінічні та клінічні питання» СТ-Н МОЗУ 427.4:2015, затверджена наказом МОЗ України від 21.05.2015 № 295.
Конституція України (1996 р.) проголошує:
- принцип пріоритету людини, її життя і здоров’я, честі і гідності, недоторканості і безпеки, захисту прав і свобод (стаття 3);
- право на повагу її гідності — це положення, крім всього, включає заборону на проведення медичних, наукових і інших досліджень на людині без її добровільної згоди (стаття 28);
- недопущення збору, зберігання, використання і розповсюдження конфіденційної інформації про людину без її згоди (стаття 32).
В статті 45 Закону України «Основи законодавства України про охорону здоров’я» зазначається, що «застосування медико-біологічних експериментів на людях допускається із суспільно корисною метою за умови їх наукової обґрунтованості, переваги можливого успіху над ризиком спричинення тяжких наслідків для здоров’я або життя, гласності застосування експерименту, повної інформованості і вільної згоди повнолітньої дієздатної фізичної особи, яка підлягає експерименту, щодо вимог його застосування, а також за умови збереження в необхідних випадках лікарської таємниці. Забороняється проведення науково-дослідного експерименту на хворих, ув’язнених або військовополонених, а також терапевтичного експерименту на людях, захворювання яких не має безпосереднього зв’язку з метою досліду».
Одним з основних законодавчих актів, що регулює клінічні випробування лікар-
- ських засобів в Україні, є Закон України «Про лікарські засоби», 1996. Відповідно до ст. 7 Закону України «Про лікарські засоби», 1996: «Клінічні випробування лікарських засобів проводяться після обов’язкової оцінки етичних та морально-правових аспектів програми клінічних випробувань комісіями з питань етики, які створюються і діють при лікувально-профілактичних закладах, де проводяться клінічні випробування».
Тобто, вимога щодо етичної оцінки матеріалів КВ комісіями з питань етики при ЛПЗ, закріплена на законодавчому рівні.
Стаття 8 Закону України «Про лікарські засоби», 1996 повністю присвячена захисту прав пацієнта (добровольця): «Клінічні випробування лікарських засобів проводяться при наявності письмової згоди пацієнта (добровольця) на участь в клінічному дослідженні або письмової згоди його законного представника на участь у клінічному дослідженні за участю неповнолітнього або недієздатного пацієнта. Пацієнт (доброволець) або його законний представник повинен отримати інформацію по суті і можливих наслідків дослідження, властивостям лікарського засобу, очікуваної ефективності, ступеніми ризиками».
На виконання Закону України «Про лікарські засоби», наказом МОЗ України від 23.09.2009 р. № 690 (зареєстровано в Міністерстві юстиції України 29.10.2009 за № 1010/17026 та за № 1011/17027) зі змінами були затверджені Порядок проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань (далі-Порядок) та Типове положення про комісії з питань етики при лікувально-профілактичних закладах, в яких проводять клінічні випробування (далі-Типове положення про комісії з питань етики).
Цей Порядок розроблений відповідно до статей 3, 44, 45 Закону України «Основи законодавства України про охорону здоров’я», статей 7, 8 Закону України «Про лікарські засоби», Закону України «Про захист персональних даних», з урахуванням вимог Директиви Європейського Парламенту та Ради 2001/20/ЄС від 04 квітня 2001 року, 2001/83/ЄС від 06 листопада 2001 року, Постанов Європейського Парламенту та Ради 1901/2006 від 12 грудня 2006 року та 1902/2006 від 20 грудня 2006 року, ICH GCP, міжнародних етичних принципів біомедичних досліджень із залученням людини та етичного кодексу лікаря.
Відповідно до пункту 3.5. Розділу ІІІ Порядку: «Усі клінічні випробування розпочинаються після прийняття рішення ЦОВВ, отримання протоколу(ів) комісії(й) з питань етики при ЛПЗ, де безпосередньо має проводитися клінічне випробування щодо погодження цього клінічного випробування, що є підставою для початку його проведення, та укладення договору про страхування життя і здоров’я пацієнта (добровольця) у порядку, передбаченому законодавством».
Порядком та Типовим положенням про комісії з питань етики визначено, що: «Комісії з питань етики при лікувально-профілактичному закладі (далі — Комісії з питань етики) — незалежний орган, що діє при закладі охорони здоров’я (лікувально-профілактичному закладі), де проводяться клінічні випробування, який включає медичних/ наукових спеціалістів, осіб інших спеціальностей, представників громадськості, які здійснюють нагляд за дотриманням прав, безпекою, благополуччям досліджуваних пацієнтів (здорових добровольців), етичних та морально-правових принципів проведення клінічного дослідження».
Комісія з питань етики при закладі охорони здоров’я (лікувально-профілактичному закладі) погоджує проведення клінічного випробування безпосередньо у місці його проведення, що розташовується на базі лікувально-профілактичного закладу, при якому створена і діє ця комісія, на підставі оцінки етичних та морально-правових принципів.
Таке визначення повністю відповідає загальновизнаним міжнародним принципам проведення клінічних випробувань лікарських засобів, суб’єктом яких є людина, зокрема:
- Правилам Належної клінічної практики (ICH GCP), 1996;
- Директиві Європейського Парламенту та Ради 2001/20/ЄС від 04 квітня 2001 року «Про наближення законів, підзаконних актів та адміністративних положень держав-членів стосовно запровадження належної клінічної практики при проведенні клінічних випробувань лікарських засобів для вживання людиною»;
- Ґельсінської Декларації Всесвітньої Медичної Асоціації «Етичні принципи проведення медичних досліджень за участю людини», 1964–2013;
- Міжнародним керівним принципам етики для біомедичних досліджень за участю людини, CIOMS, 2016.
Всі вищезазначені документи підкреслюють незалежність провадження діяльності Комісіями з питань етики при ЛПЗ для можливості прийняття ними неупереджених рішень. Такий принцип, прийнятий в загальновизнаних міжнародних документах, покладений в основу регулювання клінічних випробувань в Україні. Комісії з питань етики повинні бути незалежними і здатними приймати рішення без політичного, професійного, організаційного або маркетингового впливу. Ця найважливіша вимога має бути відображена в процедурах призначення членів Комісії з питань етики, у вимогах до членства в Комісії з питань етики і в процедурах, передбачених для управління потенційними конфліктами інтересів (члени Комісії з питань етики повинні заявляти про можливі конфлікти інтересів), а також в джерелах фінансування Комісії з питань етики.
Комісії з питань етики надають незалежне рішення про ступінь відповідності біомедичного дослідження визнаним етичним стандартам, а також його відповідність національному законодавству. Оцінювати наукову обґрунтованість і законність дослідження можуть, як самі Комісії з питань етики, так і інші компетентні органи.
Комісії з питань етики відіграють все більш важливу роль в спілкуванні з суспільством щодо етичних аспектів біомедичних досліджень.
Згідно з п. 3.1. Розділу ІІІ Порядку: «Усі клінічні випробування проводяться відповідно до міжнародних етичних принципів із забезпеченням захисту прав, безпеки та благополуччя досліджуваних. Клінічне випробування може проводитись тільки в тому випадку, якщо очікувана користь виправдовує ризик».
Відповідно п. 3.7. Розділу ІІІ Порядку: «Планування, проведення та звітність усіх клінічних випробувань, у тому числі досліджень з оцінки біоеквівалентності, здійснюються із дотриманням вимог та принципів Належної клінічної практики (GCP)». Тобто, Комісія з питань етики в своїй діяльності має дотримуватися вимог та принципів GCP.
Без наявної діючої Комісії з питань етики неможливе проведення жодного клінічного випробування в лікувально-профілактичному закладі (ЛПЗ): «ЛПЗ залучається до проведення клінічного випробування у разі, якщо: при ЛПЗ створена та діє комісія з питань етики» (пункт 5.2. Розділу V Порядку).
3. СКЛАД ТА ДІЯЛЬНІСТЬ ЛЕК
Вимоги до складу ЛЕК визначені Належною клінічною практикою (GCP) (розділ 3) та Типовим положенням про комісії з питань етики (затверджено наказом МОЗ України від 23.09.2009 р. № 690 зі змінами).
Склад Комісії з питань етики при ЛПЗ (ЛЕК) повинен включати достатню кількість осіб, колективні знання і досвід яких в етичних і наукових питаннях, а також методи роботи і загальне функціонування повинні забезпечити надійність і здатність ефективним і незалежним чином виконувати свої обов’язки.
До складу ЛЕК мають входити не менше 5 членів, з них не менше одного члена, який не є науковим співробітником та не менше одного члена, який не залежить від закладу у якому має проводитись дослідження. ЛЕК повинні мати у своєму складі достатню кількість медичних працівників, а також представників громадськості (наприклад, юристів, релігійних діячів, представників громадських організацій та ін.), які мають необхідний досвід та кваліфікацію для оцінки наукових, медичних і етичних аспектів КВ.
Члени ЛЕК повинні отримувати належну початкову та подальшу підготовку, необхідну для виконання своїх функцій в ЛЕК, шляхом участі у семінарах з КВ, школах та конференціях, на яких обговорюються питання КВ.
В своїй діяльності ЛЕК керується нормативно-законодавчими вимогами та має переконатися в науковій цінності дослідження та його відповідності національному законодавству.
При проведенні етичної та морально-правової оцінки матеріалів клінічного випробування лікарського засобу, комісія з питань етики визначає етичну та наукову обґрунтованість клінічного випробування, кваліфікацію дослідників, можливості залучення місця проведення дослідження, методи та процедури отримання від досліджуваних інформованої згоди на підставі ознайомлення та її документального оформлення, повноту письмової інформації для пацієнтів або здорових добровольців стосовно дослідження, яке планується, вивчає всю інформацію, яка стосується процедури залучення суб’єктів в дослідження, механізми компенсації та відшкодування збитків досліджуваним.
Як зазначено в Додатковому Протоколі до Конвенції Ов’єдо про біомедичні дослідження (1998р), будь-який учасник дослідження, який зазнав збитків у результаті участі в дослідженні, має право на справедливу компенсацію відповідно до національного законодавства. Умови та порядок компенсації можуть бути різними в різних країнах, але у всіх випадках дослідники повинні представити в Комісію з питань етики детальну інформацію про будь-яке страхування або гарантії відшкодування шкоди, що з’явилася в ході дослідження.
Члени ЛЕК повинні оцінити можливості мінімізації ризиків та очікувану користь від участі в випробуванні. Зокрема, приділяти увагу таким проблемним питанням як планування плацебо-контрольованих досліджень, включення в клінічні випробування вразливих суб’єктів, зокрема, дітей, недієздатних осіб, осіб в критичних та невідкладних станах, а також аналізу умов страхування досліджуваних на випадок завдання шкоди їх життю та здоров’ю у клінічному випробуванні.
Комісії з питань етики мають бути впевнені, що дослідники не чинитимуть небажаний вплив на людей, спонукаючи їх прийняти участь в дослідженні. Тиск може мати фінансовий характер, але може приймати й інші форми. Наприклад, хворі і ослаблені люди можуть відчути, що вони повинні погодитися на участь, навіть, якщо це суперечить їхнім бажанням. Віра пацієнтів в лікарів та інших працівників охорони здоров’я також може призвести до неналежного впливу, особливо якщо лікуючий лікар є дослідником.
Деякі групи людей можуть бути особливо вразливі щодо примусу участі у КВ-наприклад, хворі на невиліковні/інвалідизуючі захворювання, військовослужбовці або особи, які чутливі в даному суспільстві через домінуючу соціальну ієрархію. Ці особи відносяться до уразливої категорії, тому Комісія з питань етики повинна приділяти увагу різним джерелам неналежного впливу.
Наприклад, якщо співробітників (дослідників) змусили відчути, що можливість продовження трудової діяльності залежить від їх участі в дослідженні, або якщо молодших лікарів змусили відчути, що їх кар’єрне зростання залежить від набору пацієнтів для дослідження старшого колеги.
Комісії з питань етики оцінюють етичну прийнятність матеріалів КВ з двох основних точок зору:
- потенційних учасників дослідження-захисту їх прав, гідності, безпеки та благополуччя;
- проведення дослідження, очікуваних результатів дослідження та можливих наслідків результатів дослідження для суспільства.
Термін «суспільство» може тлумачитися як у вузькому, так і в більш широкому сенсі, і може включати в себе потенційні інтереси майбутніх поколінь.
Комісії з питань етики повинні оприлюднювати достатньо інформації про свою роботу, а саме: про етичну експертизу, супровід дослідження і про інші види діяльності, за допомогою структурованих належним чином періодичних звітів, в яких не повинні розкриватися конфіденційні дані, що стосуються дослідження або його учасників. Такі звіти, у вичерпній формі або у формі робочого резюме, повинні перебувати у відкритому доступі, наприклад, на веб-сайті Комісії з питань етики, ЛПЗ, тощо.
Також існують взаємодоповнюючі види діяльності Комісій з питань етики, такі як публічне спілкування, семінари, що стосуються етичних проблем, просвітницька діяльність стосовно підвищення обізнаності щодо етичних проблем в галузі біомедичних досліджень.
Комісії з питань етики забезпечують публічні гарантії недопущення неетичних досліджень і заохочення етично прийнятних досліджень належного рівня якості. Таким чином, Комісії з питань етики відіграють одну з головних ролей в процесі оцінки матеріалів КВ та їх проведення.
Комісії з питань етики в своїй діяльності повинні керуватись принципами незалежності, компетентності, справедливості та прозорості.
4. АЛГОРИТМ ДІЯЛЬНОСТІ КОМІСІЇ З ПИТАНЬ ЕТИКИ ПРИ ЛПЗ
Основні вимоги до оцінки етичних і морально-правових аспектів КВ, які можуть проводитись за участю пацієнтів (здорових добровольців), та здійснення нагляду за забезпеченням їх прав, безпеки, благополуччя під час участі у КВ лікарських засобів, визначені «Порядком проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань» і «Типовим положенням про комісії з питань етики», затвердженими наказом МОЗ України від 23.09.2009 р. за № 690 із змінами та доповненнями.
Так, обов’язково при кожному ЛПЗ, де планується проведення КВ повинна бути організована ЛЕК.
Персональний склад Комісії з питань етики формує та затверджує керівник ЛПЗ (пункт 3.2 Розділу ІІІ Типового положення). Тобто, керівник ЛПЗ видає наказ, яким затверджує склад ЛЕК.
Наступним кроком організації діяльності Комісії з питань етики є перше засідання, на якому члени ЛЕК обирають Голову, його заступника та відповідального секретаря. Відповідно до пункту 3.3. Розділу ІІІ Типового положення: «Комісію з питань етики очолює Голова. Голова, його заступник та відповідальний секретар обираються на першому засіданні відкритим голосуванням простою більшістю голосів». Це зазначається у відповідному протоколі засідання ЛЕК.
На засіданні визначаються першочергові завдання в подальшій організації діяльності, зокрема, створення Положення ЛЕК та стандартних операційних процедур (СОП). При розробці Положення конкретної ЛЕК необхідно взяти за основу «Типове положення про Комісію з питань етики». Відповідно до пункту 3.4. Розділу ІІІ Типового положення: «Комісії з питань етики діють відповідно до законодавства, Положення та стандартних операційних процедур, що затверджуються на засіданні Комісії з питань етики при ЛПЗ».
Діяльність комісії має бути описана в СОП — це детальні письмові інструкції, що забезпечують однаковість виконання певних функцій.
СОП забезпечує процес проведення етичної експертизи послідовним, узгодженим, передбаченим та відтвореним. Безперечні переваги, які досягаються при застосуванні СОП: чіткий розподіл завдань за компетенцією, забезпечення якістю та логічною послідовністю дій.
СОП корисні для навчання нових членів ЛЕК, використовуються в якості керівництва для перевірки на відповідність, дають можливість чітко працювати членам ЛЕК в умовах, які визначені особливостями роботи ЛЕК.
Створення СОП входить до сфери відповідальності спеціально призначених кваліфікованих членів ЛЕК. З метою уніфікації та дотримання належного формату викладення, при написанні СОП потрібно керуватися наступними правилами:
- застосування стандартних визначень та обов’язкове включення трактування терміну в зміст СОП;
- застосування стандартного формату з послідуючим викладенням змісту;
- визначення приналежності СОП до діяльності конкретної ЛЕК;
- обов’язкове обговорення змісту СОП членами ЛЕК та їх схвалення на засіданні.
СОП повинні уніфікувати методику роботи членів ЛЕК. Тому, важливо, щоб знали та діяли у відповідності до СОП.
На початковому етапі створення СОП, доцільно створити план (список) процедур, які будуть використовуватися в процесі роботи ЛЕК. Цей план повинен включати в себе поступовий опис процедур. Процедури повинні бути написані доступною мовою, уникаючи складних висловлювань та термінів.
Доцільно:
- використання коротких речень, простих слів і термінів;
- написання у вигляді інструкції: «зробити чи забезпечити»;
- обмеження кількості інформації на сторінці. Наприклад, максимум до 10 дій;
- використання схем, таблиць, де це можливо;
- посилання на використані джерела в кінці СОП.
СОП повинні бути затверджені на засіданні ЛЕК, що зазначається у відповідному протоколі. Процедура внесення змін/виправлень в СОП повинна відображатися в окремому СОП. Також повинні бути визначені часові обмеження переглядів СОП. Написання та затвердження СОП-це початок розбудови чіткої, систематичної роботи ЛЕК. Після розробки та затвердження СОП необхідно проводити тренінги співробітників щодо СОП, інакше витрачені зусилля на їх написання будуть даремні, оскільки СОП не будуть виконуватись.
СОП корисні тільки в тому випадку, коли всі члени ЛЕК їх розуміють та діють у відповідності до цих письмових процедур та є обов’язковими для виконання.
Організацію роботи ЛЕК забезпечує керівник ЛПЗ (визначає постійне місце проведення засідань, зберігання документації, архівування документації, тощо). У ЛЕК в окремому користуванні має бути телефон/факс (окремий телефонний номер) та інтернет.
Засідання є найбільш важливою складовою діяльністю ЛЕК. На цих засіданнях члени ЛЕК аналізують матеріали досліджень та обговорюють їх етичну прийнятність. План засідань повинен оголошуватися заздалегідь, а у членів Комісії з питань етики має бути достатньо часу для того, щоб проаналізувати відповідні документи до початку кожного такого засідання.
На засіданні ЛЕК може бути присутнім Дослідник, який приймає участь у дослідженні, для надання пояснень щодо особливостей проведення КВ, якщо виникнуть питання щодо таких особливостей під час обговорення.
Дослідник, якщо він є членом ЛЕК може приймати участь в обговоренні матеріалів, але в голосуванні участі не приймає.
Рішення ЛЕК приймається за наявності кворуму та зазначається у протоколі засідань. Можливі варіанти прийнятих рішень:
- схвалення матеріалів КВ або суттєвої поправки;
- необхідність внесення змін до матеріалів КВ для отримання схвалення;
- відмова у схваленні матеріалів КВ з обґрунтуванням причин;
- відкликання раніше виданого схвалення матеріалів КВ (наприклад, у разі зміни співвідношення користь/ризик застосування досліджуваного лікарського засобу).
Після винесення рішення, ЛЕК готує витяг з протоколу засідання, який надається заявнику/спонсору, досліднику.
Відповідно до п. 8.7. Розділу VII Порядку: «Під час здійснення оцінки морально-етичних та правових аспектів матеріалів клінічних випробувань комісія з питань етики одноразово може запитати в письмовій формі у заявника додаткові матеріали та/або запросити на засідання комісії представника заявника для надання додаткових пояснень. Строк для надання додаткових матеріалів/пояснень не повинен перевищувати 30 календарних днів.
Строк для оцінки етичних та морально-правових аспектів не повинен перевищувати 30 календарних днів з дати отримання комісією заяви (без врахування часу, необхідного для підготовки запитаних додаткових матеріалів/пояснень)».
Згідно п. 3.3. Розділу Х Порядку: «Комісія з питань етики розглядає суттєві поправки протягом 15 календарних днів з дати отримання повного пакета документів.»
Всі матеріали, що отримує ЛЕК чи видає зацікавленим особам, необхідно реєструвати у відповідних журналах (вхідної та вихідної документації).
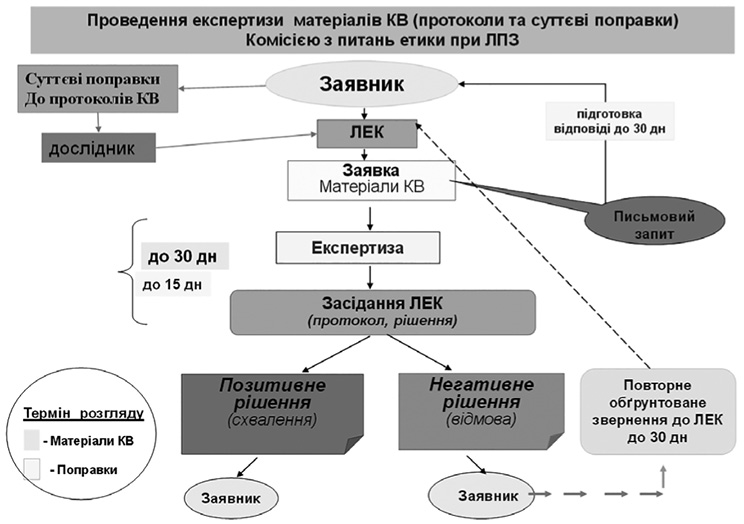
Експертиза матеріалів КВ та суттєвих поправок проводиться у відповідності до загальних принципів GCP, але обов’язково з урахуванням чинної законодавчої бази країни, де проводиться дане КВ.
5. ІНФОРМОВАНА ЗГОДА
Особлива увага під час експертизи матеріалів КВ повинна приділятися Інформованій згоді досліджуваного.
Згідно Статті 7 «Міжнародного пакту про громадянські та політичні права», яка проголошує: «Жодна людини не повинна без її вільної згоди піддаватися медичним, науковим чи іншим експериментам».
«Будь-яке втручання, що стосується здоров’я людини, може здійснюватися тільки після отримання її вільної та інформованої згоди», — зазначено в Європейській Конвенції з захисту людських прав і гідності в галузі біології і медицини.
Інформована згода — рішення взяти участь у клінічному випробуванні, яке має бути складено в письмовій формі, датоване та підписане, приймається добровільно після належного поінформування про характер клінічного випробування, його значення, вплив та ризик, відповідним чином документально оформляється особою, яка спроможна дати згоду, або її законним представником/близьким родичем; у виняткових випадках, якщо відповідна особа неспроможна писати, вона може дати усну згоду в присутності щонайменше одного свідка, який засвідчує згоду суб’єкта дослідження в письмовій інформованій згоді.
Отримання Інформованої згоди — це процедура, за допомогою якої досліджуваний добровільно підтверджує свою згоду на участь у певному клінічному випробуванні після ознайомлення з усіма особливостями дослідження, які можуть вплинути на його рішення. Особливу увагу ЛЕК має приділяти способу надання інформації потенційним учасникам дослідження.
Учасники біомедичних досліджень з самого початку повинні розуміти, що вони абсолютно вільні у своєму рішенні щодо участі в дослідженні, та можуть відмовитися від участі (і згодом відкликати свою згоду) в ньому без пояснення причини. Особа, що не дала згоду або відкликала свою згоду в будь-який момент часу, не повинна піддаватися ніякій формі дискримінації, зокрема, щодо права на медичну допомогу.
5.1. Вимоги до оформлення та змісту інформації для пацієнта
та форми згоди
У письмовій та усній інформації, що надається пацієнту (здоровому добровольцю) або законному представнику/близькому родичу має бути відображена інформація щодо:
- дослідницького характеру клінічного випробування;
- завдань клінічного випробування;
- досліджуваного лікарського засобу, в тому числі стосовно вірогідності побічних реакцій та можливості залучення до однієї з груп клінічного випробування;
- процедур проведення клінічного випробування;
- прав та обов’язків досліджуваного;
- незручностей для досліджуваного, а також очікуваного ризику та очікуваної користі;
- клінічного випробування, яке не має лікувального характеру;
- інших видів медикаментозного або немедикаментозного лікування, що можуть бути призначені досліджуваному;
- компенсації та/або лікування, на які досліджуваний може розраховувати у разі завдання шкоди його здоров’ю під час клінічного випробування, включаючи інформацію про договір страхування життя та здоров’я пацієнта (добровольця) із зазначенням найменування та місцезнаходження страхової компанії, номера та дати укладення договору;
- розміру та умов виплат досліджуваному, якщо такі передбачені (окрім страхових);
- витрат досліджуваного, якщо такі очікуються, пов’язаних з його участю в клінічному випробуванні;
- добровільної участі у клінічному випробуванні і, що досліджуваний може відмовитися від неї в будь-який момент без пояснень, без будь-яких санкцій або обмежень його прав;
- прав представників Центру, Комісії з питань етики та спонсора стосовно безпосереднього доступу до інформації у первинній медичній документації досліджуваного для перевірки процедур та/або даних клінічного випробування та/або її обробки, не порушуючи при цьому анонімності досліджуваного;
- своєчасності ознайомлення досліджуваного або його законного представника з новою інформацією, що може вплинути на бажання досліджуваного продовжити участь у клінічному випробуванні;
- використання персональних даних досліджуваного для проведення цього клінічного випробування відповідно до Закону України «Про захист персональних даних», а саме: мети обробки персональних даних, складу та змісту персональних даних, володільця персональних даних пацієнта, доступу до персональних даних третіх осіб. Відомості про досліджуваного зберігаються в таємниці та обробляються в межах клінічного випробування у знеособленому вигляді;
- осіб, до яких можна звернутися для одержання додаткової інформації про клінічне випробування і права досліджуваного, а також фізичних та/або юридичних осіб, з якими досліджуваний може зв’язатися у разі завдання шкоди його здоров’ю під час клінічного випробування;
- обставин та/або причин, через які участь досліджуваного у клінічному випробуванні може бути припинена;
- тривалості участі досліджуваного у клінічному випробуванні;
- приблизної кількості досліджуваних, що братимуть участь у клінічному випробуванні.
Письмові матеріали та усна інформація про клінічне випробування не повинні містити спеціальних термінів і мають бути зрозумілі пацієнту (здоровому добровольцю) або законному представнику/близькому родичу.
Ні усна, ні письмова інформація про випробування, включаючи форму письмової інформованої згоди, не повинні містити будь-якого формулювання, що прямо змушує суб’єкта чи законного представника суб’єкта відмовитися від своїх законних прав або допускає подібне тлумачення. Вони також не повинні містити заяв, що звільняють дослідника, медичний заклад, спонсора чи його представників від відповідальності за заподіяну шкоду, або формулювань, що дозволяють подібну інтерпретацію.
В інтересах досліджуваного інформована згода має бути доповнена іншою протокол-специфічною інформацією.
Сфера дії одержаної згоди повинна бути чітко викладена для Комісії з питань етики та в загальному, повинна стосуватися конкретно даного проекту дослідження. Слід уникати необґрунтованого прагнення отримати згоду для використання в майбутніх дослідженнях. Наприклад, біологічні зразки та пов’язані з ними дані повинні бути анонімними в тій мірі, в якій це відповідає дослідженню.
Необхідно звертати увагу, що в формі інформованої згоди має бути передбачено місце для внесення інформації про страхову компанію та контактну особу, яку вносить дослідник власноруч.
5.2. Умови отримання інформованої згоди від пацієнтів та здорових добровольців
До початку проведення клінічного випробування лікарського засобу, Комісія з питань етики оцінює етичні та морально-правові аспекти матеріалів проекту клінічного випробування, включаючи отримання інформованої згоди від пацієнтів та здорових добровольців. Нижче перелічені основні умови отримання інформованої згоди від пацієнтів та здорових добровольців:
1. До початку участі в клінічному випробуванні пацієнт (здоровий доброволець) або його законний представник/близький родич, а також відповідальний дослідник/дослідник, відповідальний за отримання інформованої згоди, підписують та власноручно датують два примірники інформованої згоди, один з яких залишається відповідальному досліднику/досліднику, а другий передається досліджуваному.
2. При отриманні та документальному оформленні інформованої згоди дослідник повинен дотримуватися діючих регуляторних вимог, правил належної клінічної практики і етичних принципів Гельсінської декларації.
3. Перед початком випробування дослідник повинен мати письмове схвалення/ позитивне рішення експертної ради незалежної Комісії з питань етики та форми письмової інформованої згоди та будь-якої іншої письмової інформації’, що надається суб’єктам випробування.
4. Пацієнти (здорові добровольці), яких планується залучити до клінічного випробування, мають отримати достатньо інформації про мету та суть клінічного випробування. Відповідальний дослідник/дослідник, відповідальний за отримання інформованої згоди, інформує пацієнта (здорового добровольця), а якщо той неспроможний самостійно дати інформовану згоду, його законного представника/близького родича, щодо всіх аспектів клінічного випробування.
5. Дослідник надає пацієнту (здоровому добровольцю) або законному представнику/близькому родичу достатню кількість часу для прийняття рішення про участь у клінічному випробуванні.
6. Пацієнт (здоровий доброволець) або законний представник/близький родич повинен одержати вичерпні відповіді на всі питання щодо клінічного випробування.
7. Рішення пацієнта (здорового добровольця) або його законного представника/ близького родича щодо прийняття участі або продовження участі в клінічному випробуванні має бути прийняте ним самостійно й вільно, без будь-якого тиску на нього. Ні дослідник, ні співробітники, які беруть участь у випробуванні, не повинні чинити тиску на суб’єкта або викликати в нього необґрунтовану зацікавленість в участі чи продовженні участі у випробуванні.
8. Якщо суб’єкт або його законний представник не вміють або не можуть читати, протягом всієї бесіди про інформовану згоду має бути присутній незаінтересований свідок. Після того як суб’єкту чи законному представнику суб’єкта прочитали й роз’яснили форму письмової інформованої згоди та інші письмові інформаційні матеріали, що надаються суб’єкту, суб’єкт або законний представник суб’єкта дають усну згоду на участь у випробуванні та, якщо спроможні, особисто підписують і датують форму інформованої згоди. Свідок також повинен підписати й особисто датувати форму письмової інформованої згоди, цим він підтверджує, що інформація, яка міститься в цьому документі й будь-яких інших письмових інформаційних матеріалах, була точно роз’яснена й, очевидно, зрозуміла суб’єкту чи законному представникові суб’єкта, і що згода на участь у дослідженні дана добровільно суб’єктом або законним представником суб’єкта.
9. Інформація щодо договору страхування та страхової компанії (найменування, адреса, контактна особа, телефон/факс, електронна пошта тощо) заповнюється лікарем-дослідником безпосередньо в лікувально-профілактичному закладі при проведенні процедури підписання інформованої згоди пацієнтом на підставі відповідної інформації від заявника клінічного випробування.
10. Суб’єкт або законний представник суб’єкта/близький родич повинні бути вчасно ознайомлені з новою інформацією, здатною вплинути на бажання суб’єкта продовжити участь у випробуванні. Факт повідомлення цієї інформації має бути документально підтвердженим.
11. Форму письмової інформованої згоди та іншу письмову інформацію, що надається суб’єктам, слід переглядати з появою нової важливої інформації, здатної вплинути на згоду суб’єктів. Інформація щодо розгляду відповідних змін до інформованої згоди (суттєві поправки) розглядається на засіданні ЛЕК.
12. У разі участі у клінічному випробуванні малолітніх (до 14 років) та неповнолітніх (від 14 до 18 років) дітей необхідно отримувати інформовану згоду обох батьків. Неповнолітній пацієнт особисто підписує та датує інформовану згоду, яка розроблена у відповідності до його віку та розуміння.
13. У разі залучення недієздатного досліджуваного, який неспроможний самостійно дати інформовану згоду, відповідно до специфіки дослідження інформована згода має бути отримана у його законного представника / близького родича. За необхідності до підписання форми інформованої згоди може бути залучений свідок — це особа, яка не приймає участь у КВ. Процедура включення свідка до підписання інформованої згоди має бути зазначена у протоколі КВ.
14. Досліджуваний (законний представник/близький родич) у разі порушення прав досліджуванного під час проведення клінічного випробування може звернутися до спонсора, Центру, Комісії з питань етики при ЛПЗ, ЦОВВ або до суду в установленому законодавством порядку.
15. Інтереси досліджуваних (пацієнтів/здорових добровольців) завжди переважають над інтересами науки і суспільства.
6. ЕТИЧНІ ПИТАННЯ КЛІНІЧНИХ ВИПРОБУВАНЬ ЗА УЧАСТЮ МАЛОЛІТНІХ ТА НЕПОВНОЛІТНІХ ДІТЕЙ
Незважаючи на те, що дитяча популяція представляє значну частину всього населення, більшість лікарських засобів (ЛЗ) розробляються і досліджуються виключно для застосування у дорослих.
Вважається, що від 50% до 75% всіх ЛЗ (в залежності від галузі медичного застосування), які використовуються в педіатрії, проводили клінічні випробування тільки із залученням дорослої популяції. Однак, дитину не можна розглядати як зменшену копію дорослої людини. Діти мають ряд фізіологічних та психологічних особливостей, пов’язаних з ростом і розвитком організму. У дітей раннього віку можуть виникати особливі реакції на лікарські речовини, як кількісні, так і якісні. Ефективність одних і тих же ЛЗ у дітей може виявитися різною в залежності від віку дитини та методів лікування.
Відомо ряд захворювань, які не мають аналогів у дорослих, наприклад, хвороби перинатального періоду. Деякі з них швидко прогресують і можуть призводити до незворотних змін в організмі дитини. Відмінності між дитячою та дорослою популяцією, а також між дітьми різних вікових груп дозволяють говорити про необхідність проведення клінічних випробувань ЛЗ в педіатрії, оскільки існує ризик неефективності ЛЗ при їх медичному застосуванні у дітей.
Світові організації такі як ВООЗ, FDA, EMA, стимулюють світових виробників ліків до розробки лікарських засобів та проведення клінічних випробувань та, в подальшому, реєстрації лікарських засобів для дітей.
Тому, в усьому світі, і в Україні в тому числі, гостро стоїть питання необхідності розробки нових та докладного вивчення вже існуючих на ринку лікарських засобів для використання їх в педіатричній практиці.
В законодавчо-нормативних документах України визначені ті особливості, які мають бути враховані при плануванні та проведенні КВ лікарських засобів із залученням дітей.
У 8 статті Закону України «Про лікарські засоби» зазначено:
«Клінічні випробування лікарських засобів за участю малолітньої або неповнолітньої особи можуть проводитися лише у разі, якщо відповідний лікарський засіб призначений для лікування дитячих захворювань або якщо метою клінічних випробувань є оптимізація дозування чи режиму застосування лікарського засобу відповідно для малолітніх або неповнолітніх осіб.
Клінічні випробування лікарських засобів за участю малолітньої або неповнолітньої особи, метою яких є оптимізація дозування чи режиму застосування лікарського засобу для таких осіб, проводяться після завершення клінічних випробувань відповідних лікарських засобів за участю повнолітніх дієздатних осіб.
Клінічні випробування лікарських засобів за участю малолітньої особи можуть проводитися у порядку, передбаченому Законом України «Про лікарські засоби», за наявності письмової згоди її батьків та за умови надання малолітній особі відповідної інформації в доступній для неї формі, а за участю неповнолітньої особи -у разі наявності її письмової згоди та письмової згоди її батьків.
У разі проведення клінічних випробувань за участю малолітніх та неповнолітніх осіб відповідна інформація направляється до органів опіки та піклування за місцем постійного проживання таких осіб у порядку, встановленому центральним органом виконавчої влади у галузі охорони здоров’я. Забороняється проведення клінічних випробувань лікарських засобів за участю малолітньої або неповнолітньої особи, яка позбавлена батьківського піклування, усиновленої дитини або дитини-сироти».
В Україні проведення КВ регламентується Порядком, який визначає особливості проведення КВ із залученням дітей.
Клінічні випробування за участю малолітніх (до 14 років) та неповнолітніх (від 14 до 18 років) дітей проводяться у разі:
а) отримання письмової інформованої згоди обох батьків;
б) надання доступної для розуміння малолітньою та неповнолітньою дитиною письмової та усної інформації про клінічне дослідження. Якщо малолітній спроможний, він усно дає свою згоду на участь у клінічному дослідженні.
Неповнолітній пацієнт особисто підписує та датує інформовану згоду.
Відповідальний дослідник/дослідник, відповідальний за отримання інформованої згоди, враховує явне бажання малолітньої та неповнолітньої дитини взяти участь, або відмовитися від участі в клінічному дослідженні, або вийти з нього в будь-який час;
в) відсутності використання будь-яких заохочень або стимулів, крім компенсації у разі завдання шкоди здоров’ю під час клінічного випробування;
г) малолітні та неповнолітні діти одержать безпосередню користь від участі у клінічному випробуванні, якщо:
- інших клінічних випробувань, які проводились на дорослих, або для підтвердження даних, отриманих за допомогою інших методів дослідження;
- клінічне випробування стосується захворювань, від яких страждають малолітні та неповнолітні діти;
- клінічне випробування має такі особливості, що його можна проводити тільки за участю малолітніх та неповнолітніх дітей;
ґ) планування клінічних випробувань здійснено таким чином, що мінімізовані біль, дискомфорт, страх і ризик. Поріг ризику і ступінь дискомфорту, болю чітко визначаються і постійно відстежуються;
Інформація про залучення до клінічних випробувань малолітніх та неповнолітніх дітей в довільній формі, але з обов’язковим зазначенням прізвища, імені та по батькові, віку дитини, прізвищ, імен та по батькові батьків, місце проживання дитини, наявності інформованої згоди, коротке викладення суті клінічного випробування та з обов’язковою позначкою, що інформація, яка надається, є конфіденційною. Ця інформація направляється відповідальним дослідником/дослідником до органів опіки та піклування за місцем постійного проживання дитини протягом доби з дати отримання інформованої згоди на участь у клінічному випробуванні.
Інтереси малолітніх та неповнолітніх дітей переважають над інтересами науки і суспільства.
Діти відносяться до особливо вразливих груп, які можуть залучатися до КВ. Тому, особливе місце у КВ педіатричних лікарських засобів відводиться дотриманню етичних норм. Члени комісії з питань етики при лікувально-профілактичному закладі, де планується проведення КВ повинні володіти достатнім об’ємом знань у галузі педіатрії.
Найважливішими цілями діяльності ЛЕК при розгляді протоколу КВ лікарських засобів за участю дітей є:
- оцінка наукової і клінічної обґрунтованості дослідження та важливості передбачуваних результатів;
- захист здоров’я і прав дітей при проведенні досліджень.
Один з перших моментів, на який повинні звертати увагу члени ЛЕК, стосується ініціювання клінічного дослідження за участю дітей. Це можливо лише в тому випадку, коли необхідні дані неможливо отримати за участю пацієнтів інших вікових груп.
Дослідження нових методів терапії повинні проводитися тільки на тих групах пацієнтів, для яких ці методи розробляються.
При цьому особлива увага приділяється оцінці ризик/користь.
Існує декілька категорій досліджень із залученням дітей, що відрізняються співвідношенням рівня ризику та користі.
1. Дослідження з ризиком не більше ніж мінімальний.
2. Дослідження з більш ніж мінімальним ризиком, але з перспективою прямої користі для досліджуваного. Випробування, що відносяться до цієї категорії, схвалюються в таких випадках:
- ризик виправдовується очікуваною користю для суб’єкта;
- співвідношення ризик /користь, щонайменше, є настільки ж сприятливим, як і при будь-якому іншому можливому підході.
3. Дослідження з більш ніж мінімальним ризиком, без перспективи прямої користі для досліджуваних, спрямоване на узагальнення знань щодо даної патології. Дослідження, що відносяться до цієї категорії’, схвалюються в таких випадках:
- ризик лише злегка перевищує мінімальний, тобто ризик, який цілком можна порівняти з тим реальним чи очікуваним ризиком, з яким пацієнти стикаються при звичайному лікуванні (наприклад, стандартне лікування);
- КВ, швидше за все, дасть можливість узагальнення і поглиблення знань в області досліджуваної патології, що матиме велике значення для інших пацієнтів в майбутньому.
У КВ за участю дітей, при відсутності істотних протипоказань, необхідно намагатися здійснювати набір пацієнтів з урахуванням демографічної картини суспільства.
Особи, які не досягли повноліття, залежать багато в чому від своїх батьків. Батьки несуть відповідальність за прийняття рішення про участь у випробуванні. Письмову згоду на участь дитини у КВ, які не досягли 18-річного віку, підписують батьки. їм надається для ознайомлення інформація про випробування та форма згоди на участь дитини у випробуванні.
Принцип інформованої згоди, який закладений в GCP, передбачає добровільну згоду обох батьків дитини та самої дитини на участь у КВ на основі інформування про всі аспекти випробування. Гельсінська декларація Всесвітньої медичної асоціації, 1964-2013 рр. передбачає, що «згоду дитини потрібно отримати на додаток до дозволу її батьків».
При проведенні КВ, так само як і в ході медичного лікування, основоположним принципом є максимально можливе залучення та активна участь дитини в процесі прийняття рішення. Інформація про КВ повинна бути передана дітям мовою, доступною для їхнього розуміння з урахуванням віку.
Діти, які досягли 14-річного віку, також як і батьки, власноруч підписують і датують інформовану згоду (форму передбачену для даного віку). Дитина має право погоджуватися чи не погоджуватися брати участь у КВ, або відмовитися від участі в ньому в будь-який момент (п. 3, р.№ Порядку).
Включення учасників КВ має проводитись без неправомірного спонукання до цього батьків чи самих учасників.
У процесі отримання інформованої згоди особи, яка не досягла повноліття, повинні бути присутні батьки. Під час цього процесу обговорюють:
- відомості про КВ, доступні для розуміння дитини; розповідь про те, чого він може чекати від діагностичного або лікувального втручання;
- клінічна оцінка розуміння ситуації дитиною і факторів, що впливають на його відповідь (в тому числі, чи не було надано тиску для отримання згоди на проведення діагностичного або лікувального втручання);
- прохання до пацієнта висловити його готовність прийняти умови випробування.
Отримання згоди дитини являє собою активне спілкування, при якому сторони діляться інформацією та її оцінкою і приймають спільне рішення.
Можливі ситуації, коли пацієнт наполегливо відмовляється від втручання. Найчастіше це буває у разі клінічного обстеження. Лікар повинен поважати бажання пацієнта припинити або тимчасово відмовитися від участі у випробуванні, щоб обміркувати своє рішення або впоратися зі своїми побоюваннями. Тиск і примус до діагностики та лікування є неприпустимими.
КВ, що проводяться за участю дітей, які є фізично або розумово неповноцінними, повинні бути обмежені. Планування таких випробувань може бути обумовлено гострою медичною необхідністю. Вони повинні бути науково і клінічно обґрунтованими і проводитися за суворими показаннями та у відповідності до нормативно-правових вимог. Таким випробуванням ЛЕК приділяють особливу увагу.
До початку випробування дослідник повинен бути повністю поінформований про всі існуючі побічні ефекти досліджуваного лікарського засобу. При плануванні КВ в педіатрії необхідно ретельно проаналізувати всі доклінічні дані та дані з клінічної безпеки досліджуваних методів діагностики і лікування на прикладі дорослих пацієнтів.
Для того, щоб звести до мінімуму ризик в педіатричних КВ, дослідники повинні мати відповідну підготовку та досвід проведення КВ в педіатрії. КВ повинні проводитися в ЛПЗ, які визначені для лікування дітей (педіатричних клініках, відділах або відділеннях), що передбачають наявність медичного персоналу, компетентного в медичних та психосоціальних питаннях при лікуванні дітей.
Зменшити дискомфорт та розвиток негативних емоцій у дітей, викликаних процедурами, дозволяє кваліфікована робота досвідченого персоналу, раціональна мінімізація кількості інвазивних процедур.
Таким чином, КВ із залученням дітей може розглядатися як етично прийнятне, якщо:
1. Необхідні дані не можуть бути отримані на дорослих пацієнтах;
2. Випробування раціонально сплановано з урахуванням мінімізації дискомфорту та інвазивних процедур;
3. Дослідження передбачає отримання важливих результатів, спрямованих на вдосконалення діагностики та лікування або сприяють узагальненню і систематизації даних про дитячі захворювання;
4. Дослідження базується на основі вже отриманих та проаналізованих результатів доклінічних досліджень та КВ із залученням дорослих пацієнтів, поглибленому знанні історії проблеми. Очікувані результати КВ мають лише підтвердити обґрунтованість КВ;
5. Очікувана користь від дослідження перевищує потенційний ризик, а потенційний ризик є мінімальним, чи не більшим, ніж при виконанні звичайних лікувальних і діагностичних процедур при даній патології;
6. Дослідник володіє достатньою інформацією про передбачуваність будь-яких можливих несприятливих наслідків дослідження;
7. Досліджуваним і їх батькам надана вся інформація, необхідна для отримання їх усвідомленої і добровільної згоди.
Клінічне випробування повинно проводитись відповідно до затвердженого протоколу: тільки організоване належним чином, з дотриманням етичних норм, спрямованих на захист кожної дитини, яка бере участь у випробуванні.
Проведення КВ із залученням дітей різної вікової категорії дозволяє забезпечити їх ефективними та безпечними лікарськими засобами у відповідних лікарських формах, що буде сприяти удосконаленню раціональної терапії дитячих захворювань.
7. КЛІНІЧНІ ВИПРОБУВАННЯ ЗА УЧАСТЮ НЕДІЄЗДАТНИХ ОСІБ (В ТОМУ ЧИСЛІ, ОСІБ, ЯКІ ПЕРЕБУВАЮТЬ В КРИТИЧНИХ ТА НЕВІДКЛАДНИХ СТАНАХ)
Принцип добровільної інформованої згоди учасників є центральним для етичного проведення біомедичних досліджень. Проведення досліджень на людях, не здатних дати свою згоду, є важливим для поліпшення діагностики, лікування і профілактики захворювань в цих групах осіб. Тому, за умови дотримання необхідних заходів захисту, законодавча база дозволяє такі дослідження. Особи, нездатні дати свою згоду, включаючи дітей, не повинні виключатися з участі у відповідних дослідженнях.
Перш ніж дозволити таке дослідження, Комісія з питань етики повинна впевнитися, що КВ є науково обґрунтованим і не може бути рівноцінним при проведенні дослідження на особах, здатних дати свою згоду. Загалом, дослідження повинно бути потенційно корисним для здоров’я учасників (пряма користь) і будь-які очікувані ризики, включаючи ризик щодо недоторканності приватного життя, не повинні перевищувати потенційну користь.
У разі, якщо відсутня ймовірність прямої користі, дослідження можуть проводитися із застосуванням додаткових заходів захисту, тільки якщо це дозволяється національним законодавством, включаючи:
- дослідження, націлене на розширення наукового розуміння хвороби або порушення у людини, яке може принести суттєву користь учаснику або іншим людям з таким або подібним захворюванням або порушенням;
- дослідження тягне за собою мінімальний ризик і мінімальне навантаження для учасника.
Нездатність дати свою згоду може бути частковою або повною, тимчасовою або постійною. Важливо, що багато людей, які не можуть дати свою згоду особисто, однак можуть зрозуміти деяку інформацію щодо пропонованого втручання. Здатність до розуміння інформації, що буде надана потенційному досліджуваному, повинна бути оцінена дослідником.
Інформація щодо КВ повинна бути представлена потенційним учасникам, щоб дізнатися про їх готовність до співпраці або їх заперечення проти участі в дослідженні.
Необхідний правовий захист зазвичай забезпечує законний представник, який повинен отримати всю належну інформацію щодо пропонованого дослідження.
При подачі матеріалів КВ до ЛЕК заявники повинні подати документи, які вони мають намір надати законному представнику, ступінь детальності викладу повинен бути таким, як при наданні інформації дієздатній особі, потенційному учаснику дослідження. Згода законного представника, яка має бути в письмовій формі, враховує раніше виражене потенційним досліджуваним бажання або заперечення щодо участі в КВ.
Законний представник може не надати згоду на участь в дослідженні особи, інтереси якої він представляє, або відкликати її в будь-який час без збитку для досліджуваного. Крім того, законні представники не повинні отримувати ніяку особисту вигоду від згоди або незгоди на участь потенційного учасника в КВ.
Обов’язки законного представника полягають в тому, щоб представляти інтереси відповідної особи, але законний представник не є особистим адвокатом цієї особи.
Відповідно до Порядку: «Законні представники-батьки, усиновлювачі, батьки-вихователі, опікуни, піклувальники, представники закладів, які виконують обов’язки опікунів та піклувальників».
Клінічні випробування за участю недієздатних пацієнтів проводяться, якщо (пункт 4.1. Розділу IV Порядку):
- отримано інформовану згоду законного представника/близького родича (згода може бути в будь-який момент відкликана без негативних для досліджуваного наслідків);
- недієздатним пацієнтом одержано інформацію про клінічне випробування, пов’язані з ним ризик і користь у формі, доступній для його розуміння;
- відповідальним дослідником/дослідником, відповідальним за отримання інформованої згоди, враховано бажання недієздатного пацієнта узяти участь, або відмовитися від участі в клінічному випробуванні, або вийти з нього в будь-який момент;
- не використовуються будь-які заохочення або стимули, крім компенсації у разі завдання шкоди здоров’ю досліджуваного під час клінічного випробування;
- клінічне випробування має значення для підтвердження інформації, отриманої під час інших клінічних випробувань або отриманої іншими методами дослідження;
- клінічне випробування безпосередньо стосується захворювання, на яке страждає досліджуваний;
- клінічне випробування сплановане таким чином, щоб мінімізувати біль, дискомфорт, страх і ризик. Поріг ризику і ступінь дискомфорту, болю повинні бути чітко визначені і постійно відстежуватися;
- є підстави очікувати, що застосування досліджуваного лікарського засобу принесе досліджуваному користь, що перевищує ризик або не призведе до жодного ризику.
Нижче представлені ключові питання, які ЛЕК повинна розглянути при проведенні етичної експертизи дослідження за участю осіб, які неспроможні дати свою згоду:
- Чи дозволяє нормативна база проводити дослідження за участю осіб, які неспроможні дати свою згоду?
- Чи задовольняє дослідження усі відповідні умови для проектів досліджень за участю осіб, здатних дати свою згоду?
Крім того:
- Чи обгрунтував спонсор наукову необхідність проведення дослідження за участю осіб, які неспроможні дати свою згоду?
- Чи є які-небудь альтернативи для дослідження в плані наукової ефективності, які можна було б провести за участю осіб, здатних дати згоду?
- Яка причина нездатності надати згоду?
- Як будуть оцінювати відсутність здатності дати свою згоду?
Дослідження з потенційною прямою користю для учасника:
- Чи є ризики і навантаження для досліджуваного прийнятними в порівнянні з очікуваною користю для учасника?
Дослідження без потенційної прямої користі:
- Чи обґрунтував спонсор наукову необхідність даного типу дослідження?
- Як будуть оцінювати мінімальний ризик та мінімальне навантаження?
- Чи є які-небудь спеціальні заходи захисту, передбачені нормативними документами, і як вони будуть дотримуватися?
- У чому полягають плани дій дослідників в разі непередбачених результатів дослідження?
Питання, стосовно законних представників, які ЛЕК повинна розглянути при проведенні етичної експертизи:
- Хто є законним представником, який має право вирішувати питання щодо участі суб’єкта в КВ?
- Яку інформацію отримає законний представник щодо пропонованого дослідження?
- Як потенційні учасники дослідження братимуть участь в процедурі прийняття рішення щодо участі в КВ?
- Як буде реєструватися незгода на участь потенційного учасника, та як будуть повідомляти про них законного представника?
- Чи є призначена особа, яка буде відповідати на будь-які питання, які можуть виникнути в учасників щодо дослідження або процедури дозволу?
- Чи передбачено відкликання згоди на участь в КВ, як учасники дослідження братимуть участь в наданні згоди/відкликанні згоди?
8. КЛІНІЧНІ СИТУАЦІЇ, ЩО ВИМАГАЮТЬ НЕВІДКЛАДНОЇ МЕДИЧНОЇ ДОПОМОГИ
Клінічні ситуації, що вимагають невідкладної медичної допомоги-це такі випадки, коли необхідність невідкладної допомоги є непередбачуваною, а швидкі дії необхідними, наприклад, зупинка серця, важкий напад, травми голови, небезпечні для життя, тощо. Ефективні види лікування багатьох станів, що призводять до ситуацій, що вимагають невідкладної допомоги, вельми обмежені, тому дослідження є дуже важливими для розвитку доказової медицини. Однак проведення досліджень в клінічних ситуаціях, що вимагають невідкладної медичної допомоги є етично проблемним, оскільки неможливо дотримуватися основних етичних і правових вимог отримання інформованої згоди пацієнта.
У міжнародних документах, що стосуються біомедичних досліджень-Директива 2001/20 / ЄС та Додатковий протокол Ради Європи до Конвенції Ов’єдо про біомедичні дослідження-тільки в Протоколі розглядаються дослідження в невідкладних ситуаціях. Додатковим Протоколом до Конвенції Ов’єдо про біомедичні дослідження (Стаття 19) передбачені наступні умови захисту:
- дослідження не може бути проведено з аналогічною ефективністю в ситуаціях, які не є невідкладними;
- проект був спеціально дозволений стосовно невідкладних ситуацій;
- будь-якому раніше вираженому запереченні учасників, про який відомо дослідникам, має поважатися.
Додатковим протоколом Ради Європи про біомедичні дослідження передбачена вимога, що в найкоротші терміни учасникам дослідження або, у відповідних випадках, їх представникам, повинна бути представлена вся належна інформація про участь в дослідженні і повинно бути поставлено питання про їхню згоду або дозвіл на продовження участі в КВ. Якщо згода не надається, у досліджуваного учасника/законного представника повинна бути можливість вимагати виключення з дослідження всіх отриманих персональних даних.
Ключові питання, які повинні розглянути члени ЛЕК при етичній експертизі матеріалів КВ, що стосуються клінічних ситуацій, що вимагають невідкладної медичної допомоги:
- Чи можливо отримати подібні результати при проведенні дослідження за участю людей, які не перебувають в ситуаціях, що вимагають невідкладної медичної допомоги?
- Чи будуть потенційні учасники дослідження в стані, який не дозволить їм надати інформовану згоду?
- Наскільки екстреною є ситуація? Чи є ліміт часу настільки жорстким, що не дозволить знайти законних представників для отримання згоди?
- Чи може дослідження принести потенційну пряму користь учасникам дослідження?
- Якщо потенційної прямої користі немає, чи націлене дослідження на отримання результатів, які можуть принести користь іншим учасникам дослідження або іншим людям з аналогічним порушенням / станом?
- У чому полягають ризики і навантаження, пов’язані з дослідженням?
- При відсутності потенційної прямої користі чи є ризики і навантаження мінімальними?
- Які процедури передбачені дослідниками для гарантування того, що:
– дозвіл буде отримано від законних представників потенційних учасників дослідження відповідно до нормативних вимог?
– вся належна інформація, що стосується участі в проекті дослідження, буде надана досліджуваним або, у відповідних випадках, їх представникам у найкоротші терміни після включення їх у дослідження?
– питання про згоду на участь / продовження участі буде поставлено в найкоротші терміни після включення досліджуваного у КВ?
Таким чином, клінічне випробування може проводитися у разі, коли:
- очікуваний ризик і незручності були зважені стосовно очікуваної користі для досліджуваних (пацієнтів/здорових добровольців);
- досліджуваний або, якщо він не здатний особисто дати інформовану згоду, його законний представник, а в разі відсутності законного представника у пацієнтів, які перебувають у критичному та невідкладному станах-близький родич мав можливість під час розмови з відповідальним дослідником/ дослідником, відповідальним за отримання інформованої згоди, зрозуміти мету, ризик і незручності клінічного випробування, а також умови, за яких воно буде проводитися;
- права досліджуваного на фізичне і психічне благополуччя, таємницю особистого життя і захист персональних даних забезпечені відповідно до вимог законодавства;
- недієздатним пацієнтом одержано інформацію про клінічне випробування, пов’язані з ним ризик і користь у формі, доступній для його розуміння;
- відповідальним дослідником/дослідником, відповідальним за отримання інформованої згоди, враховано бажання недієздатного пацієнта узяти участь, або відмовитися від участі в клінічному випробуванні, або вийти з нього в будь- який момент;
- не використовуються будь-які заохочення або стимули, крім компенсації у разі завдання шкоди здоров’ю досліджуваного під час клінічного випробування;
- клінічне випробування має значення для підтвердження інформації, отриманої під час інших клінічних випробувань або отриманої іншими методами дослідження;
- клінічне випробування безпосередньо стосується захворювання, на яке страждає досліджуваний;
- клінічні випробування сплановані таким чином, щоб мінімізувати біль, дискомфорт, страх і ризик. Поріг ризику і ступінь дискомфорту, болю повинні бути чітко визначені і постійно відстежуватися;
- у виняткових випадках, якщо особа не в змозі писати/читати, то нею може бути дана усна згода в присутності мінімум одного свідка, який письмово засвідчує згоду пацієнта в інформованій згоді;
- за бажанням досліджуваний (законний представник/близький родич, що підписав інформовану згоду) у будь-який час може без будь-якої шкоди для себе (досліджуваного) припинити участь у клінічному випробуванні.
У разі неможливості одержання інформованої згоди у пацієнта і відсутності його законного представника/близького родича залучення таких пацієнтів до клінічного випробування не допускається, їм має надаватися медична допомога у встановленому порядку.
9. ЕТИЧНІ ПИТАННЯ ВИКОРИСТАННЯ ПЛАЦЕБО
Плацебо можна застосовувати в якості методу контролю тільки в строго певних умовах, а саме, якщо відсутні методи з доведеною ефективністю або якщо відмову або утримання від застосування таких методів не тягне за собою неприйнятний ризик або навантаження. Отже, ЛЕК повинен приділяти підвищену увагу щодо очікуваного ризику або навантаження під час використання плацебо.
Перелік конкретних питань, необхідних для розгляду ЛЕК плацебо-контрольованих досліджень:
- Чи є незаперечна наукова підстава для проведення плацебо- контрольованого дослідження?
- Чи є відомий метод лікування з доведеною ефективністю?
- Якщо так, то чи знаходження без такого лікування протягом періоду, необхідного для проведення КВ є безпечним для пацієнтів? Іншими словами, чи є додатковий ризик прийнятним?
- Чи прийнятне додаткове навантаження на пацієнта внаслідок відсутності лікування?
- Чи будуть пацієнти поінформовані про ймовірність розподілу в групу, яка буде отримувати плацебо?
- Чи передбачено включення пацієнтів, нездатних надати інформовану згоду?
- Чи передбачені заходи для раннього виявлення серйозної несприятливої динаміки хвороби у пацієнтів групи плацебо?
- Чи передбачено проведення належного, своєчасного проміжного аналізу?
- Чи отримають учасники дослідження інформацію про те, в яку групу вони були розподілені, коли дослідження завершиться?
10. МОНІТОРИНГ ЩОДО ЗАБЕЗПЕЧЕННЯ ЗАХИСТУ ПРАВ, БЕЗПЕКИ, БЛАГОПОЛУЧЧЯ ДОСЛІДЖУВАНИХ, ЕТИЧНИХ ТА МОРАЛЬНО-ПРАВОВИХ АСПЕКТІВ У ПРОЦЕСІ ПРОВЕДЕННЯ КЛІНІЧНОГО ВИПРОБУВАННЯ
Члени ЛЕК виконують конкретні функції до, під час і після проведення КВ. Тому відповідальність і практичні обов’язки членів ЛЕК охоплюють повний цикл від планування до закінчення дослідження.
До початку дослідження проводиться етична експертиза його матеріалів.
Основна задача ЛЕК полягає у тому, щоб гарантувати, що матеріали, які розглядаються для проведення КВ є етично прийнятними. Таким чином, Комісії з питань етики забезпечують публічні гарантії недопущення неетичних досліджень і заохочення етично прийнятних досліджень належного рівня якості.
З метою забезпечення захисту прав, безпеки, благополуччя пацієнтів (здорових добровольців), досліджуваних, етичних та морально-правових аспектів у процесі проведення КВ в ЛПЗ Комісія з питань етики повинна здійснювати моніторинг клінічного випробування.
Особливо це важливо у тих випадках, коли дослідження має такий рівень ризику, який не може бути проігнорований, або коли очікується, що у дослідженні буде отримана клінічно значуща інформація, яка може вплинути-позитивно або негативно на безпечність, здоров’я або благополуччя досліджуваних. Особлива увага приділяється залученню до КВ малолітніх та неповнолітніх осіб, недієздатних осіб, пацієнтів у критичному та невідкладному станах.
Порядок здійснення моніторингу клінічного випробування Комісією з питань етики повинен бути передбачений у Положенні та чітко прописаний у СОПах.
СОП повинна визначити загальний порядок проведення моніторингу щодо забезпечення захисту прав, безпеки, благополуччя досліджуваних, етичних та морально-правових аспектів у процесі проведення КВ в конкретному лікувально-профілактичному закладі, а також оцінка результату проведеного моніторингу. Проведення моніторингу передбачає контакти з відповідальним дослідником та дослідниками, а також доступ до конфіденційної інформації досліджуваних (наприклад, матеріали КВ та історії хвороби пацієнтів).
Підходи щодо здійснення моніторингу можуть бути різними, тому СОП необхідно передбачити як аналіз інформації, яка надходить до ЛЕК на будь-якому етапі КВ, так і безпосередньої її перевірки в місці проведення дослідження.
Також у СОП необхідно передбачити періодичність проведення безпосередньої перевірки клінічного випробування в місці його проведення, що може в кожному конкретному випадку відрізнятися.
10.1. Аналіз наявної в ЛЕК інформації щодо КВ
Під час аналізу в ЛЕК особлива увага приділяється повідомленням, які може отримувати ЛЕК в ході дослідження, наприклад:
- про відхилення від протоколу;
- про обставини, що підвищують ступінь ризику для суб’єктів і/чи істотно впливають на проведення клінічного випробування;
- про всі серйозні непередбачувані побічні реакції;
- про появу нових даних, які можуть свідчити про несприятливий вплив на безпеку досліджуваних або вплинути на хід дослідження в цілому;
- щодо порушення процедури отримання інформованої згоди.
Результати розгляду такої інформації повинні бути документально зафіксовані.
Також під час здійснення моніторингу клінічного випробування ЛЕК повинна бути переконана, що договір страхування чинний, покриває всю кількість досліджуваних, залучених до КВ, та весь період КВ (з дати включення першого пацієнта до дати останнього візиту останнього досліджуваного).
ЛЕК контролює вчасність надання періодичного звіту про стан проведення КВ та перевіряє інформацію щодо затверджених суттєвих поправок, кількості залучених досліджуваних, інформацію щодо суттєвих відхилень від протоколу КВ, інформацію щодо підозрюваних непередбачуваних серйозних побічних реакцій.
10.2. Проведення моніторингу (перевірки) КВ в місці його проведення, що проводить ЛЕК
СОП проведення моніторингу щодо забезпечення захисту прав, безпеки, благополуччя досліджуваних, етичних та морально-правових аспектів може визначати об’єкт перевірки, осіб, які можуть здійснювати перевірку, терміни проведення моніторингу, порядок комунікацій, обсяг перевірки, порядок документального оформлення, обговорення та оцінки результатів перевірки, також порядок можливих дій у разі виявлення недоліків, що можуть негативно вплинути або вплинули на права, безпеку та здоров’я досліджуваних.
10.3. Планування проведення моніторингу (перевірки)
Необхідно визначити:
- місце, обсяг, дату та термін проведення перевірки;
- осіб, що будуть здійснювати перевірку;
- об’єм документів, що будуть перевірятись.
10.4. Підготовка до моніторингу (перевірки)
Особи, які здійснюватимуть моніторинг (перевірку), можуть брати з собою необхідні матеріали КВ, які знаходиться у їх розпорядженні, наприклад, для порівняння комплектації документів.
10.5. Інформування дослідника про моніторинг
(перевірку)
Сповіщення відповідального дослідника про майбутню перевірку (повідомити, яке дослідження буде перевірено, в якому об’ємі, хто буде перевіряти) та узгодити час перевірки.
10.6. Безпосереднє проведення моніторингу
(перевірки)
Моніторингу (перевірці) підлягають документи, записи, приміщення, устаткування та обладнання, які зберігаються у дослідника, що мають стосунок до клінічного випробування, що перевіряється.
Члени Комісії з питань етики, які здійснюють моніторинг в місці проведення дослідження (ЛПЗ), діють у відповідності до своїх повноважень та на підставі наданого досліджуваним доступу до його персональних даних під час підписання ним інформованої згоди.
«У письмовій та усній інформації, що надається пацієнту (здоровому добровольцю) або законному представнику / близькому родичу, зазначається інформація щодо:… прав… комісії з питань етики… стосовно безпосереднього доступу до інформації у первинній медичній документації досліджуваного для перевірки процедур та/або даних клінічного випробування та/або її обробки, не порушуючи при цьому анонімності досліджуваного» (додаток 2 до Порядку).
Членам Комісії з питань етики, які здійснюють моніторинг КВ необхідно переконатися, що:
- клінічне випробування почалось (дата підписання форми інформованої згоди першим пацієнтом в МПВ) після отримання позитивного рішення ЦОВВ та витягу з протоколу комісії з питань етики;
- форма інформованої згоди та інша письмова інформація, що надавалася досліджуваним, була погоджена Комісією з питань етики та затверджена ЦОВВ;
- процедура отримання інформованої згоди проводилась відповідним чином та чітко зазначена в історії хвороби/амбулаторній картці пацієнта.
- забезпечено страховий захист досліджуваних.
Комісія з питань етики повинна переконатися, що:
- відповідальний дослідник/дослідник(и)/співробітники, які беруть участь у випробуванні, не чинили тиску на суб’єктів випробування;
- пацієнту (здоровому добровольцю) або законному представнику/ близькому родичу надано вичерпну інформацію про суть, значимість, значення і ризик клінічного випробування, у якому вони беруть участь, та достатню кількість часу для прийняття рішення щодо участі у клінічному випробуванні;
- пацієнт (здоровий доброволець) його законний представник/близький родич одержав вичерпні відповіді на всі питання щодо клінічного випробування;
- особи, на участь у клінічних випробуваннях яких обов’язково потрібна згода їх законного представника, поінформовані в межах їх розуміння про клінічне випробування, та прийнято до уваги їхнє бажання чи небажання участі в запропонованому клінічному випробуванні.
Крім того, під час проведення моніторингу (перевірки) клінічного випробування, у яке залучаються малолітні або неповнолітні особи, члени Комісії з питань етики повинні переконатися, що інформація про залучення до клінічного випробування малолітніх та неповнолітніх дітей була направлена відповідальним дослідником/дослідником до органів опіки та піклування за місцем постійного проживання дитини протягом доби з дати отримання інформованої згоди на участь у клінічному випробуванні. Ця інформація повинна містити прізвище, ім’я та по батькові, вік дитини, прізвища, імена та по батькові батьків, місце проживання дитини, наявність інформованої згоди, коротке викладення суті клінічного випробування та обов’язково повинна мати позначку, що ця інформація є конфіденційною (пункт 3.2. р. IV Порядку).
Комісія з питань етики повинна перевіряти усі методи інформування та залучення пацієнтів (здорових добровольців) до клінічних випробувань лікарських засобів у лікувально-профілактичному закладі та у разі виявлення порушень надає відповідну інформацію до Центру (пункт 2.5 Типового положення).
Під час перевірки Комісія з питань етики може запитати у дослідника інформацію про відхилення від протоколу, наявність побічних явищ та реакцій, конфліктних ситуацій, пов’язаних з порушенням прав, безпеки, благополуччя досліджуваних пацієнтів (здорових добровольців), етичних та морально-правових аспектів у процесі проведення клінічного випробування (пункт 2.8.2 Типового положення).
Крім того, ЛЕК може запитати у дослідника інформацію про побічні реакції, які сталися в місці проведення дослідження та, що може розцінюватися як страховий випадок, та перевірити чи відповідальний дослідник/дослідник вчасно інформував про це спонсора (пункт 1.4 розділ IV Порядку).
У разі виникнення в місці проведення дослідження, що перевіряється, підозрюваних непередбачуваних серйозних побічних реакцій ЛЕК може перевірити вчасність надання такої інформації. Терміни надання інформації щодо підозрюваних непередбачуваних серйозних побічних реакцій повинні відповідати розділу ХІІ Порядку.
Комісія з питань етики може перевірити порядок надання інформації про клінічне випробування пацієнту (здоровому добровольцю)/законному представнику/близькому родичу, а також отримання інформованої згоди від нього.
ЛЕК може запитати у відповідального дослідника / дослідника щодо усної інформації про клінічне випробування, яку він надає досліджуваному, та переконатися, що висловлювання не змушують пацієнта (здорового добровольця), а також його законного представника/близького родича відмовитись від своїх прав.
Також Комісія з питань етики може перевірити файл дослідника, комплектація якого повинна відповідати додатку 1 Порядку.
Перевірка та її обсяг повинні бути документально зафіксовані.
10.7. Представлення результатів моніторингу (перевірки)
Особи, які проводять моніторинг (перевірку), доводять до відома дослідників свої спостереження, зауваження, вказують виявлені недоліки (якщо є).
Обговорюються відповідні рекомендації щодо усунення недоліків (якщо виявлені) та визначається термін їх усунення (якщо необхідно).
Результати моніторингу (перевірки) разом з іншою інформацією повинні бути документально зафіксовані та оформлені у вигляді звіту.
З метою поширення інформації серед членів усієї ЛЕК результати перевірки мають бути представлені на засіданні ЛЕК.
10.8. Тимчасове або повне зупинення клінічного випробування
При виявленні під час здійснення моніторингу КВ порушень прав, безпеки, благополуччя досліджуваних, етичних та морально-правових аспектів у процесі проведення КВ ЛЕК, після прийняття рішення на засіданні, подає письмові пропозиції до Центру про можливість розгляду питання щодо тимчасового або повного зупинення клінічного випробування, а також інформує спонсора/заявника.
Відновлення проведення КВ, що було тимчасово зупинено, можливе у разі повного усунення причин, що обумовили тимчасове зупинення, та інформування про це ЛЕК для прийняття відповідного рішення на засіданні.
У разі повного зупинення клінічного випробування його відновлення можливе за наявності повторного погодження ЛЕК.
11. ЗАСОБИ САМООЦІНКИ КОМІСІЇ З ПИТАНЬ ЕТИКИ ПРИ ЛПЗ
Крім незалежного аудиту або інспекційних перевірок, які можуть проводитись у відповідності до нормативних вимог, ЛЕК повинні мати механізми для періодичної оцінки якості своєї роботи та функціонування з метою знаходження можливостей удосконалення. Така оцінка ЛЕК може проводитись шляхом, наприклад, анкетування.
Типові засоби самооцінки роботи ЛЕК:
- вільна дискусія серед членів ЛЕК в спеціально відведений час на засіданнях;
- підготовка та обговорення щорічних звітів ЛЕК;
- заповнення та оцінка анкет по самооцінці ЛЕК;
- структуровані завдання, націлені на самооцінку ЛЕК.
Відповідальність і завдання Голови та членів Комісії з питань етики повинні бути чітко прописані, наприклад, в Положенні або стандартних операційних процедурах.
Такий аналіз дозволить удосконалити роботу ЛЕК, а також буде забезпечувати підвищення сукупності знань членів ЛЕК, які так необхідні для проведення експертизи матеріалів КВ.
Слід відзначити, що виникла необхідність створення єдиних підходів та критеріїв до проведення етичної експертизи, оскільки відсутнє методологічне забезпечення діяльності етичних комісій при лікувально-профілактичних закладах, не налагоджено взаємодію та координацію роботи цих комісій для обміну досвідом щодо дотримання етичних та нормативно-правових аспектів проведення КВ.
Для вирішення цієї проблеми в структурі ДП «Державний експертний центр МОЗ України» був створений Сектор координації діяльності ЛЕК, завданнями і функціями якого є саме організаційно-методичне забезпечення взаємодії етичних комісій при лікувально-профілактичних закладах, визначення стандартів діяльності ЛЕК, сприяння удосконаленню експертизи в країні, а також забезпечення постійного навчання членів комісій.
Необхідність створення цього Сектору значною мірою посилюється міжнародними рекомендаціями щодо забезпечення системності та комплексного підходу до етичної експертизи клінічних випробувань.
Комісії з питань етики повинні надавати достатню інформацію про свою роботу, а саме-про етичну експертизу, супровід ходу дослідження і про інші види діяльності організації, яка заснувала їх, або компетентному органу за допомогою структурованих належним чином періодичних звітів, в яких не повинні розкриватися конфіденційні деталі, що стосуються дослідження або його учасників. Такі звіти, у вичерпний формі або у формі робочого резюме, повинні перебувати у відкритому доступі, наприклад, на веб-сайті Комісії з питань етики, організації або регіонального компетентного органу.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим