|
Расследование показало, что в Китае на ранних стадиях производства в лекарственное средство (ЛС) был добавлен сульфатированный хондроитин сульфат, который, как и гепарин, обладает антикоагулянтными свойствами, но в 100 раз дешевле в производстве (не 2000, а 20 дол. США за 1 кг). Контроль ЛС на наличие этого вещества не был предусмотрен спецификацией качества на гепарин, и предполагают, что постороннее вещество могло быть добавлено в ЛС намеренно [2]. Итог трагичен — по меньшей мере 81 пациент в США умер по причине побочных реакций на препарат. Другие источники сообщают о более чем 100 смертельных случаях в США и сотнях серьезных побочных реакций в Европе [3] или о более 200 случаев смерти в разных странах [4]. Мощнейшее регуляторное агентство, Управление по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA), точнее его Центр по оценке и исследованию лекарственных средств (Center for Drug Evaluation and Research — CDER), который на своей веб-страничке назван сторожевым псом американских потребителей, не защитил этих людей от смертельной опасности. Д-р Джанет Вудкок (Janet Woodcock), директор CDER, отметила по этому поводу на слушаниях в Конгрессе, что было бы лукавством со стороны FDA утверждать, будто оно может инспектировать каждое производство, каждый продукт и что такая инспекция может выявить любую проблему.
FDA не может следить за качеством продуктов на всех этапах их обращения, — это неоднократно подчеркивала Дж. Вудкок [5]. Кроме того, по ее словам, основную ответственность за качество ЛС на разных этапах его обращения несут производители, импортеры, посредники и дистрибьюторы. Уровень требований к ним как со стороны властей, так и широкой общественности будет постоянно расти. Гепариновый кризис, когда к конечному потребителю, пройдя проверку на этапах лекарственной субстанции и готового ЛС, попал препарат, содержащий большое (по массе) количество контаминанта, должен рассматриваться всеми как грозное предупреждение, — отметила директор CDER.
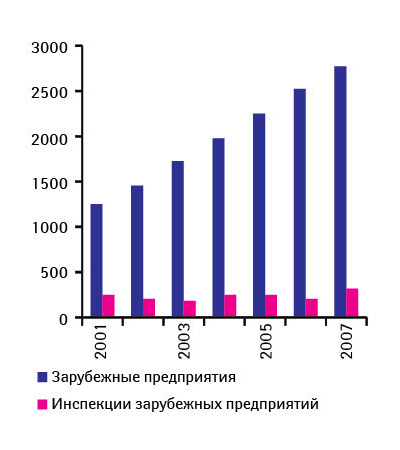
Контроль именно зарубежных производств и экспортируемых препаратов — больной вопрос для управления. После гепаринового скандала его обсуждают особенно активно. Ведь согласно данным Правительственного управления по отчетности (Government Accountability Office) 80% лекарственных субстанций, используемых производителями в США, импортируются, причем преимущественно из Китая [6]. Кроме того, 40% готовых ЛС в США завозят из-за рубежа. Подсчитано, что при существующем положении вещей для проведения разовых инспекционных проверок каждого зарубежного производителя потребуется не меньше 13 лет [7]. Но такое возможно, если бы все оставалось на своих местах. На деле же количество лекарственных продуктов (готовых ЛС, субстанций и вспомогательных веществ), импортируемых в США, как отметила Дж. Вудкок, с 2001 г. более чем удвоилось. То же касается и зарубежных производителей, выпускающих одобренные FDA продукты. Количество инспекций, конечно, увеличивается, но далеко не пропорционально росту числа производителей. Поэтому частота инспекций, как сообщила директор CDER, с 2001 по 2007 г. снизилась на 41% (рис. 1). Система обеспечения качества ЛС, по словам Дж. Вудкок, сегодня стоит перед лицом ряда нерешенных проблем:
|
- глобализация производства;
- усиление угроз злоупотреблений и преступлений (фальсификаций, биотерроризма и т.д.);
- продолжающееся разобщение и фрагментированность регуляторной сферы в глобальном масштабе;
- резкое сокращение охвата предприятий инспекциями в последнее время;
- недостаточное внедрение современных информационных технологий (например автоматизированный учет разрешений на маркетинг, лицензий, хода инспекций и т.д.).
Стратегические планы FDA по их решению представлены в ряде программ, в том числе «Фармацевтическое качество ХХI столетию» (Pharmaceutical Quality for the 21st Century). В 2002 г. FDA выступило с инициативой, которая нашла отражение в поистине революционном документе, получившем название «Технологии анализа процессов» (Process Analytical Technology — PAT) [8]. Инициативы РАТ в первую очередь позиционируются как интегрированная система для планирования, анализа и контроля наиболее критических характеристик исходного сырья, компонентов процесса, а также параметров самого процесса с целью обеспечения необходимого качества готового ЛС РАТ предполагает создание системы контроля качества ЛС в реальном времени (то есть в момент выпуска). Один из ключевых элементов РАТ — сделать качество неотъемлемым свойством как самого препарата, так и процесса его производства. Для поддержки инициативы РАТ в CDER создан специальный комитет, разработано руководство. Особое внимание управление уделяет не только качеству, но и безопасности препаратов. «Safety first» (приоритет безопасности) — такую задачу ставит FDA перед отраслью. Управление привлекает новые ресурсы: 600 специалистов были приняты на работу в CDER в 2008 г. В ближайшее время управление рассчитывает нанять еще примерно столько же, так что количество его штатных сотрудников превысит 11 тыс. [9].
Но все же: «Мы не можем быть службой, контролирующей качество, для всего мира», — заявила Дж. Вудкок на вышеупомянутых слушаниях в Конгрессе по поводу инспекций производств, выпускающих за рубежом одобренные FDA продукты. То есть о качестве должен печься прежде всего производитель. Если он не сможет убедить FDA на этапе получения разрешения на маркетинг, ну что же — в США ему путь заказан. Большинства других государств это, к сожалению, не касается. Некоторое представление о проблемах развивающихся стран можно получить, ознакомившись с бюллетенем «Matrix of Drug Quality Reports Affecting USAID-assisted Countries», который готовят в рамках Программы качества ЛС и информации Фармакопеи США (USP Drug Quality and Information Program) [10]. «Согласно источникам из Великобритании в 2003 г. в Китае около 100 000 человек умерли от незаконных ЛС», — сообщается в этом источнике со ссылкой, почему-то, на сайт , где к тому же (возможно по причине реконструкции сайта) соответствующую статью обнаружить не удалось. Откуда такая цифра, непонятно, но в том, что субстандартных, фальсифицированных, в том числе смертельно опасных, ЛС в странах третьего мира очень и очень много, сомневаться не приходится. Достаточно хотя бы полистать вышеупомянутый бюллетень.
Многоуровневая система контроля качества ЛС — как компьютерное антивирусное программное обеспечение — нуждается в постоянном совершенствовании. Размещая здесь кое-какую информацию, почерпнутую из общедоступных источников, мы не предполагаем, что специалисты именно в этой сфере найдут здесь для себя что-то новое. Просто маховик государственной системы крутится тогда, когда общество зорко следит за тем, чтобы не было остановок. Поэтому все мы должны знать, и требовать, и участвовать…
ГОСУДАРСТВЕННАЯ СИСТЕМА КОНТРОЛЯ КАЧЕСТВА ЛС США
Система сообщений о качестве ЛС (Drug Quality Reporting System — DQRS) функционирует в США с начала 1970-х годов. В настоящее время сообщения о подозреваемых проблемах качества ЛС посредством телефонной связи или других быстрых коммуникаций (к примеру системы репортирования о побочных реакциях — MedWatch) поступают в Отдел соответствия, управления рисками и надзора CDER (The Division of Compliance Risk Management and Surveillance). Подходы к расследованию случаев детально описаны в презентации «Drug Quality Reporting System» (датирована 18.01.2007 г.) [11]. Соответствующие обязанности разделяют владелец разрешения на маркетинг, региональное подразделение FDA и CDER.
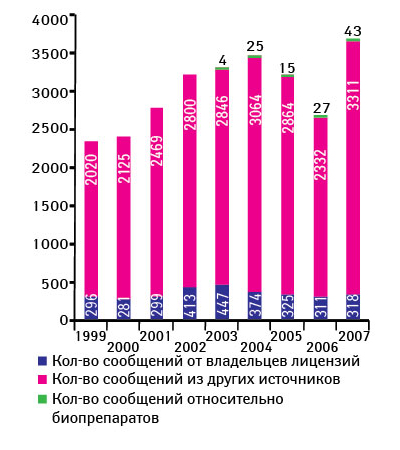
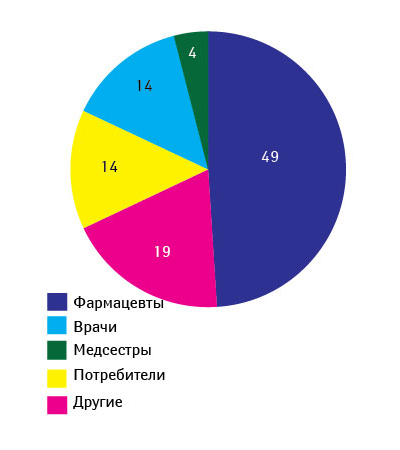
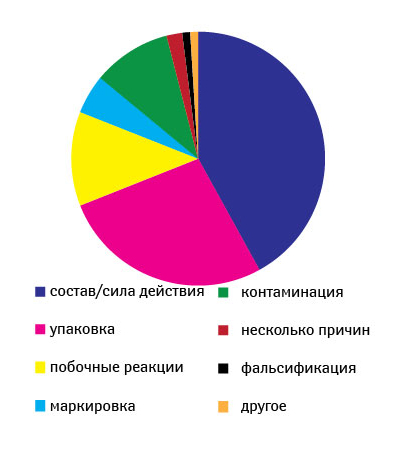
Ежегодно CDER получает около 2,5–3,5 тыс. сообщений о проблемах качества ЛС (период 1999–2007 гг.) (рис. 2) [12]. О возможных проблемах с качеством препаратов от специалистов здравоохранения и потребителей FDA в 2007 г. получило 3311 сообщений, от производителей — 318, а также 43 сообщения, касающихся биопрепаратов. Стоит отметить, что фармацевты отправляют примерно половину сообщений от специалистов здравоохранения и потребителей (рис. 3). В 2007 г. наибольшее количество сообщений было вызвано отклонениями в составе препаратов (42%), состоянии упаковки (27%), побочными реакциями (12%) (рис. 4) [12].
|
|
|
Вообще анализ результатов инспекций производств и проверка качества препаратов, находящихся в обращении, путем отбора и анализа проб — основные стратегические направления надзора FDA за соответствием препаратов установленным стандартам [12]. Выбор препаратов и производств для проверки (как в стране, так и за рубежом) осуществляется на основании оценки рисков. В случае обнаружения отклонений региональные подразделения FDA проводят инспекции для выяснения их причин и разработки корректирующих действий совместно с компанией. Выделяют следующие критерии для отбора проб на основании оценки рисков:
- опасность микробного/эндотоксинового заражения;
- проблемы стабильности;
- проблемы стерильности;
- отклонения по результатам теста растворения;
- превышение допустимого уровня примесей/продуктов распада;
- «скомпрометированная» история препарата;
- подозрение на фальсификацию;
- прежние случаи нарушений.
Рецептурные и безрецептурные препараты в США согласно федеральному законодательству должны соответствовать стандартам Фармакопеи США (United States Pharmacopeia — USP), если таковые существуют. Кстати, USP — организация неправительственная, неприбыльная, в которой работают эксперты-волонтеры в соответствии со строгими правилами, предупреждающими конфликт интересов. Несоответствие компендиальным стандартам качества, силы действия или чистоты, выявленное при тестировании предписанным USP методом, позволяет считать ЛС фальсифицированным, если в инструкции по медицинскому применению эти отличия четко не обозначены (Федеральный закон о продуктах питания, лекарственных и косметических средствах (Federal Food, Drug, and Cosmetic Act). Если же ЛС не описано в официальном компендиуме, фальсифицированным оно считается в случае несоответствия стандартам качества, указанным в инструкции, спецификациях производителя или заявке на получение разрешения на маркетинг. Согласно закону таковым оно будет считаться, если отклонения выявлены любым научно обоснованным методом [13].
В 2007 фискальном году FDA провело 1119 пострегистрационных внутренних (в результате была приостановлена работа 1 предприятия и выпущено 14 предупредительных писем) и 289 предрегистрационных инспекций (проводятся в обязательном порядке при подаче заявок на получение разрешения на маркетинг оригинальных или генерических препаратов), а также 333 зарубежные инспекции [12].
Интересно, что по закону FDA может своим решением отзывать биопрепараты и медицинское оборудование, но не ЛС, и большинство отзывов проводят по инициативе производителей. Это случается, когда либо само предприятие обнаружило дефекты (нарушения, отклонения), либо их выявило FDA и рекомендовало отзыв. При этом обычно компания следует рекомендации. В противном случае FDA может требовать наложения санкций в предусмотренном Федеральным законом о продуктах питания, лекарственных и косметических средствах порядке. На ЛС может быть наложен арест, против компании выдвинуто обвинение, а препарат может быть отозван по решению суда [14; 15].
Динамика количества отзывов (recalls) (применяют в отношении нескольких серий) за год, отмечает CDER, непоказательна, поскольку рекорд 2007 г. вызван 670 отзывами со стороны одного упаковщика, так что общее количество отозванных рецептурных препаратов составило 851, безрецептурных — 136 [12].
Основными причинами отзыва ЛС с рынка были следующие:
- несоответствие требованиям к упаковке;
- нарушение температурных условий хранения;
- уменьшенное количество действующего вещества;
- химическая контаминация;
- превышение допустимого уровня примесей/продуктов распада;
- несоответствие требованиям теста растворения;
- повреждение маркировки;
- маркетинг в отсутствие одобренной заявки на получение разрешения на маркетинг;
- недостатки в обеспечении стерильности;
- приводящее к ошибкам сходство маркировок.
Последний пункт особенно обращает на себя внимание, так как у нас подобное практически никогда не становилось причиной запрещения обращения ЛС. В США же сходство маркировки с таковой у жизненно важного препарата становится причиной отзыва I класса [16]. Стоит напомнить, что отзыв I класса, как и II, осуществляется до уровня аптек включительно, кроме того, предусматривает обязательное извещение всех пациентов, получающих ЛС.
Удалено с рынка (withdraw) в связи с проблемами безопасности в 2007 г. всего 2 препарата — перголид и тегасерода малеат [12].
По инициативе FDA USP приняло ряд изменений к монографии на гепарин. Первый пересмотр был произведен в феврале 2008 г. с введением новых тестов (при помощи электрофореза и ядерно-магнитного резонанса) для идентификации сульфатированного хондроитин сульфата. Исчерпывающая модификация этой статьи с включением методов и технологий, уже широко используемых производителями, должна вступить в силу в августе 2009 г. [4].
Информации непосредственно о лабораторных проверках маловато, но, в частности, удалось выяснить, что частота отклонений нормативных показателей при этом составляет около 2%. Интересно, что аналогичный показатель у изготовляемых по индивидуальному требованию ЛС составил 34% (по результатам ограниченной проверки, проведенной FDA в 2001 г. [17]).
О количестве ЛС, тестируемых FDA за год, не удалось найти информации. Вот только сообщают, что с 1996 по 2001 г. проанализировано свыше 3000 препаратов разных производителей [17].
О ЧЕМ СООБЩАЕТ БРИТАНСКИЙ РЕГУЛЯТОР?
Зато Агентство по регулированию лекарственных средств и продуктов для здравоохранения Великобритании (Medicines and Healthcare Products Regulatory Agency — MHRA) сообщает на своем сайте, что в рамках программы проверки ЛС (Medicines testing scheme — MTS) ежегодно отбирают и анализируют 2000–2500 образцов препаратов [18]. Целью программы является подтверждение качества ЛС, представленных на рынке Великобритании. Препараты для анализа могут быть взяты либо по требованию других подразделений MHRA, либо в рамках национальной программы надзора за ЛС. По требованию могут отбирать пробы в рамках расследований, инспекций, плановых мероприятий постмаркетингового надзора, перед выдачей разрешений на маркетинг (таблица) [19]. По инициативе сотрудников MTS пробы на этапах производства, хранения, дистрибьюции или розничной продажи отбирают инспекторы Королевского фармацевтического общества (Royal Pharmaceutical Society). Это могут быть новые химические вещества (New chemical entity — NCE), генерики, не получающие разрешение на маркетинг специальные ЛС (см. ниже) и препараты, получившие разрешение на маркетинг по централизованной процедуре.
|
Таблица 1 |
Источники образцов препаратов, проверенных в рамках системы MTS |
|
Источники |
2004/05 |
2005/06 |
2006/07 |
2007/08 |
|
На основании DMRC |
44 |
44 |
70 |
83 |
|
Инспекторат по ЛС |
26 |
16 |
27 |
25 |
|
Плановые проверки |
54 |
552 |
619 |
406 |
|
Предрегистрационные |
75 |
194 |
225 |
163 |
|
Надзор за продуктами на рынке |
2273 |
2055 |
1363 |
1347 |
|
ЕМЕА*-централизованная процедура |
17 |
16 |
15 |
9 |
|
Другие |
64 |
31 |
35 |
176 |
Для подтверждения соответствия спецификациям положено в течение первых двух лет маркетинга проанализировать 3 серии новых препаратов, относящихся к NCE. Принято также ежегодно на основании анализа рисков производить отбор и исследование ряда генерических препаратов [20]. С 2002 г. MHRA располагает собственной лабораторией.
Для подтверждения минимальных требований в отношении спецификаций согласно Британской Фармакопее** анализируют не получающие разрешение на маркетинг (специальные) ЛС, изготовленные по специальной лицензии для удовлетворения специфических нужд конкретных пациентов. В период 2000–2005 гг. 56% из 79 таких препаратов оказались несоответствующими регуляторным требованиям. У получивших разрешение на маркетинг препаратов частота отклонений ниже. Правда, большинство выявленных недостатков у не получающих разрешение на маркетинг ЛС не влияли на их качество и безопасность и заключались в нарушениях маркировки, спецификаций и контроля качества производителем. На основании результатов MTS предпринимают более обширные расследования. Их приоритетными направлениями называют:
- выявление источников фальсифицированных ЛС;
- выявление в составе растительных препаратов рецептурных ЛС (глибенкламид, фенфлурамин, сибутрамин, сильденафил, кортикостероиды, дисульфирам, тадалафил);
- выявление в составе растительных препаратов запрещенных или неразрешенных компонентов (эфедра, аристолохия, гидрохинон);
- экспертиза диетических добавок (для уточнения принадлежности продуктов к этой группе);
- токсические примеси в растительных продуктах (тяжелые металлы, нитрозофенфлурамин).
Производители, импортеры и дистрибьюторы обязаны информировать MHRA о любых заподозренных дефектах качества ЛС, которые потенциально требуют отзыва или приостановления поставок. Информирование о возможных дефектах качества ЛС входит также в обязанности специалистов здравоохранения. Потребителям в аналогичных ситуациях MHRA советует по возможности для начала проконсультироваться с врачом или фармацевтом; но если такая возможность отсутствует — сообщать непосредственно в агентство.
В 2007/2008 финансовом году MHRA получило 465 сообщений о проблемах качества ЛС и в поддержку действий производителей, отзывающих препараты или их отдельные серии, издало 34 срочных предупреждения, из которых 8 — класса I (требующие немедленных действий со стороны участников обращения ЛС), 14 класса II (действия в течение 48 ч), 2 класса III (действия в течение 5 дней) и 1 — класса IV (о необходимости осторожного применения). Одно из предупреждений извещало об отзыве препарата, еще пять — о фальсифицированных препаратах [19].
ПОВТОРЕНИЕ ГЕПАРИНОВОЙ ТРАГЕДИИ ВОЗМОЖНО?
Усилия, прикладываемые регуляторными органами для выявления субстандартных и фальсифицированных ЛС, главный специалист по науке USP Даррелл Абернети (Darrell Abernethy) сравнивает с работой Олимпийского комитета по выявлению случаев употребления допинга спортсменами. Ясно, что пока комитет ищет подход к распознаванию одного запрещенного препарата, на свет появляется следующий, выявлять который еще предстоит научиться, — отмечает Д. Абернети. По мнению некоторых экспертов, мы стоим на пороге других страшных последствий применения субстандартных ЛС. Чтобы этого не случилось, предприятия отрасли должны взять на себя всю полноту ответственности за производственный цикл, начиная с сырья для выпуска ЛС, ибо, как с грустной иронией говорит Уоррен Перри (Warren Perry), эксперт из «Qumas Consulting» (Сан-Франциско): «Пока производители греются у очага регуляторов, ничего не изменится» [21]. Когда же отрасль предпримет решительные меры, чтобы минимизировать опасность наступления очередного кризиса? К сожалению, прежде может понадобиться еще несколько тяжелых уроков, цена которых — жизни людей.
Дарья Полякова
*Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA);
**всемирно известная Британская Фармакопея является единственным в стране полным собранием стандартов на ЛС в любую фазу их жизненного цикла.
ЛИТЕРАТУРА
- Harris G. Heparin Contamination May Have Been Deliberate, F.D.A. Says. // New York Times. April 30, 2008. Available at: http://www.nytimes.com/2008/04/30/health/policy/30heparin.html _r=2.
- Committee on Energy and Commerce Subcommittee on Oversight and Investigations. The heparin disaster: Chinese counterfeits and American failures. April 29, 2008. Available at: http://energycommerce.house.gov/cmte_mtgs/110-oi-hrg.042908.Heparin.shtml.
- Annual report of activities of the EDQM — 2008. Available at: http://www.edqm.eu/medias/fichiers/Annual_Report_Activitie.pdf.
- The Standard. 2009. Vol. 6, Issue 3. Available at: http://www.usp.org/pdf/EN/aboutUSP/theStandard2009Winter.pdf.
- Janet Woodcock. Director, Center for Drug Evaluation and Research, FDA. CDER Priorities for 2009. Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm117684.pdf.
- Drug Safety: Preliminary Findings Suggest Weaknesses in FDA’s Program for Inspecting Foreign Drug Manufacturers, GAO-08-224T, November 1, 2007. Available at: http://www.gao.gov/new.items/d08224t.pdf.
- It’s Still the Economy, Stupid. Eagle ForumVol. 41, No. 7 February 2008. Available at: http://www.eagleforum.org/psr/2008/feb08/psrfeb08.html.
- OPS Process Analytical Technology —(PAT) Initiative . Available at: http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm088828.htm.
- Vivian J.C. FDA Inspection of Foreign Drug Companies US Pharmacist. 2008;33(6):53-57 6/19/2008. Available at: www.uspharmacist.com/content/d/pharmacy_law/c/9789.
- Matrix of Drug Quality Reports Affecting USAID-assisted Countries By the U.S. Pharmacopeia Drug Quality and Information Program. Created by: Joyce Primo-Carpenter, M.D., BSc. Pharm. USP DQI Associate Director, 2003–2008. Updated: June 1, 2009.
- Johnson J. Drug Quality Reporting System Presentation (updated 1/18/2007). Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm102838.pdf.
- CDER 2007 Report (update): Improving Public Health Through Human Drugs. Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/WhatWeDo/UCM121704.pdf.
- CPG Sec. 420.100 Adulteration of Drugs Under Section 501(b) and 501(c) of the Act. Direct Reference Seizure Authority for Adulterated Drugs Under Section 501(b) (CPG 7132a.03) Available at: http://www.fda.gov/ICECI/ComplianceManuals/CompliancePolicyGuidanceManual/ucm074367.htm.
- Federal Food, Drug, and Cosmetic Act (FD&C Act). Available at: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm.
- Public Health Service Act. Available at: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm.
- Debra B.F. Pharmacy law. McGraw-Hill Professional. 2007.
- Report: Limited FDA Survey of Compounded Drug Products. Available at: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/PharmacyCompounding/ucm155725.htm.
- Medicines testing. Available at: http://www.mhra.gov.uk/Howweregulate/Medicines/Inspectionandstandards/Medicinestesting/index.htm.
- MHRA annual statistics 2007/08. Available at:http://www.mhra.gov.uk/Publications/Corporate/AnnualReports/CON020817.
- Lee G, Charvill A, Heddell G. The MHRA medicines testing scheme: working to protect the public. Pharmaceutical Journal. 2005.Vol. 275.
- Shanley A, Thomas P., Vaccarello M., Ciurczak E. Lessons from Heparin Available аt: http://www.pharmamanufacturing.com/articles/2008/123.html page=print.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим