ПОВІДОМЛЕННЯ ПРО ОПРИЛЮДНЕННЯ
проекту наказу міністерства охорони здоров’я України «Про внесення змін до деяких наказів Міністерства охорони здоров’я України»
З метою забезпечення вивчення та врахування думки громадськості Міністерство охорони здоров’я України на своєму офіційному веб-сайті пропонує для публічного обговорення доопрацьований проект наказу Міністерства охорони здоров’я України «Про внесення змін до деяких наказів Міністерства охорони здоров’я України « (далі — Проект наказу).
Проект наказу розроблено з метою оптимізації нагляду за безпекою та ефективністю лікарських засобів в Україні шляхом удосконалення вітчизняного законодавства щодо здійснення фармаконагляду, його гармонізації з нормативною базою Європейського Союзу з урахуванням рекомендацій Всесвітньої організації охорони здоров’я (далі — ВООЗ).
Проект наказу розроблено відповідно до законів України «Про лікарські засоби», «Про захист населення від інфекційних хвороб», «Про забезпечення санітарного та епідемічного благополуччя населення», «Про загальнодержавну програму адаптації законодавства України до законодавства Європейського Союзу», Порядку державної реєстрації (перереєстрації) лікарських засобів і Розмірів збору за державну реєстрацію (перереєстрацію) лікарських засобів, затвердженого постановою Кабінету Міністрів України від 26 травня 2005 року № 376, пункту 8 Положення про Міністерство охорони здоров’я України, затвердженого постановою Кабінету Міністрів України від 25 березня 2015 року № 267, доручення Прем’єр-міністра України Яценюка А.П.від 17 квітня 2015 року№ 14335/1/1-15, наказу Міністерства охорони здоров’я від 25 червня 2015 року № 390 «Про створення робочих груп з питань удосконалення законодавства».
Проект наказу, аналіз регуляторного впливу та пояснювальна записка оприлюднені шляхом розміщення на офіційному веб-сайті Міністерства охорони здоров’я України в мережі Інтернет http://www.moz.gov.ua.
Пропозиції та зауваження від фізичних та юридичних осіб щодо проекту наказу просимо надсилати протягом місяця з дня опублікування у письмовому та/або електронному вигляді за адресою:
– Міністерство охорони здоров’я України 01601, м. Київ-601, вул. Михайла Грушевського, 7
ПОЯСНЮВАЛЬНА ЗАПИСКА
до проекту наказу Міністерства охорони здоров’я України «Про внесення змін до деяких наказів Міністерства охорони здоров’я України»
1. Обґрунтування необхідності прийняття акта
Проект наказу Міністерства охорони здоров’я України «Про внесення змін до деяких наказів Міністерства охорони здоров’я України» (далі — Проект наказу) розроблено відповідно до законів України «Про лікарські засоби», «Про захист населення від інфекційних хвороб», «Про забезпечення санітарного та епідемічного благополуччя населення», «Про загальнодержавну програму адаптації законодавства України до законодавства Європейського Союзу», Порядку державної реєстрації (перереєстрації) лікарських засобів і Розмірів збору за державну реєстрацію (перереєстрацію) лікарських засобів, затвердженого постановою Кабінету Міністрів України від 26 травня 2005 року № 376, пункту 8 Положення про Міністерство охорони здоров’я України, затвердженого постановою Кабінету Міністрів України від 25 березня 2015 року № 267, доручення Прем’єр-міністра України Яценюка А.П. від 17 квітня 2015 року № 14335/1/1-15, наказу Міністерства охорони здоров’я України від 25 червня 2015 року № 390 «Про створення робочих груп з питань удосконалення законодавства».
Проект наказу з метою удосконалення законодавства щодо здійснення фармаконагляду затверджує Порядок здійснення фармаконагляду у новій редакції (далі — Порядок).
Станом на сьогодні здійснення нагляду за безпекою лікарських засобів в Україні врегульовано низкою наказів Міністерства охорони здоров’я України. Основним серед них є наказ Міністерства охорони здоров’я України від 27 грудня 2006 року № 898 «Про затвердження Порядку здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування», зареєстрований у Міністерстві юстиції України 29 січня 2007 року за № 73/13340 (далі — наказ МОЗ України № 898).
Крім цього, у чинному наказі МОЗ України № 898 не повною мірою відображені зміни, яких зазнала Директива Європейського Парламенту та Ради ЄС №2001/83/EU, внесені до неї Директивою Європейського Парламенту та Ради ЄС № 2010/84/EU (ст. 1 п.11, п.15, п. 28b-28e; ст.22; ст.22 a; ст.23 (4); розділ ІХ: ст.101-107q; ст. 108; ст. 111(8); ст. 116; ст. 117). Також у чинному наказі № 898 не відображені зміни, внесені до Регламенту (ЄС) № 726/2004 Європейського Парламенту та Ради ЄС (ст.9; ст.10а; ст.10b; ст.14b; ст.15; розділ 3 (ст. 21–29); ст.87).
Чинним наказом МОЗ України № 898 врегульовано взаємодію усіх зацікавлених сторін, що регулюють обіг лікарських засобів з питань безпеки. Однак проведений аналіз результатів взаємодії між Державною службою України з лікарських засобів (далі — Держлікслужби) та Державним підприємством «Державний експертний центр Міністерства охорони здоров’я України» за період 2012–2015 рр., а також врахування європейських та світових підходів щодо управління інформацією про летальні наслідки побічних реакцій, непередбачені побічні реакції, відсутність ефективності лікарських засобів та несприятливі події після імунізації дозволили дійти висновку про необхідність внесення змін до Порядку встановлення заборони (тимчасової заборони) та поновлення обігу лікарських засобів на території України, затвердженого наказом Міністерства охорони здоров’я України від 22 листопада 2011 року № 809, щодо дій Держлікслужби стосовно зазначених вище ситуацій.
2. Мета і шляхи її досягнення
Проект Порядку розроблено відповідно до вимог Загальнодержавної програми адаптації законодавства України до законодавства Європейського Союзу, затвердженої Законом України від 18 березня 2004 року № 1629-IV, з метою оптимізації нагляду за безпекою та ефективністю лікарських засобів в Україні для убезпечення їх застосування у післяреєстраційному періоді. Досягнути цієї мети можливо, зокрема, шляхом досягнення відповідності правової системи України щодо здійснення фармаконагляду acquis communautaire з урахуванням критеріїв, що висуваються ЄС до держав, які мають намір вступити до нього.
В основу проекту Порядку покладено європейський досвід та положення Директиви Європейського Парламенту та Ради ЄС № 2001/83/ЄС та Регламенту (ЄС) № 726/2004 Європейського Парламенту та Ради ЄС, а також рекомендації ВООЗ щодо здійснення фармаконагляду.
3. Правові аспекти
Правовими підставами розроблення цього проекту наказу є:
– Закон України «Про лікарські засоби»;
– постанова Кабінету Міністрів України від 26 травня 2005 року № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)»;
– постанова Кабінету Міністрів України від 25 травня 2015 № 267 «Про затвердження Положення про Міністерство охорони здоров’я України»;
– наказ Міністерства охорони здоров’я України від 25 червня 2015 року № 390 «Про створення робочих груп з питань удосконалення законодавства»;
– наказ Міністерства охорони здоров’я України від 13 жовтня 2010 року № 769 «Про затвердження Концепції розвитку фармацевтичного сектору галузі охорони здоров’я України на 2011 — 2020 роки»;
– зміни до Директиви Європейського Парламенту та Ради ЄС № 2001/83/ЄС, регламентовані Директивою Європейського Парламенту та Ради ЄС N 2010/84/ЄС;
– зміни до Регламенту (ЄС) №726/2004 Європейського Парламенту та Ради ЄС.
4. Фінансово-економічне обґрунтування
Введення в дію цього Проекту наказу не потребує додаткових матеріальних та інших витрат з Державного бюджету України.
5. Позиція заінтересованих органів
Проект наказу потребує погодження з Державною службою України з питань регуляторної політики та розвитку підприємництва та державної реєстрації в Міністерстві юстиції України.
6. Регіональний аспект
Проект наказу не стосується питання розвитку адміністративно-територіальних одиниць.
61. Запобігання дискримінації
У Проекті наказу відсутні положення, які містять ознаки дискримінації. Проект наказу не потребує проведення громадської антидискримінаційної експертизи.
7. Запобігання корупції
Проект наказу не містить ризиків вчинення корупційних правопорушень і не потребує проведення громадської антикорупційної експертизи.
8. Громадське обговорення
Проект наказу потребує проведення консультацій з громадськістю. Проект наказу оприлюднено на офіційному веб-сайті Міністерства охорони здоров’я України.
9. Позиція соціальних партнерів
Проект наказу не стосується соціально-трудової сфери.
10. Оцінка регуляторного впливу
Проект наказу є регуляторним актом та відповідає принципам державної регуляторної політики.
Прийняття наказу не потребує додаткових фінансових витрат з Державного бюджету України. Можливої шкоди у разі настання очікуваних наслідків дії акта не передбачається.
10.1 Вплив акта на ринок праці
Проект наказу на ринок праці не впливає.
11. Прогноз результатів
Прийняття запропонованого проекту наказу дозволить удосконалити та гармонізувати нормативну базу України щодо фармаконагляду відповідно до вимог Європейського Союзу, оптимізувати його здійснення і результативність.
| В.о. Міністра охорони здоров’я України |
Уляна Супрун |
Проект
оприлюднений на офіційному сайті МОЗ України 25.08.2016 р.
МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ
НАКАЗ
Про внесення змін до деяких наказів Міністерства охорони здоров’я України
Відповідно до законів України «Про лікарські засоби», «Про захист населення від інфекційних хвороб», «Про забезпечення санітарного та епідемічного благополуччя населення», «Про загальнодержавну програму адаптації законодавства України до законодавства Європейського Союзу», Порядку державної реєстрації (перереєстрації) лікарських засобів і Розмірів збору за державну реєстрацію (перереєстрацію) лікарських засобів, затвердженого постановою Кабінету Міністрів України від 26 травня 2005 року № 376, пункту 8 Положення про Міністерство охорони здоров’я України, затвердженого постановою Кабінету Міністрів України від 25 березня 2015 року № 267,
НАКАЗУЮ:
1. У заголовку та тексті наказу Міністерства охорони здоров’я України від 27 грудня 2006 року № 898 «Про затвердження Порядку здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування», зареєстрованого в Міністерстві юстиції України 29 січня 2007 року за № 73/13340, а також у заголовку та тексті Порядку здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування, затвердженого наказом Міністерства охорони здоров’я України від 27 грудня 2006 року № 898, зареєстрованим в Міністерстві юстиції України 29 січня 2007 року за № 73/13340, слова та знаки «Порядок здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування» у всіх відмінках замінити словами «Порядок здійснення фармаконагляду».
2. Внести зміни до Порядку здійснення фармаконагляду, затвердженого наказом Міністерства охорони здоров’я України від 27 грудня 2006 року № 898, зареєстрованого в Міністерстві юстиції України 29 січня 2007 року за № 73/13340, виклавши його у новій редакції, що додається.
3. В абзаці шостому пункту 5.2 розділу 5 Правил зберігання та проведення контролю якості лікарських засобів у лікувально-профілактичних закладах, затверджених наказом Міністерства охорони здоров’я України від 16 грудня 2003 року № 584, зареєстрованих в Міністерстві юстиції України 03 березня 2004 року за № 275/8874, слова: «, та побічні реакції або загибель людей при застосуванні серії(й) лікарських засобів» виключити.
4. Пункт 3.2.5 розділу 3 Порядку встановлення заборони (тимчасової заборони) та поновлення обігу лікарських засобів на території України, затвердженого наказом Міністерства охорони здоров’я України від 22 листопада 2011 року № 809, зареєстрованого в Міністерстві юстиції України 30 січня 2012 року за № 126/20439, викласти у такій редакції:
«негативні висновки щодо якості зразків серії або серій підозрюваного у виникненні смерті, непередбаченої побічної реакції та/або відсутності ефективності лікарського засобу за наявності причинно-наслідкового зв’язку на підставі надходження повідомлень від МОЗ України та/або територіальних органів Держлікслужби;».
5. Визнати такими, що втратили чинність:
наказ Міністерства охорони здоров’я України від 15 червня 2007 року № 325 «Про затвердження показників якості імунобіологічних препаратів (вакцин та анатоксинів)», зареєстрований в Міністерстві юстиції України 07 липня 2007 року за № 773/14040;
наказ Міністерства охорони здоров’я України від 24 липня 2009 року № 531 «Про затвердження Порядку проведення моніторингу безпеки та ефективності лікарських засобів у стаціонарах закладів охорони здоров’я», зареєстрований в Міністерстві юстиції України 17 серпня 2009 року за № 774/16790.
6. Виключити:
підпункти 1.4, 1.5, 1.6 пункту 1 наказу Міністерства охорони здоров’я України від 16 вересня 2011 року № 595 «Про порядок проведення профілактичних щеплень в Україні та контроль якості й обігу медичних імунобіологічних препаратів», зареєстрованого в Міністерстві юстиції України 10 жовтня 2011 року за № 1159/19897;
у зв’язку з цим підпункти 1.7, 1.8 пункту вважати відповідно 1.4, 1.5;
підпункт 3.1.3 пункту 3.1 розділу ІІІ Порядку встановлення заборони (тимчасової заборони) та поновлення обігу лікарських засобів на території України, затвердженого наказом Міністерства охорони здоров’я України від 22 листопада 2011 року № 809, зареєстрованим в Міністерстві юстиції України 30 січня 2012 року за № 126/20439;
у зв’язку з цим підпункти 3.1.4, 3.1.5 пункту 3.1 розділу ІІІ вважати відповідно 3.1.3, 3.1.4.
7. Управлінню фармацевтичної діяльності та якості фармацевтичної продукції забезпечити подання цього наказу на державну реєстрацію до Міністерства юстиції України в установленому порядку.
8. Контроль за виконанням цього наказу залишаю за собою.
9. Цей наказ набирає чинності з дня його офіційного опублікування.
| В.о. Міністра охорони здоров’я |
Уляна Супрун |
ЗАТВЕРДЖЕНО
Наказ Міністерства охорони здоров’я України
від 27 грудня 2006 року № 898
ПОРЯДОК ЗДІЙСНЕННЯ ФАРМАКОНАГЛЯДУ
Порядок здійснення фармаконагляду (далі — Порядок) розроблено відповідно до законів України «Про лікарські засоби», «Про захист населення від інфекційних хвороб», «Про забезпечення санітарного та епідемічного благополуччя населення», «Про загальнодержавну програму адаптації законодавства України до законодавства Європейського Союзу», Порядку державної реєстрації (перереєстрації) лікарських засобів і Розмірів збору за їх державну реєстрацію (перереєстрацію) лікарських засобів, затверджених постановою Кабінету Міністрів України від 26 травня 2005 року № 376, з урахуванням Концепції розвитку фармацевтичного сектору галузі охорони здоров’я України на 2011 — 2020 роки, затвердженої наказом Міністерства охорони здоров’я України від 13 вересня 2010 року № 769 (у редакції наказу Міністерства охорони здоров’я України від 27 березня 2013 року № 242), положень Директиви Європейського Парламенту та Ради ЄС № 2001/83 ЄС, Регламенту (ЄС) № 726/2004 Європейського Парламенту та Ради ЄС, що встановлюють основні правила та вимоги щодо здійснення фармаконагляду.
І. ВИЗНАЧЕННЯ ТЕРМІНІВ
1. У цьому Порядку терміни вживаються у такому значенні:
Вакцинація (щеплення, активна/пасивна імунізація) — створення штучного імунітету у людини до певних інфекційних хвороб шляхом введення вакцини чи імуноглобуліну.
Відсутність ефективності лікарського засобу — відсутність сприятливої діагностичної, лікувальної чи профілактичної дії лікарського засобу на встановлення характеру захворювання, його перебіг, тривалість або корекцію стану чи фізіологічних функцій організму людини відповідно до показань до застосування, зазначених в інструкції для медичного застосування.
Групові несприятливі події після імунізації — два або більше випадків несприятливих подій після імунізації, що мають подібні клінічні прояви, пов’язані за часом, місцем проведення імунізації/туберкулінодіагностики та типом введеної вакцини, туберкуліну.
Групові побічні реакції — два або більше випадки побічних реакцій, пов’язаних із застосуванням лікарського засобу, вакцини та туберкуліну, що мають подібні клінічні прояви, пов’язані за часом, місцезнаходження закладу охорони здоров’я та/або серією лікарського засобу.
Ефективність вакцин — зниження відсотку керованих інфекційних захворювань у групі щеплених осіб в порівнянні з групою нещеплених осіб.
Ефективність лікарського засобу — сприятлива діагностична, лікувальна чи профілактична дія лікарського засобу на встановлення характеру захворювання, його перебіг, тривалість або корекцію стану чи фізіологічних функцій організму людини відповідно до показань до застосування, зазначених в інструкції для медичного застосування.
Загальний звіт по картах епідрозслідування — звіт про всі зареєстровані випадки захворювання на інфекційні хвороби, що керуються засобами специфічної імунопрофілактики у щеплених, складений на підставі даних карт епідрозслідування.
Зафіксована побічна реакція лікарського засобу, вакцини та туберкуліну — побічна реакція, характер або тяжкість проявів якої відповідає наявній інформації, що міститься в основній інформації з безпеки заявника.
Зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну — звіт, що узагальнює інформацію про побічні реакції згідно з кодами клінічних проявів побічних реакцій та/або інструкціями про застосування вакцин, туберкуліну за звітний період.
Звіт про післяреєстраційне дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну — надані в письмовій формі результати післяреєстраційного дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну та їх аналіз.
Звіт про випадки побічних реакцій та/або відсутності ефективності лікарських засобів, вакцини та туберкуліну та/або несприятливих подій після імунізації у закладах охорони здоров’я — щорічний звіт про всі випадки побічних реакцій та/або відсутності ефективності лікарських засобів, вакцини та туберкуліну та/або несприятливі події після імунізації складають та подають усі заклади охорони здоров’я незалежно від відомчого підпорядкування та форм власності (далі — заклади охорони здоров’я), структурні підрозділи з питань охорони здоров’я обласних, Київської міської державних адміністрацій (далі — структурні підрозділи з питань охорони здоров’я).
Імуногенність вакцини — здатність вакцини утворювати гуморальний (визначені рівні антитіл у відсотках) і/або клітинно-опосередкований імунітет. Цей показник, за даними клінічних досліджень, має бути більше 90% (крім вакцин для профілактики грипу); для комбінованих вакцин кожен з компонентів повинен відповідати даному показнику.
Інформація, що ідентифікує випадок побічної реакції та/або відсутності ефективності лікарського засобу, вакцини та туберкуліну та/або несприятливу подію після імунізації — відомості про джерело отримання інформації, підозрюваний лікарський засіб, вакцину та туберкулін, пацієнта (хворого), опис побічної реакції/зазначення про відсутність ефективності/опис несприятливої події після імунізації.
Інша важлива медична оцінка — випадок побічної реакції, що не становить безпосередньої загрози для життя пацієнта або не призводить до смерті, або госпіталізації, але піддає пацієнта небезпеці або потребує втручання для запобігання прояву побічної реакції, що розвинулась.
Карта епідрозслідування — форма згідно з якою відповідальна особа районного чи обласного підрозділу охорони здоров’я надає інформацію про випадок захворювання на інфекційні хвороби, що керуються засобами специфічної імунопрофілактики у щеплених.
Карта–повідомлення про побічну реакцію та/або відсутність ефективності лікарського засобу, вакцину та туберкулін та/або несприятливу подію після імунізації (далі — карта-повідомлення) — форма, за якою лікарі, провізори, фельдшери, акушери, фармацевти, медичні сестри (далі — медичні працівники) усіх закладів охорони здоров’я незалежно від форм власності повідомляють про будь-які випадки побічних реакцій, відсутності ефективності лікарських засобів, вакцину та туберкулін та несприятливі події після імунізації.
Комбіновані вакцини — вакцини призначені для захисту від декількох інфекційних хвороб, що можуть бути викликані різними штамами, різними мікроорганізмами чи серотипами мікроорганізмів.
Користь — сукупність ступенів позитивного впливу лікарського засобу, вакцини та туберкуліну на зменшення тяжкості перебігу або зниження інтенсивності проявів симптомів захворювання та інтенсивності позитивної фармакологічної реакції на введення лікарського засобу, вакцини та туберкуліну та її тривалості.
Майстер–файл системи фармаконагляду (далі — МФСФ) — документ, що містить опис системи фармаконагляду, що використовується заявником лікарського засобу, вакцини та туберкуліну (далі — заявником) щодо одного або декількох лікарських засобів, вакцин та туберкуліну.
Мета–аналіз — метод отримання інформації, у тому числі про побічні реакції лікарських засобів, вакцини та туберкуліну, в якому використовується статистичний аналіз для інтеграції даних декількох незалежних досліджень для моніторингу лікарських засобів, вакцин та туберкуліну і побічних реакцій, зокрема тих, що виникають через тривалий період часу. При цьому враховуються всі медичні записи про хворого, зроблені протягом усього його життя з різних джерел інформації (лікарні, де він лікувався, пологового будинку, виписані рецепти тощо), що є підґрунтям для створення досьє пацієнта та наступного аналізу.
Міжнародна дата народження лікарського засобу, вакцини та туберкуліну — дата видачі заявнику першої ліцензії на продаж лікарського засобу, вакцини та туберкуліну у будь-якій країні світу.
Моніторинг рецептів — метод отримання інформації про побічні реакції лікарських засобів, заснований на обліку призначень препарату, коли за встановлений період часу визначається кількість зареєстрованих побічних реакцій і кількість хворих, які застосовували препарат, що дозволяє виявити взаємозв’язок між побічною реакцією і застосуванням лікарського засобу, за допомогою обліку виписаних рецептів.
Моніторинг стаціонару(ів) — метод отримання інформації про побічні реакції лікарських засобів, вакцини та туберкуліну, що дозволяє визначити частоту побічних реакцій та виявити особливості взаємодії лікарських засобів у хворих одного чи декількох стаціонарів, коли протягом певного періоду часу під контролем знаходяться усі хворі стаціонару(ів), враховуються всі лікарські засоби, вакцини та туберкулін, що призначаються, і усі побічні реакції, що виникають.
Неефективність вакцинації — визначається на основі клінічних проявів інфекційної хвороби, що лабораторно підтверджено відсутністю захисних маркерів від даної хвороби. Первинна неефективність вакцинації може бути наслідком недосягнення запланованого ефекту від щеплення (відсутність сероконверсії або серопротекції), вторинна — вроджених та/або набутих дефектів імунної системи або коли призначена вакцина вводилась неналежним чином.
Незафіксована побічна реакція лікарського засобу, вакцини та туберкуліну — побічна реакція, характер або тяжкість проявів якої не відповідає наявній інформації, що міститься в основній інформації з безпеки лікарського засобу, вакцини та туберкуліну заявника. До таких побічних реакцій належать ті, які за характером, тяжкістю проявів, специфічністю або наслідками не відповідають інформації, що міститься в основній інформації з безпеки лікарського засобу заявника, включаючи реакції, властиві певній фармакологічній групі лікарських засобів, вакцин та туберкуліну, що не виникали при застосуванні даного лікарського засобу, вакцини та туберкуліну.
Непередбачена побічна реакція — побічна реакція, характер або тяжкість проявів якої не узгоджується з наявною інформацією про лікарський засіб, вакцину та туберкулін в інструкції для його медичного застосування/короткій характеристиці лікарського засобу, вакцини та туберкуліну.
Несерйозна побічна реакція — будь-яка побічна реакція, що не призводить до смерті, не становить загрози для життя, не вимагає госпіталізації або подовження терміну госпіталізації, не викликає стійкої або значної непрацездатності чи інвалідності та вроджених аномалій чи вад розвитку, та не має іншої важливої медичної оцінки.
Несприятлива подія після імунізації (далі — НППІ) — будь-яка несприятлива з медичної точки зору подія, що спостерігається після імунізації/туберкулінодіагностики, та не обов’язково має причинно-наслідковий зв’язок з використанням вакцини та/або туберкуліну. Несприятливою подією може бути будь-яка несприятлива або ненавмисна ознака, відхилення у результатах лабораторних досліджень, симптомів або захворювання.
Основна інформація з безпеки заявника (далі — ОІБЗ) — документ, складений заявником, що містить усю відповідну інформацію з безпеки, що є складовою переліку основних даних заявника. ОІБЗ використовується при складанні звітності для визначення, чи зафіксована побічна реакція в переліку основних даних заявника чи не зафіксована. Дані ОІБЗ не використовуються для визначення, чи є побічна реакція передбаченою чи непередбаченою, для подання повідомлення про випадок побічної реакції (Company Core Safety Information (CCSI).
Первинні документи — вихідні документи, дані і записи (наприклад, історії хвороби, амбулаторні карти, лабораторні записи, службові записки, щоденники досліджуваних або опитувальники, журнали видачі лікарських засобів, роздруківки приладів, верифіковані та засвідчені копії або розшифровки фонограм, мікрофіші, фотографічні негативи, мікроплівки або магнітні носії, рентгенівські знімки, адміністративні документи, записи, що зберігаються в аптеці, лабораторії та у відділенні інструментальної діагностики закладів охорони здоров’я).
Передбачена побічна реакція — побічна реакція, характер або тяжкість проявів якої узгоджується з наявною інформацією про лікарський засіб, вакцину та туберкулін в інструкції для медичного застосування зареєстрованого лікарського засобу/короткій характеристиці лікарського засобу, вакцини та туберкуліну.
Перелік основних даних заявника (далі — ПОДЗ) — документ, складений заявником, що містить інформацію про лікарський засіб, вакцини та туберкуліну щодо безпеки, пропонованих показань до застосування, сили дії, особливостей застосування, фармакологічних властивостей тощо (Company Core Data Sheet (CCDS).
Період після застосування вакцини, туберкуліну — проміжок часу після проведеної імунізації/туберкулінодіагностики, що коливається у межах від першої доби до 24 місяців та залежить від типу вакцини, туберкуліну і для переважної більшості з них становить 30 діб.
Підозрюваний лікарський засіб, вакцина та туберкулін — лікарський засіб, вакцина та туберкулін, при призначенні якого існує причинно- наслідковий зв’язок між клінічними проявами будь-якої побічної реакції та/або відсутністю ефективності та/або НППІ та його застосуванням.
Післяреєстраційне дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну — будь-яке післяреєстраційне дослідження з безпеки та ефективності дозволеного до медичного застосування лікарського засобу, вакцини та туберкуліну, що проводиться з метою визначення, характеристики чи оцінки загрози безпеки, підтвердження профілю безпеки лікарського засобу, вакцини та туберкуліну та/або оцінки ефективності заходів з управління ризиками.
Післяреєстраційне неінтервенційне дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну — вид післяреєстраційного дослідження з безпеки та ефективності лікарських засобів, вакцин та туберкуліну, в якому лікарські засоби, вакцини та туберкулін призначаються звичайним способом відповідно до умов, зазначених в інструкції для медичного застосування лікарського засобу, вакцини та туберкуліну. Залучення пацієнта до групи з визначеним методом лікування в протоколі дослідження заздалегідь не передбачено, а призначення лікарського засобу, вакцини та туберкуліну диктується сучасною практикою та не залежить від рішення включити пацієнта у дослідження. При проведенні такого дослідження не застосовують додаткових діагностичних або моніторингових процедур щодо пацієнтів, а для аналізу зібраних даних використовують епідеміологічні методи.
Післяреєстраційне неінтервенційне дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну «випадок- контроль» — вид післяреєстраційного дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну, що проводиться на двох групах пацієнтів, в однієї з яких присутні конкретні ятрогенні захворювання чи побічні реакції, а в другої — немає подібних захворювань чи побічних реакцій, з метою виявлення факторів ризику розвитку ятрогенних захворювань та побічних реакцій.
Післяреєстраційне неінтервенційне когортне дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну — вид післяреєстраційного дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну, при проведенні якого протягом певного часу ведеться спостереження за двома підібраними великими групами хворих, одна з яких отримує досліджуваний препарат, а друга — його не отримує, з метою виявлення побічних реакцій, факторів ризику виникнення побічних реакцій, тощо.
План управління ризиками — детальний опис системи управління ризиками.
Побічна реакція — будь–яка ненавмисна і шкідлива реакція на:
лікарський засіб;
вакцину, туберкулін, у разі її спричинення або прискорення активним компонентом або одним із інших компонентів, або вона пов’язана з дефектом якості вакцини, туберкуліну.
Причинно-наслідковий зв’язок між клінічними проявами будь–якої побічної реакції/НППІ та застосуванням лікарського засобу, вакцини та туберкуліну — ступінь, який визначається прийнятним методом (якісна методика Всесвітньої організації охорони здоров’я (далі — ВООЗ), шкала Наранжо, бінарний метод тощо) за певними критеріями та вказує на взаємопов’язаність/взаємозв’язок побічної реакції/НППІ, що спостерігається, із застосуванням лікарського засобу, вакцини та туберкуліну.
Протокол післяреєстраційного дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну — документ, що містить опис, завдання, методологію, процедури, статистичні аспекти та організацію післяреєстраційного дослідження з безпеки та ефективності лікарського засобу, вакцини та туберкуліну, а також, за необхідності, раніше отримані дані щодо досліджуваного лікарського засобу, вакцини та туберкуліну та обґрунтування цього дослідження.
Профіль безпеки — сукупність показників застосування лікарського засобу, вакцини та туберкуліну, що дозволяють визначити співвідношення користь/ризик лікарського засобу, вакцини та туберкуліну.
Регулярно оновлюваний звіт з безпеки лікарського засобу, вакцини та туберкуліну — письмовий звіт, що містить регулярно оновлювану інформацію з безпеки лікарського засобу, вакцини та туберкуліну (далі — регулярний звіт).
Ризик, пов’язаний із застосуванням лікарського засобу, вакцини та туберкуліну — будь-який ризик, пов’язаний з якістю, безпекою чи ефективністю лікарського засобу, вакцини та туберкуліну, що стосується здоров’я пацієнтів або суспільного здоров’я, чи будь-який ризик небажаних ефектів на довкілля.
Розподіл випадків побічних реакцій лікарського засобу, вакцини та туберкуліну за частотою їх виникнення:
понад 10% — дуже часті;
1 10% — часті;
0,1 — 1% — нечасті;
0,01 — 0,1% — поодинокі;
менше 0,01% — рідкісні.
Серйозна побічна реакція — будь-яка побічна реакція, що призводить до смерті, становить загрозу для життя, вимагає госпіталізації або подовження терміну госпіталізації, викликає стійку або значну непрацездатність чи інвалідність, або є вродженою аномалією чи вадою розвитку, або має іншу важливу медичну оцінку.
Сероконверсія — продукування або збільшення концентрації антитіл, що розглядається як перехід від серонегативної до серопозитивної реакції або як клінічно значиме збільшення заданого рівня антитіл, що демонструє імуногенність вакцини.
Серопротекція — титр антитіл, що захищає від визначеної(их) інфекції(ій).
Сигнал — інформація, що походить з одного або декількох джерел (у тому числі спостережень і досліджень), що свідчить про виявлений новий потенційний зв’язок або новий аспект відомого зв’язку між лікарським засобом, вакциною та туберкуліном і явищем або сукупністю взаємопов’язаних явищ, як несприятливих, так і сприятливих, і яка вважається достатньо достовірною, щоб обґрунтувати її перевірку.
Система управління ризиками — комплекс процесів та заходів із фармаконагляду, спрямованих на виявлення, характеристику, запобігання або мінімізацію ризиків, пов’язаних із застосуванням лікарського засобу, вакцини та туберкуліну, що включає оцінку їх ефективності.
Система фармаконагляду — система, що використовується державою та заявником для здійснення фармаконагляду з метою моніторингу безпеки та ефективності лікарських засобів, вакцин та туберкуліну та визначення будь- яких змін співвідношення користь/ризик.
Співвідношення користь/ризик лікарського засобу, вакцини та туберкуліну — оцінка позитивних терапевтичних ефектів лікарського засобу, вакцини та туберкуліну відносно будь-яких ризиків, пов’язаних з якістю, безпекою чи ефективністю лікарського засобу, вакцини та туберкуліну, що стосуються здоров’я пацієнтів чи суспільного здоров’я.
Термінове повідомлення — це повідомлення про випадок серйозної (передбаченої або непередбаченої) побічної реакції лікарського засобу, що стався на території України, наслідком якого є смерть пацієнта.
Узагальнюючий звіт — письмовий звіт, що узагальнює інформацію з безпеки лікарського засобу, вакцини та туберкуліну, що міститься у двох або більше регулярно оновлюваних звітах з безпеки лікарського засобу, вакцини та туберкуліну.
Фармаконагляд — процес, пов’язаний із виявленням, збором, оцінкою, вивченням та запобіганням виникненню побічних реакцій, НППІ та будь-яких інших проблем, зумовлених впливом лікарських засобів, вакцини та туберкуліну на стан здоров’я пацієнтів чи суспільного здоров’я.
Частота випадків побічних реакцій лікарського засобу, вакцини та туберкуліну — співвідношення кількості пацієнтів за певний проміжок часу, в яких виникла побічна реакція при застосуванні лікарського засобу, вакцини та туберкуліну до кількості пацієнтів за цей же проміжок часу, які застосовували цей лікарський засіб, вакцину та туберкулін виражене у відсотках.
2. Інші терміни вживаються в цьому Порядку у значенні, визначеному законодавством України.
ІІ. ЗАГАЛЬНІ ПОЛОЖЕННЯ ЩОДО ЗДІЙСНЕННЯ ФАРМАКОНАГЛЯДУ
1. Здійснення фармаконагляду в Україні забезпечується за застосування міжнародних стандартів, виконання правил і вимог, встановлених цим Порядком, та передбачає створення і функціонування системи фармаконагляду.
2. Система фармаконагляду створюється у системі охорони здоров’я на загальнодержавному рівні та у заявників лікарських засобів, вакцин та туберкуліну (далі — лікарські засоби).
3. Система фармаконагляду у системі охорони здоров’я використовується з метою:
1) збору інформації про ризики лікарських засобів, щодо здоров’я пацієнтів чи суспільного здоров’я, зокрема, для збору інформації про побічні реакції, відсутність ефективності лікарських засобів, НППІ (класифікація НППІ за випадком наведена у додатку 1 до цього Порядку) та про інші проблеми з безпеки, пов’язані із застосуванням лікарських засобів;
2) обґрунтованої оцінки усієї інформації про ризики лікарських засобів, щодо здоров’я пацієнтів чи суспільного здоров’я;
3) розробки заходів для запобігання чи мінімізації ризиків, пов’язаних із застосуванням лікарських засобів;
4) застосування регуляторних заходів впливу щодо дії реєстраційного посвідчення на лікарський засіб у разі необхідності.
4. Для здійснення фармаконагляду Міністерство охорони здоров’я України (далі — МОЗ України):
1) залучає медичних працівників, заявників, пацієнтів та/або їх представників, організацій, що представляють інтереси пацієнтів, тощо;
2) встановлює порядок дій керівників структурних підрозділів з питань охорони здоров’я, керівників закладів охорони здоров’я, медичних працівників, а також — правила, вимоги та зобов’язання до заявників лікарських засобів;
3) регулює взаємовідносини між структурами, що мають відношення до процесу регулювання обігу лікарських засобів.
5) Здійснення фармаконагляду на загальнодержавному рівні покладено МОЗ України на Державне підприємство «Державний експертний центр Міністерства охорони здоров’я України» (далі — Центр).
6. Центр для виконання покладених на нього повноважень щодо фармаконагляду:
1) здійснює заходи щодо оптимізації подання медичними працівниками інформації про побічні реакції, відсутність ефективності, НППІ та інші проблеми з безпеки, пов’язані із застосуванням лікарських засобів до Центру, у тому числі шляхом залучення до цього процесу співробітників із фармаконагляду Центру в регіонах;
2) сприяє поданню пацієнтами та/або їх представниками, організаціями, що представляють інтереси пацієнтів, повідомлень про побічні реакції та/або відсутність ефективності лікарського засобу, НППІ, та інші проблеми з безпеки, пов’язані із застосуванням лікарського засобу, усіма доступними способами;
3) здійснює необхідні заходи для отримання точних даних, що піддаються перевірці, для оцінки повідомлень про побічні реакції та/або відсутність ефективності лікарського засобу, НППІ та інші проблеми з безпеки, пов’язані із застосуванням лікарського засобу;
4) інформує громадськість про важливі дані щодо безпеки застосування лікарських засобів усіма доступними методами;
5) проводить перевірку системи фармаконагляду у заявників;
6) вживає необхідних заходів для забезпечення того, щоб до заявників, які не виконують правила, вимоги та зобов’язання, викладені у цьому Порядку, були застосовані ефективні, пропорційні та стримуючі санкції.
7. Заявник для виконання покладених на нього правил, вимог та зобов’язань із фармаконагляду створює та керує системою фармаконагляду. Заявник може мати більше ніж одну систему фармаконагляду, наприклад, специфічні системи для різних видів лікарських засобів (наприклад, вакцини, безрецептурні лікарські засоби, тощо).
8. Заявник, здійснюючи фармаконагляд:
1) постійно і безперервно має у штаті уповноважену особу, відповідальну за фармаконагляд (далі — УОВФ);
2) створює, підтримує і надає на вимогу Центру МФСФ;
3) керує системою управління ризиками для лікарського засобу, у разі необхідності;
4) здійснює моніторинг результатів заходів з мінімізації ризиків, що містяться в плані управління ризиками, та тих, що є умовою видачі реєстраційного посвідчення;
5) регулярно оновлює систему управління ризиками та проводить моніторинг даних з фармаконагляду для виявлення нових ризиків, змін відомих ризиків та змін співвідношення користь/ризик лікарських засобів;
6) регулярно проводить аудит своєї системи фармаконагляду.
9. Кожна система фармаконагляду заявника може мати лише одну УОВФ. Якщо УОВФ не проживає в Україні, то на території України призначається єдина контактна особа з фармаконагляду (далі — КОВФ), яка проживає і працює в Україні та підзвітна УОВФ.
10. Заявник надає до Центру інформацію про УОВФ, КОВФ, а саме: прізвище, ім’я, по батькові, контактні дані та документи (копію диплому) про наявність медичної або фармацевтичної освіти.
ІІІ. ДЖЕРЕЛА ОТРИМАННЯ ТА ОБМІН ІНФОРМАЦІЄЮ З БЕЗПЕКИ ТА ЕФЕКТИВНОСТІ ЛІКАРСЬКИХ ЗАСОБІВ
1. Інформація з безпеки та ефективності лікарських засобів надходить до Центру від:
1) медичних працівників;
2) фізичних осіб-підприємців, які здійснюють господарську діяльність з медичної практики;
3) закладів охорони здоров’я;
4) заявників;
5) пацієнтів та/або їх представників, організацій, що представляють інтереси пацієнтів, за формою, наведеною у додатку 2 до цього Порядку;
6) Державної служби України з лікарських засобів (далі — Держлікслужба) та територіальних органів Держлікслужби;
7) співробітників із фармаконагляду Центру в регіонах;
8) ВООЗ, міжнародних агентств (EMA (Європейське медичне агентство), FDA (Агентство з контролю за лікарськими засобами та харчовими продуктами США), MHRA (Агентство з регулювання обігу лікарських засобів і виробів медичного призначення Великобританії), Health Canada (Міністерство охорони здоров’я Канади), TGA (Агентство з контролю за лікарськими засобами та виробами медичного призначення Австралії), Swissmedic (Агентство з лікарських засобів Швейцарії), PMDA (Агентство з контролю за лікарськими засобами та виробами медичного призначення Японії) та інших міжнародних організацій;
9) офіційних інформаційних джерел та періодичних видань;
10) інших джерел.
2. Центр надає інформацію з безпеки та ефективності лікарських засобів:
1) Держлікслужбі (у паперовому та/або електронному вигляді):
копії отриманих карт-повідомлень про непередбачені побічні реакції, побічні реакції, що призвели до смерті пацієнта, про групові побічні реакції та/або відсутність ефективності лікарських засобів, за виключенням вакцин, туберкуліну, за наявності причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу, за виключенням вакцин, туберкуліну, — не пізніше 48 годин з моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу, за виключенням вакцин, туберкуліну. У разі якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
копії отриманих карт-повідомлень про групові побічні реакції та/або серйозні побічні реакції, пов’язані з дефектом якості вакцини, туберкуліну за наявності причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну — не пізніше 48 годин від моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну. У разі якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
2) заявнику — стосовно лікарських засобів, які він представляє на фармацевтичному ринку України:
про непередбачені побічні реакції, побічні реакції, що призвели до смерті пацієнта, про групові побічні реакції та/або відсутність ефективності лікарських засобів, за виключенням вакцин, туберкуліну, за наявності причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу, за виключенням вакцин, туберкуліну, — не пізніше 48 годин з моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу за виключенням вакцин, туберкуліну. У разі якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
про випадки групових побічних реакцій та/або серйозних побічних реакцій, пов’язаних з дефектом якості вакцини, туберкуліну за наявності причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну — не пізніше 48 годин від моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну. У разі якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
про випадки побічних реакцій та/або відсутності ефективності лікарського засобу та /або НППІ, інформація про які надійшла до Центру протягом певного періоду — на його вимогу;
3) МОЗ України:
на вимогу;
щороку у вигляді звіту — 1 березня наступного року. У випадку, якщо цей строк надання звіту припадає на вихідний або святковий день, звіт надається у перший після нього робочий день;
щороку у вигляді звіту про зареєстровані випадки захворювання на інфекційні хвороби, що керуються засобами специфічної імунопрофілактики у щеплених — до 20 березня наступного року. У випадку, якщо цей строк надання звіту припадає на вихідний або святковий день, звіт надається у перший після нього робочий день;
щоквартально у вигляді звіту щодо зведених даних про випадки побічних реакцій після застосування вакцин, туберкуліну — до 30 числа наступного за звітним місяця. У випадку, якщо цей строк припадає на вихідний або святковий день, зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну надаються у перший після нього робочий день;
копії (у паперовому та/або електронному вигляді) отриманих карт-повідомлень про непередбачені побічні реакції, побічні реакції, що призвели до смерті пацієнта, про групові побічні реакції та/або відсутність ефективності лікарських засобів, за виключенням вакцин, туберкуліну, за наявності причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу, за виключенням вакцин, туберкуліну, — не пізніше 48 годин з моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу за виключенням вакцин, туберкуліну. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
копії (у паперовому та/або електронному вигляді) отриманих карт-повідомлень про групові побічні реакції та/або серйозні побічні реакції, пов’язані з дефектом якості вакцини, туберкуліну за наявності причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну, протоколу розслідування серйозних та/або групових НППІ після застосування вакцини, туберкуліну та визначення причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну (далі — протокол розслідування НППІ) — не пізніше 48 годин від моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
копії (у паперовому та/або електронному вигляді) отриманих карт-повідомлень про НППІ, що пов’язані з програмною помилкою імунізації/туберкулінодіагностики при застосуванні вакцини, туберкуліну за наявності інформації згідно протоколу розслідування НППІ — не пізніше 48 годин від моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
копії (у паперовому та/або електронному вигляді) отриманих карт- повідомлень про НППІ (летальний випадок), що зареєстрований протягом 30 діб після імунізації/туберкулінодіагностики — не пізніше 48 годин від моменту отримання Центром карти-повідомлення. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
4) Центральній групі оперативного реагування:
копії (у паперовому та/або електронному вигляді) отриманих карт- повідомлень про НППІ, що містять інформацію про клінічні прояви, що не зазначені в інструкції для медичного застосування або переліку клінічних проявів побічних реакцій при застосуванні вакцин, туберкуліну, що зазначені у додатку 11 до цього Порядку, карт-повідомлень, що стали підставою для формування сигналів — не пізніше 48 годин з моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між такими НППІ та застосуванням серії/серій вакцини, туберкуліну. У разі якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
копії (у паперовому та/або електронному вигляді) отриманих карт- повідомлень про НППІ (летальний випадок), що зареєстрований протягом 30 діб після імунізації/туберкулінодіагностики — не пізніше 48 годин від моменту отримання Центром карти-повідомлення. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
копії (у паперовому та/або електронному вигляді) отриманих карт- повідомлень про НППІ, що пов’язані з програмною помилкою імунізації/туберкулінодіагностики при застосуванні вакцини, туберкуліну за наявності інформації згідно протоколу розслідування НППІ — не пізніше 48 годин від моменту отримання Центром інформації, що підтверджує наявність причинно-наслідкового зв’язку між ними та застосуванням вакцини, туберкуліну. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
5) ВООЗ;
6) іншим організаціям у межах законодавства України.
3. Держлікслужба надає Центру копії (у паперовому та/або електронному вигляді) усіх отриманих карт-повідомлень, включаючи НППІ та інформацію про летальний випадок незалежно від джерела їх надходження — не пізніше 48 годин від моменту отримання цієї інформації. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
4. Центр аналізує інформацію, надану Держлікслужбою, відповідно до пункту 3 цього розділу, та надає результати цього аналізу Держлікслужбі не пізніше 48 годин від моменту отримання такої інформації. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
5. Територіальні органи Держлікслужби надають співробітникам із фармаконагляду Центру в регіонах та Держлікслужбі копії (у паперовому та/або електронному вигляді) усіх отриманих карт-повідомлень — не пізніше 48 годин від моменту отримання цієї інформації територіальними органами Держлікслужби. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
6. Співробітники із фармаконагляду Центру в регіонах надають:
1) до Центру карти-повідомлення та/або їх копії (у паперовому та/або електронному вигляді):
про випадки побічних реакцій, що призвели до смерті пацієнта, та/або відсутності ефективності лікарського засобу — не пізніше 48 годин від моменту їх надходження до співробітників із фармаконагляду Центру в регіонах. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
про усі інші випадки побічних реакцій — не пізніше 15 днів від моменту їх надходження до співробітників із фармаконагляду Центру в регіонах. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
2) до структурних підрозділів з питань охорони здоров’я копії (у паперовому та/або електронному вигляді) отриманих карт-повідомлень про побічні реакції та/або відсутність ефективності лікарського засобу, що призвели до смерті пацієнта, — не пізніше 48 годин від моменту їх надходження до співробітників із фармаконагляду Центру в регіонах. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
7. Структурні підрозділи з питань охорони здоров’я при отриманні карт- повідомлень або їх копій (у паперовому та/або електронному вигляді) про побічні реакції та/або відсутність ефективності лікарського засобу, що призвели до смерті пацієнта, надають цю інформацію до відповідної клініко-експертної комісії, що здійснює контроль якості медичної допомоги — не пізніше 48 годин від моменту отримання такої інформації. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
8. Контроль якості медичної допомоги щодо випадків побічних реакцій та/або відсутності ефективності лікарського засобу, що призвели до смерті пацієнта, здійснюється клініко-експертною комісією із залученням співробітників із фармаконагляду Центру в регіонах та інших фахівців за згодою.
9. За результатами роботи клініко-експертної комісії складаються протокол та висновок клініко-експертної комісії щодо випадків побічних реакцій та/або відсутності ефективності лікарського засобу, що призвели до смерті пацієнта, що надаються до МОЗ України, Центру та Держлікслужби не пізніше 24 годин від моменту їх складання. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
ІV. ПОРЯДОК ДІЙ КЕРІВНИКІВ СТРУКТУРНИХ ПІДРОЗДІЛІВ З ПИТАНЬ ОХОРОНИ ЗДОРОВ’Я, КЕРІВНИКІВ ЗАКЛАДІВ ОХОРОНИ ЗДОРОВ’Я ТА МЕДИЧНИХ ПРАЦІВНИКІВ У КОНТЕКСТІ ЗДІЙСНЕННЯ ФАРМАКОНАГЛЯДУ
1. Керівники структурних підрозділів з питань охорони здоров’я:
1) призначають відповідальну особу за складання зведеного звіту за формою № 69 «Звіт про випадки побічних реакцій та/або відсутності ефективності лікарських засобів, НППІ у закладах охорони здоров’я за 20____ рік» (далі — форма № 69), за формою, наведеною у додатку 3 до цього Порядку, у термін, визначений підпунктом 2 пункту 1 розділу ІV цього Порядку. Зведений звіт за формою № 69 складається на підставі даних форми № 69, що подаються відповідальними особами з питань фармаконагляду підзвітних закладів охорони здоров’я за формою, наведеною у додатку 4 до цього Порядку, у термін, визначений підпунктом 2 пункту 3 розділу ІV цього Порядку;
2) забезпечують своєчасне подання до Центру зведеного звіту за формою № 69 до 30 січня наступного за звітним року за підписом керівника відповідного структурного підрозділу з питань охорони здоров’я у паперовому та електронному вигляді (таблиця Excel). На титульній сторінці форми № 69 вказуються поштова та електронна адреси. Зведений звіт за формою № 69 у паперовому вигляді подається до Центру разом із супровідним листом на адресу, що зазначена у формі № 69. Зведений звіт у електронному вигляді (таблиця Excel) подається на електронну адресу, що зазначена у формі № 69;
3) забезпечують своєчасне подання до Центру протоколів розслідування та встановлення причинно-наслідкового між НППІ та вакциною, туберкуліном, за формою, наведеною у додатку 5 до цього Порядку, та карт-повідомлень, за формою, наведеною у додатку 6 до цього Порядку;
4) призначають відповідальну особу за складання загального звіту по картах епідрозслідування за формою, наведеною у додатку 7 до цього Порядку, яка складає загальний звіт по картах епідрозслідування на підставі даних карт епідрозслідування за формою, наведеною у додатку 8 до цього Порядку;
5) забезпечують своєчасне подання загального звіту по картах епідрозслідування до Центру у електронному вигляді у термін до 30 січня наступного за звітним року. Загальний звіт у електронному вигляді подається на електронну адресу, що зазначена у вимогах до заповнення загального звіту по картах епідрозслідування, наведених у додатку 7 до цього Порядку;
6) забезпечують подання до Центру зведених даних про випадки побічних реакцій після застосування вакцин, туберкуліну щоквартально у електронному та паперовому вигляді за формою, наведеною у додатку 11 до цього Порядку у термін до 10 числа наступного за звітним місяця. У разі якщо цей строк припадає на вихідний або святковий день, зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну надаються у перший після нього робочий день. Зведені дані у паперовому вигляді подаються до Центру разом із супровідним листом на адресу, що зазначена вимогах до складання зведених даних, наведених у додатку 11 до цього Порядку. Зведені дані у електронному вигляді подаються на електронну адресу, що зазначена у вимогах до складання зведених даних, наведених у додатку 11 до цього Порядку;
7) заслуховують щопівроку питання про стан подання медичними працівниками інформації про випадки побічних реакцій, відсутності ефективності лікарських засобів, НППІ на засіданнях колегій структурних підрозділів з питань охорони здоров’я;
8) включають до складу атестаційних комісій при підпорядкованих структурних підрозділах з питань охорони здоров’я співробітників із фармаконагляду Центру в регіонах;
2. Керівник Державного підприємства «Укрвакцина» МОЗ України (далі — Укрвакцина) призначає відповідальну особу за складання та подання звіту щодо кількісного розподілу вакцин, анатоксинів, туберкуліну по регіонам України за формою, наведеною у додатку 9 до цього Порядку, до Центру на вимогу.
3. Керівники закладів охорони здоров’я призначають заступника головного лікаря з лікувальної роботи або іншу особу, яка має вищу медичну та/або фармацевтичну освіту, відповідальною особою з питань фармаконагляду та забезпечують:
1) своєчасне подання медичними працівниками інформації про випадки побічних реакцій, відсутності ефективності лікарських засобів, НППІ та інших проблем, пов’язаних із застосуванням лікарських засобів, за формою, наведеною у додатку 6 до цього Порядку, у термін, визначений пунктом 9 розділу ІV цього Порядку;
2) своєчасне подання до відповідного структурного підрозділу з питань охорони здоров’я звіту за формою № 69 (за виключенням таблиці 1001) у термін не пізніше 20 січня наступного за звітним роком у паперовому та електронному вигляді (таблиця Excel). Звіт за формою № 69 у паперовому вигляді подається разом із супровідним листом за підписом керівника закладу охорони здоров’я на адресу відповідних структурних підрозділів з питань охорони здоров’я;
3) своєчасне подання до структурних підрозділів з питань охорони здоров’я форми № 058/о «Екстрене повідомлення про інфекційне захворювання, харчове, гостре професійне отруєння, незвичайну реакцію на щеплення» (далі — повідомлення про інфекційне захворювання), затвердженої наказом МОЗ України від 10 січня 2006 року № 1 «Про затвердження Форм первинної облікової документації з інфекційної, дерматовенерологічної, онкологічної захворюваності та інструкцій щодо їх заповнення» у випадку інфекційного захворювання у хворого, щепленого проти відповідної інфекції, у термін не пізніше 12 годин після виявлення такого хворого.
4. Відповідальна особа з питань фармаконагляду закладу охорони здоров’я:
1) створює стандартні операційні процедури щодо порядку дій у разі виникнення побічних реакцій, відсутності ефективності лікарського засобу, НППІ та ознайомлює з ними медичних працівників;
2) веде облік карт-повідомлень;
3) складає звіт за формою, наведеною у додатку 4 до цього Порядку (за виключенням таблиці 1001) та надає у термін, визначений підпунктом 2 пункту 3 розділу ІV цього Порядку до відповідного структурного підрозділу з питань охорони здоров’я. Звіт у паперовому вигляді разом із супровідним листом та підписом керівника закладу охорони здоров’я подається на адресу відповідного структурного підрозділу з питань охорони здоров’я. Звіт в електронному вигляді (таблиця Excel) подається на електронну адресу відповідного структурного підрозділу з питань охорони здоров’я;
4) співпрацює зі співробітниками із фармаконагляду Центру в регіонах з питань здійснення фармаконагляду (аналізу випадків побічних реакцій, відсутності ефективності лікарських засобів, НППІ, виявлення системних помилок, навчання, тощо);
5) сприяє реалізації заходів з мінімізації ризиків, пов’язаних із застосуванням лікарських засобів.
6) веде облік побічних реакцій після застосування вакцин, туберкуліну;
7) складає та надає до відповідальної особи структурного підрозділу з питань охорони здоров’я, зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну щомісяця до 5 числа наступного за звітним місяця за формою, наведеною у додатку 11 до цього Порядку. У разі якщо цей строк припадає на вихідний або святковий день, зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну надаються у перший після нього робочий день. Зведені дані у паперовому вигляді разом із супровідним листом та підписом керівника закладу охорони здоров’ я подаються на адресу відповідного структурного підрозділу з питань охорони здоров’я. Зведені дані в електронному вигляді подаються на електронну адресу відповідного структурного підрозділу з питань охорони здоров’ я;
5. Керівники закладів охорони здоров’я забезпечують надання копій документів первинної медичної документації закладу охорони здоров’я у разі надходження запиту Центру.
6. Медичні працівники інформують:
1) пацієнтів (або їх батьків, або законних представників) про ті побічні реакції, що можуть виникнути при застосуванні лікарських засобів, та необхідність звернення за медичною допомогою до закладу охорони здоров’я у разі порушення стану здоров’я після застосування лікарського засобу;
2) осіб, які підлягають імунізації/туберкулінодіагностиці (або їх батьків, або законних представників), про ті побічні реакції, що можуть виникнути після застосування певної вакцини, туберкуліну, та необхідність звернення за медичною допомогою до закладу охорони здоров’я у разі будь-якого порушення у стані здоров’я після проведення імунізації/туберкулінодіагностики.
7. Медичні працівники виявляють:
1) побічні реакції, відсутність ефективності лікарського засобу та будь-які порушення у стані здоров’я та інші проблеми з безпеки, пов’язані із застосування лікарського засобу, при зверненні пацієнта до закладу охорони здоров’я;
2) НППІ та інші проблеми з безпеки, пов’язані із застосуванням вакцини, туберкуліну, при зверненні імунізованої особи або особи, якій було проведено туберкулінодіагностику, до закладу охорони здоров’я;
8. Медичні працівники своєчасно подають до Центру інформацію про будь-які побічні реакції, відсутність ефективності лікарського засобу та НППІ за формою та згідно з вимогами, наведеними у додатку 6 до цього Порядку, у терміни, визначені пунктом 9 розділу ІV цього Порядку.
9. Карта-повідомлення подається у паперовому та/або електронному вигляді. Електронна форма карти-повідомлення знаходиться за посиланням: https://aisf.dec.gov.ua . Копія карти-повідомлення подається відповідальній особі з питань фармаконагляду закладу охорони здоров’я для складання зведеного звіту за формою № 69.
У випадку розвитку несерйозної побічної реакції/НППІ при застосуванні лікарського засобу медичні працівники подають карту-повідомлення до Центру протягом 90 днів.
У випадку розвитку серйозної побічної реакції/НППІ при застосуванні лікарського засобу медичні працівники подають карту-повідомлення до Центру протягом 15 днів.
У випадку розвитку відсутності ефективності при застосуванні лікарського засобу медичні працівники подають карту-повідомлення до Центру протягом 48 годин.
У випадку розвитку побічної реакції та/або відсутності ефективності, НППІ при застосуванні лікарського засобу, що призвели до смерті пацієнта, медичні працівники подають карту-повідомлення до Центру протягом 48 годин.
У випадку, якщо строк подання карти-повідомлення припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
10. Інформація про побічні реакції, відсутність ефективності при застосуванні лікарського засобу, НППІ заноситься лікарем до первинної облікової медичної документації.
11. Медичні працівники своєчасно подають до Центру та групи оперативного реагування відповідних структурних підрозділів з питань охорони здоров’я (далі — регіональна група оперативного реагування) інформацію про НППІ після застосування вакцини, туберкуліну, за формою та згідно з вимогами, наведеними у додатку 6 до цього Порядку, у термін не пізніше 48 годин після реєстрації НППІ. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
12. Карта-повідомлення подається до Центру та до регіональної групи оперативного реагування у паперовому та/або електронному вигляді. Копія карти-повідомлення надається відповідальній особі з питань фармаконагляду закладу охорони здоров’я для складання зведеного звіту за формою № 69.
13. Порядок створення, склад та завдання регіональних та центральної груп оперативного реагування визначені Положенням, наведеним у додатку 10 до цього Порядку.
14. Регіональна група оперативного реагування проводить аналіз отриманих карт-повідомлень про НППІ після застосування вакцини, туберкуліну. У разі, якщо НППІ після застосування вакцини, туберкуліну є серйозною та/або груповою, регіональна група оперативного реагування проводить розслідування та встановлює причинно-наслідковий зв’язок між НППІ та вакциною, туберкуліном.
15. За результатами розслідування та встановлення причинно-наслідкового зв’язку група оперативного реагування заповнює протокол розслідування НППІ за формою, наведеною у додатку 5 до цього Порядку, з урахуванням інформації, наведеної у додатках 11 та 12 до цього Порядку, та надає його до Центру, разом з копією відповідної карти-повідомлення — не пізніше 15 днів від моменту розвитку НППІ. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
При отриманні додаткових даних щодо серйозної та/або групової НППІ, що спричиняє переоцінку причинно-наслідкового зв’язку між НППІ та вакциною, туберкуліном регіональна група оперативного реагування складає оновлений протокол розслідування НППІ за формою, наведеною у додатку 5 до цього Порядку і надає його до Центру, не пізніше 15 днів від моменту отримання додаткових даних. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
16. Центральна група оперативного реагування залучається до дій після отримання від Центру інформації, що зазначена у підпункту 4 пункту 2 розділу ІІІ. Протокол розслідування НППІ між серйозним та/або груповим НППІ та застосуванням вакцини, туберкуліну надається Центром одночасно з картами-повідомленнями.
17. Медичні працівники закладу охорони здоров’я, де проводиться щеплення, здійснюють моніторинг та надають інформацію щодо захворюваності у щеплених на інфекції, що керуються засобами специфічної імунопрофілактики.
У разі виявлення випадку інфекційного захворювання, медичні працівники заповнюють первинну облікову медичну документацію (повідомлення про інфекційне захворювання) та надають його до структурного підрозділу з питань охорони здоров’я у термін не пізніше 12 годин після виявлення такого захворювання.
18. Відповідальна особа структурного підрозділу з питань охорони здоров’я, при отриманні повідомлення про інфекційне захворювання, проводить епідеміологічне розслідування за місцем виникнення/виявлення захворювання.
У випадку, якщо під час епідеміологічного розслідування буде з’ясовано, що має місце випадок інфекційного захворювання у особи, щепленої проти відповідної інфекції, відповідальна особа структурного підрозділу з питань охорони здоров’я заповнює карту епідрозслідування, за формою, наведеною у додатку 8 до цього Порядку у термін не пізніше 48 годин з моменту з’ясування. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
19. На основі заповнених карт епідрозслідування відповідальна особа структурного підрозділу з питань охорони здоров’я складає загальний звіт по картах епідрозслідування за формою, наведеною у додатку 7 до цього Порядку.
20. Структурний підрозділ з питань охорони здоров’я надає до Центру загальний звіт по картах епідрозслідування — щороку до 30 січня наступного за звітним року. У випадку, якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день.
V. ЗДІЙСНЕННЯ ФАРМАКОНАГЛЯДУ ЗАЯВНИКОМ
V.1. Загальні вимоги до системи фармаконагляду у заявника
1. Заявник створює, забезпечує та гарантує у себе функціонування системи фармаконагляду в Україні, що є обов’язковою умовою знаходження лікарських засобів в обігу на території України.
2. Система фармаконагляду складається з елементів, що дозволяють здійснювати моніторинг безпеки лікарських засобів та визначати будь-які зміни співвідношення користь/ризик, а саме:
1) наявність УОВФ у заявника. У разі, якщо заявник знаходиться в Україні, УОВФ повинна бути в штаті заявника. У разі, якщо заявник знаходиться не в Україні, то на території України в штаті повинна бути призначена єдина КОВФ в Україні та підпорядкована УОВФ заявника. Дані про УОВФ та КОВФ в Україні повинні міститися в МФСФ заявника. УОВФ/КОВФ в Україні повинні мати вищу медичну або фармацевтичну освіту (провізор, клінічний провізор). За наявності лише вищої фармацевтичної освіти УОВФ/КОВФ в Україні повинна мати змогу звернутись до особи з вищою медичною освітою (за необхідності). Кожна система фармаконагляду може мати лише одну УОВФ/КОВФ в Україні. Має бути передбачена процедура заміщення УОВФ та/або КОВФ в разі її відсутності;
2) наявність структурованої системи організації фармаконагляду, її оновлення і підтримка;
3) документування всіх процедурних процесів;
4) створення та забезпечення функціонування баз даних, що використовуються заявником при здійсненні фармаконагляду;
5) залучення (у разі необхідності) до здійснення фармаконагляду інших осіб і організацій шляхом укладення контрактних угод;
6) забезпечення навчання персоналу заявника для виконання дій, пов’язаних із фармаконаглядом;
7) створення системи якості фармаконагляду;
8) ведення документації із фармаконагляду, включаючи її зберігання та архівування;
9) створення та підтримка системи управління ризиками.
3. УОВФ/КОВФ в Україні несе відповідальність, не обмежуючись, за:
1) створення та підтримку системи збору, оцінки та надання до Центру інформації про побічні реакції, відсутність ефективності лікарських засобів, НППІ, інших проблем, пов’язаних із застосуванням лікарських засобів, а також будь-яких інших даних, необхідних для оцінки ризику і користі при застосуванні лікарського засобу, включно із системою якості;
2) складання (для КОВФ — у разі необхідності) та/або надання регулярних звітів з періодичністю наведеною у додатку 18 до цього Порядку;
3) складання (для КОВФ — у разі необхідності) та/або надання планів управління ризиками;
4) надання на всі запити Центру додаткової інформації, необхідної для оцінки співвідношення користь/ризик лікарського засобу, термінового надання звітів, у тому числі інформації про обсяги продажу лікарського засобу, або експозицію пацієнтів, які зазнали впливу лікарського засобу;
5) забезпечення надання будь-яких даних, необхідних для оцінки співвідношення користь/ризик лікарського засобу, включаючи інформацію про післяреєстраційні дослідження з безпеки та ефективності лікарського засобу;
6) надання повідомлення до Центру, вжиття необхідних заходів та внесення відповідних змін і доповнень до інформації з безпеки лікарського засобу, відображеної в інструкції для медичного застосування/короткій характеристиці, у випадку виявлення раніше невідомих небезпечних властивостей лікарського засобу, що призвели або можуть призвести до тяжких наслідків для здоров’я і життя людей або зміни оцінки співвідношення користь/ризик у бік ризику, про які йому стало відомо,
7) забезпечення навчання персоналу заявника для виконання дій, пов’язаних із фармаконаглядом.
4. У випадку внесення змін до системи фармаконагляду, заявник оновлює її, підтримує та подає на вимогу Центру МФСФ — документ, що містить опис системи фармаконагляду заявника, що використовується заявником стосовно одного або декількох лікарських засобів. МФСФ складається відповідно до структури, наведеної у додатку 13 до цього Порядку, та містить інформацію про:
1) УОВФ/КОВФ;
2) організаційну структуру заявника, включаючи систему фармаконагляду, що забезпечує збір, визначення, оцінку та подання вірогідної інформації про побічні реакції, відсутність ефективності лікарських засобів, НППІ, а також будь-яких інших даних, необхідних для оцінки ризику і користі при застосуванні лікарського засобу;
3) фізичні та/або юридичні особи, залучені заявником до здійснення фармаконагляду;
4) джерела інформації про безпеку застосування лікарських засобів;
5) перелік та стислий функціональний опис баз даних, що використовуються заявником при здійсненні фармаконагляду;
6) процеси у фармаконагляді, включаючи перелік стандартних операційних процедур, що використовуються при здійсненні фармаконагляду, опис документації з фармаконагляду, включаючи документацію, що зберігається в архіві; інші види документації, що мають відношення до здійснення фармаконагляду;
7) результативність системи фармаконагляду;
8) систему якості у фармаконагляді, включаючи опис системи навчання персоналу заявника із зазначенням інформації про навчання та з урахуванням функціональних обов’язків персоналу заявника, із наданням стислого опису зобов’язань заявника для гарантії якості аудиту системи фармаконагляду, включаючи аудит системи фармаконагляду та аудит фізичних та/або юридичних осіб, які залучені заявником до здійснення фармаконагляду.
V.2. Вимоги до подання заявником повідомлень про побічні реакції та/або відсутність ефективності лікарських засобів
5. При поданні заявником повідомлень про побічні реакції та/або відсутність ефективності лікарських засобів, заявник:
1) своєчасно подає до Центру у будь-який спосіб достовірну інформацію про всі випадки серйозних побічних реакцій лікарських засобів, що були зафіксовані при їх застосуванні в Україні та мають медичне підтвердження і про які йому стало відомо за наявності причинно-наслідкового зв’язку між побічною реакцією та застосуванням лікарського засобу — не пізніше 15 календарних днів з моменту отримання такої інформації. Якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
2) своєчасно подає до Центру у будь-який спосіб достовірну інформацію про всі випадки несерйозних побічних реакцій лікарських засобів, що були зафіксовані при їх застосуванні в Україні, та мають медичне підтвердження і про які йому стало відомо за наявності причинно-наслідкового зв’язку між побічною реакцією та застосуванням лікарського засобу — не пізніше 90 календарних днів з моменту отримання такої інформації. Якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
3) своєчасно подає до Центру у будь-який спосіб достовірну інформацію про всі випадки серйозних непередбачених побічних реакцій лікарських засобів, що призвели до смерті або становили загрозу для життя пацієнта, про всі підозрювані випадки передавання інфекції лікарським засобом, що були зафіксовані на території іншої країни та мають медичне підтвердження і про які йому стало відомо за наявності причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу — не пізніше 15 календарних днів з моменту отримання такої інформації. Якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
4) подає до Центру інформацію про усі інші випадки побічних реакцій лікарських засобів, що були зафіксовані на території іншої країни і про які йому стало відомо — у складі чергового регулярного звіту лікарського засобу;
5) своєчасно подає до Центру у будь-який спосіб достовірну інформацію про випадки відсутності ефективності, що були зафіксовані в Україні і виникли при:
лікуванні життєво небезпечних захворювань або невідкладних станів, крім випадків, коли першоджерело повідомлення зазначає, що випадок відсутності ефективності пов’язаний із прогресуванням захворювання, а не із застосуванням лікарського засобу;
застосуванні вакцин;
застосуванні контрацептивів.
Інформація подається не пізніше 15 календарних днів з моменту її отримання інформації. Якщо цей строк припадає на вихідний або святковий день, інформація надається у перший після нього робочий день;
6) подає до Центру інформацію про всі інші ідентифіковані випадки відсутності ефективності лікарського засобу про які йому стало відомо — у складі чергового регулярного звіту;
6. Повідомлення про побічні реакції та/або відсутність ефективності лікарських засобів подається згідно з вимогами, представленими у пункті 12 розділу V цього Порядку, або інформацією, що містить розділи та графи, аналогічні викладеним у пункті 12 розділу V цього Порядку, з наданням інформації, зазначеної у пункті 12 розділу V цього Порядку, у паперовому та/або у електронному вигляді, що знаходиться за посиланням:
7. Повідомлення про випадок побічної реакції та/або відсутності ефективності лікарського засобу заявник подає до Центру у вигляді інформації про підозрювані побічні реакції та/або відсутність ефективності лікарського засобу, що виникали у одного пацієнта в певний момент часу.
8. Заявник враховує повідомлення про випадки побічних реакцій та/або відсутності ефективності лікарського засобу, отримані ним електронним шляхом або іншим методом, від пацієнтів та/або їх представників, організацій, що представляють інтереси пацієнтів або медичних працівників.
9. Заявник співпрацює з Центром у напрямку виявлення дублікатів повідомлень про випадки побічних реакцій та/або відсутності ефективності лікарського засобу.
10. Повідомлення про випадок побічної реакції та/або відсутності ефективності лікарського засобу повинне містити щонайменше інформацію, згідно з якою можна ідентифікувати репортера, пацієнта, одну підозрювану побічну реакцію/відсутність ефективності і підозрюваний(і) лікарський(і) засіб(оби).
11. При поданні повідомлення про випадок побічної реакції та/або відсутності ефективності лікарського засобу заявник надає всю наявну інформацію про кожний окремий випадок, включаючи:
1) адміністративну інформацію, а саме: вид повідомлення, дату, унікальний ідентифікаційний номер випадку, а також унікальний ідентифікатор відправника і тип відправника; дату, коли було вперше отримано інформацію від джерела, та дату отримання найновішої інформації, зазначивши точну дату; інші ідентифікатори та їх джерела, а також посилання на додаткові доступні документи, що належать відправнику повідомлення про випадок побічної реакції та/або відсутності ефективності лікарського засобу, коли це застосовано;
2) посилання відповідно до міжнародних та вітчизняних вимог для публікацій для інформації про випадки побічних реакцій та/або відсутності ефективності, за даними літературних джерел, у тому числі вичерпне резюме статті англійською або українською або російською мовою. У разі, якщо стаття викладена англійською мовою слід надати переклад резюме українською мовою. На запит Центру, заявник, який передав первинне повідомлення, надає копію відповідної статті з урахуванням обмежень авторського права, а також повний переклад цієї статті українською мовою у разі якщо стаття викладена англійською мовою;
3) вид, назву і номер, присвоєний спонсором, або реєстраційний номер дослідження з безпеки та ефективності лікарського засобу для повідомлень про випадки побічних реакцій та/або відсутності ефективності при проведенні цього неінтервенційного дослідження;
4) інформацію про першоджерело(а): інформацію, що ідентифікує джерело повідомлення, включаючи назву країни, де проживає повідомник, та його професійну кваліфікацію;
5) інформацію, що ідентифікує пацієнта (і одного з батьків у випадку повідомлення батьки-дитина), включаючи вік на момент виникнення побічної реакції/відсутності ефективності, вікову групу, гестаційний вік, коли реакція/явище спостерігалася у плода, вагу, зріст, стать, дату останнього менструального і/або гестаційного періоду на момент виникнення побічної реакції/відсутності ефективності лікарського засобу;
6) анамнез і супутні захворювання пацієнта;
7) торгівельне(і) найменування лікарського(их) засобу(ів), що підозрюється(ються) у спричиненні побічної реакції та/або відсутності ефективності, у тому числі супутні лікарські засоби, або, якщо назва не відома, то активний(і) фармацевтичний (і) інгредієнт(и) і будь-які інші дані, що дозволяють ідентифікувати лікарський(і) засіб(и), у тому числі назва заявника/виробника, номер реєстраційного посвідчення, країна, де зареєстрований лікарський засіб, форма випуску та спосіб застосування, показання для застосування у даному випадку, застосовані дози, дата початку застосування і дата закінчення застосування, вжиті заходи щодо усунення проявів побічної реакції, включаючи фармакотерапію, результат відміни та повторного призначення підозрюваного(их) лікарського(их) засобу(ів);
8) для біологічних лікарських засобів — номер серії. Для того, щоб отримати номер серії, якщо він не вказаний у первинному повідомленні, у заявника повинна бути наявна процедура збору додаткової інформації;
9) супутні лікарські засоби, що не підозрюються у спричиненні побічної реакції/відсутності ефективності, фармакотерапію пацієнта (батьків) у минулому, якщо доречно;
10) інформацію про підозрювану(і) побічну(і) реакцію(ї)/відсутність ефективності: дату початку і дату завершення або тривалість, серйозність, наслідок підозрюваної(их) побічної(их) реакції(й)/відсутності ефективності на момент останнього спостереження пацієнта, проміжок часу, що минув від початку застосування підозрюваного лікарського засобу і початком побічної(их) реакції(й)/відсутності ефективності, терміни та вислови, зазначені у документації інформації про побічну(і) реакцію(ї)/відсутність ефективності, які використовувало першоджерело для опису побічної(их) реакції(й)/відсутності ефективності та країна, на території якої виникла(и) побічна(і) реакція(ї) та/або відсутність ефективності;
11) результати тестів і процедур, що мають відношення до дослідження пацієнта;
12) дату і повідомлену причину смерті, у тому числі причину смерті, встановлену за результатами патологоанатомічного дослідження;
13) опис випадку, де це можливо — надати усю доречну інформацію щодо випадку.
Інформація подається в логічній послідовності: як ситуація розвивалася у часі, у хронології нагляду за пацієнтом, включаючи клінічний перебіг, терапію, наслідки та отриману у подальшому додаткову інформацію; будь-які доречні результати патологоанатомічного дослідження також наводяться у описі випадку; інформацію щодо причинно-наслідкового зв’язку між проявами побічної реакції/відсутності ефективності та застосуванням підозрюваного лікарського засобу; причина анулювання або внесення змін до повідомлення про побічні реакції та/або відсутність ефективності лікарського засобу.
12. Заявник реєструє відомості необхідні для отримання у подальшому додаткової інформації про побічні реакції та/або відсутність ефективності лікарських засобів. Наступні повідомлення належним чином документуються.
V.3. Вимоги до подання заявником регулярних звітів
13. Заявник подає до Центру регулярний звіт, відповідно до структури та вимог до заповнення регулярного звіту, наведених у додатку 14 до цього Порядку. Регулярний звіт подається у паперовому вигляді; додатки до регулярного звіту можуть бути подані у електронному вигляді. Повний регулярний звіт подається українською або російською чи англійською мовою з періодичністю, зазначеною у пункті 15 підрозділу У.3 розділу V цього Порядку. У разі подання регулярного звіту англійською мовою розділи 3 «Заходи з безпеки, вжиті протягом звітного періоду», 4 «Зміни у довідковій інформації з безпеки», 18 «Інтегрований аналіз користь/ризик для зареєстрованих показань» та 19 «Висновки та заходи» або інші розділи, що містять таку інформацію, подаються в перекладі українською мовою. Розділи 16 «Оцінка сигналів та ризиків» та 17 «Оцінка користі» регулярного звіту подаються в перекладі українською мовою у разі потреби, на вимогу Центру.
14. Регулярний звіт подається з такою періодичністю:
1) для лікарського засобу, що зареєстрований в Україні, як у першій країні світу, або вперше у будь-якій іншій країні світу — кожні 6 місяців протягом перших двох років (незалежно від наявності лікарського засобу в обігу), один раз на рік протягом наступних двох років та у подальшому — 1 раз кожні три роки, починаючи відлік від дати реєстрації лікарського засобу. Якщо строки надання регулярних звітів, зазначені у цьому абзаці, співпадуть із строками, вказаними у Періодичності подання регулярно оновлюваних звітів з безпеки лікарських засобів за міжнародною непатентованою назвою активного фармацевтичного інгредієнта або комбінацій активних фармацевтичних інгредієнтів (далі — Періодичність подання регулярних звітів за МНН АФІ або комбінацій АФІ) наведеній у додатку 18 до цього Порядку, — регулярний звіт подається відповідно до строків, зазначених у Періодичності подання регулярних звітів за МНН АФІ або комбінацій АФІ;
2) або відповідно до періодичності, зазначеної у реєстраційному посвідченні при його видачі, згідно з підпунктом 1 пункту 16 підрозділу У.3 розділу У цього Порядку;
3) або згідно зі строком, зазначеним у Періодичності подання регулярних звітів за МНН АФІ або комбінацій АФІ, наведеній у додатку 18 до цього Порядку;
4) для лікарських засобів, зареєстрованих після набуття чинності цього наказу — регулярний звіт подається згідно зі строками, зазначеними у Періодичності подання регулярних звітів за МНН АФІ або комбінацій АФІ, або за бажанням заявника, але не рідше встановленого законодавством строку;
5) за запитом/вимогою Центру.
15. Регулярний звіт подається на всі лікарські засоби, окрім генеричних лікарських засобів; лікарських засобів, що мають добре вивчене медичне застосування; традиційних рослинних та гомеопатичних лікарських засобів, за винятком випадків, коли надання регулярного звіту для таких лікарських засобів:
1) є умовою видачі реєстраційного посвідчення;
2) вимагає МОЗ України та/або Центр у зв’язку з проблемами, пов’язаними з даними фармаконагляду, або через відсутність регулярного звіту для активного фармацевтичного інгредієнта після видачі реєстраційного посвідчення (наприклад, коли референтний лікарський засіб більше не зареєстровано в Україні);
3) зазначено у Переліку строків подання регулярних звітів.
Заявники лікарських засобів, яким більше не потрібно подавати регулярний звіт, продовжують регулярну оцінку безпеки своїх лікарських засобів та повідомляють про будь-яку нову інформацію з безпеки, що впливає на співвідношення користь/ризик та інформацію про лікарський засіб, що зазначена в інструкції для медичного застосування/короткій характеристиці лікарського засобу.
16. Регулярний звіт складається та подається відповідно до наступних термінів:
1) протягом 70 календарних днів з дати закриття бази даних (день 0) — для регулярних звітів, що охоплюють звітний період до 12 місяців включно;
2) протягом 90 календарних днів з дати закриття бази даних (день 0) — для регулярних звітів, що охоплюють звітний період понад 12 місяців;
3) відповідно до термінів, вказаних у запитах — для регулярних звітів на запит;
4) протягом 90 календарних днів з дати закриття бази даних (день 0) — для регулярних звітів в інших випадках.
17. При зміні частоти подання та/або строках подання регулярного звіту заявник подає заяву на зміни до реєстраційних матеріалів протягом дії реєстраційного посвідчення у визначеному законодавством Порядку. Застосовується у випадках, коли заявник виявив бажання подавати регулярний звіт за іншою періодичністю, але не рідше встановленого законодавством строку.
18. При перереєстрації лікарського засобу в Україні заявник подає до Центру разом із документами, необхідними для проведення експертизи матеріалів для державної перереєстрації лікарського засобу згідно з законодавством України, зведені дані про стан безпеки медичного застосування лікарського засобу в Україні за період дії останнього реєстраційного посвідчення за встановленою формою, наведеною у додатку 15 до цього Порядку.
19. Якщо не зазначено інше у Переліку строків подання регулярних звітів та періодичність подання регулярного звіту не зазначена, як умова видачі реєстраційного посвідчення, заявник складає регулярний звіт для активного фармацевтичного інгредієнта (далі — АФІ) на всі лікарські форми. Регулярний звіт охоплює усі показання, способи застосування, форми випуску і сили дії, незалежно від того, зареєстровані чи ні такі лікарські засоби під різними торговими назвами і за різними типами заявок на реєстрацію. За необхідності, дані, що стосуються окремого показання, форми випуску, способу застосування чи сили дії, представляються в окремому розділі регулярного звіту і відповідним чином повинні бути розглянуті будь-які проблеми з безпеки.
20. Якщо не зазначено інше у Переліку строків подання регулярних звітів та періодичність подання регулярного звіту не зазначена, як умова видачі реєстраційного посвідчення, у випадку, якщо АФІ, на який поширюється дія регулярного звіту, також зареєстрована як складова фіксованої комбінації, заявник або подає окремий регулярний звіт для комбінації АФІ, що зареєстровані тим же заявником з перехресними посиланнями на регулярний(і) звіт(и) для окремого АФІ, або подає дані для комбінації в межах регулярного звіту для одного АФІ.
21. Регулярний звіт містить:
1) резюме даних щодо користі та ризиків лікарського(их) засобу(ів), включаючи результати усіх досліджень з урахуванням потенційного впливу на реєстраційне посвідчення;
2) наукову оцінку співвідношення користь/ризик лікарського(их) засобу(ів), що повинна базуватись на всіх даних, включаючи дані клінічних випробувань за незареєстрованими показаннями та популяцій;
3) всі дані щодо об’єму продажу та інші дані, що відомі заявнику щодо обсягу призначень (виписаних рецептів) лікарського(их) засобу(ів), включаючи оцінку популяції, яка зазнала впливу лікарського(их) засобу (ів).
Регулярний звіт ґрунтується на всіх доступних даних і зосереджується на новій інформації з безпеки медичного застосування лікарського засобу за звітний період. У регулярному звіті також представляється оцінка експозиції, яка піддавалась впливу лікарського засобу, в тому числі всі дані, що стосуються обсягу продажу і обсягу призначень лікарського(их) засобу(ів). Ця оцінка експозиції повинна супроводжуватися якісним і кількісним аналізом фактичного використання, яке повинно вказувати, де це доречно, як фактичне використання відрізняється від показаного застосування на основі всіх наявних у розпорядженні заявника даних, включаючи результати обсерваційних досліджень і досліджень використання лікарських засобів.
22. Регулярний звіт містить результати оцінки ефективності діяльності з мінімізації ризиків, пов’язаних з оцінкою співвідношення користь/ризик.
23. Регулярний звіт містить висновок щодо необхідності здійснення певних заходів, включаючи необхідність внесення змін та/або доповнень до інструкції для медичного застосування/короткої характеристики лікарського(их) засобу(ів), для якого(их) подається регулярний звіт.
24. У разі необхідності внесення змін до інструкції для медичного застосування/короткої характеристики лікарського засобу, заявник не пізніше 60 календарних днів подає заяву на зміни до реєстраційних матеріалів протягом дії реєстраційного посвідчення у визначеному законодавством порядку.
25. Заявник перед оприлюдненням інформації з безпеки лікарських засобів, включаючи листи-звернення до спеціалістів системи охорони здоров’я, громадськості та/або медичних та/або фармацевтичних працівників попередньо узгоджує цю інформацію з МОЗ України та/або Центром, за винятком тих випадків, що потребують термінового оприлюднення такої інформації за одночасного інформування про це МОЗ України та/або Центр.
V.4. Вимоги до системи управління ризиками у системі фармаконагляду заявника
26. Заявник створює та підтримує систему управління ризиками у фармаконагляді.
27. Заявник створює та надає до Центру плани управління ризиками з фармаконагляду:
1) для усіх нових заяв на реєстрацію лікарських засобів, у тому числі генеричних лікарських засобів (за виключенням традиційних та гомеопатичних лікарських засобів, що реєструються за спрощеною процедурою) відповідно до положень Додатку 5 до Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію(перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення, затвердженого наказом МОЗ України від 26 серпня 2005 року № 426 (у редакції наказу МОЗ України від 23 липня 2015 року № 460), зареєстрованого в Міністерстві юстиції України 07 жовтня 2015 року за № 1210/27655;
2) при змінах, що потребують нової реєстрації, зокрема нової лікарської форми, нового способу введення, нового процесу виробництва біотехнологічного лікарського засобу, педіатричних показаннях та інших істотних змінах в показаннях тощо;
3) при появі/виявленні нових даних, що впливають на співвідношення користь/ризик лікарського засобу, поточну специфікацію, план з фармаконагляду, заходи з мінімізації ризиків або їх ефективність або протягом 60 днів після досягнення важливих результатів з фармаконагляду або мінімізації ризиків;
4) при перереєстрації, якщо план управління ризиками необхідний за результатами оцінки співвідношення користь/ризик відповідно до положень Додатку 15 до Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію(перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення, затвердженого наказом МОЗ України від 26 серпня 2005 року №426 (у редакції наказу МОЗ України від 23 липня 2015 року № 460), зареєстрованого в Міністерстві юстиції України 07 жовтня 2015 року за № 1210/27655;
5) на вимогу Центру протягом 60 днів після подання запиту;
6) якщо при підготовці регулярного звіту виникає необхідність внесення змін до плану управління ризиками (внаслідок появи/виявлення нових проблем безпеки та/або нових даних), тоді разом із регулярним звітом надється оновлена версія плану управління ризиками. У цьому випадку не потрібно подавати окрему заяву на зміни для подання плану управління ризиками.
28. Для тих лікарських засобів, що не потребують створення планів управління ризиками, згідно підпункту 1 пункту 28 підрозділу V.4 розділу V цього Порядку заявник підтримує файл специфікації важливих ідентифікованих ризиків, важливих потенційних ризиків і недостатньої інформації з метою підготовки регулярного звіту.
29. Якщо інше не зазначено як умова видачі реєстраційного посвідчення та у разі доцільності план управління ризиками може бути складений для лікарських засобів, що містять однакові АФІ, належать одному й тому ж заявнику і можуть підпадати під дію (бути предметом) одного плану управління ризиками.
30. План управління ризиками подається у форматі окремого документа відповідно до структури, наведеної у додатку 16 до цього Порядку, або іншої структури, що містить модулі, частини, розділи аналогічні викладеним у додатку 16 до цього Порядку, включаючи інформацію щодо заходів з мінімізації ризиків, резюме плану управління ризиками, план з фармаконагляду. Цей документ надається у паперовому вигляді, а додатки — до плану управління ризиками можуть бути подані у електронному вигляді. При створенні плану управління ризиками слід враховувати вимоги наведені у додатку 16 до цього Порядку до заповнення в залежності від типу заяви на реєстрацію лікарського засобу. План управління ризиками подається українською, російською або англійською мовою. У разі подання плану управління ризиками англійською або російською мовою Частини V «Заходи з мінімізації ризиків» та VI «Резюме плану управління ризиками» подаються в перекладі українською мовою. Модуль СVIII «Резюме проблем з безпеки», частина ІІІ «План з фармаконагляду», частина IV «Плани щодо післяреєстраційних досліджень ефективності» подаються в перекладі українською мовою на вимогу Центру. Кожний поданий план управління ризиками повинен мати унікальний номер версії і дату його створення.
План управління ризиками повинен містити:
1) визначення і характеристику профілю безпеки лікарського(их) засобу(ів);
2) зазначення того, яким чином сприяти подальшій характеристиці профілю безпеки лікарського(их) засобу(ів);
3) документування заходів для запобігання чи мінімізації ризиків, пов’язаних з лікарським засобом, включаючи оцінку ефективності цих заходів;
4) документування післяреєстраційних зобов’язань, що були встановлені як умова видачі реєстраційного посвідчення.
31. Якщо у плані управління ризиками зазначено дані про післяреєстраційні дослідження, то необхідно вказати ким вони ініціюються, координуються: заявником на добровільних засадах чи відповідно до зобов’язань, визначених МОЗ України та/або Центром.
32. План управління ризиками повинен містити резюме плану управління ризиками.
У випадку, якщо план управління ризиками стосується більше одного лікарського засобу, резюме плану управління ризиками складається для кожного лікарського засобу.
33. Резюме плану управління ризиками загальнодоступним шляхом оприлюднюється на веб-сайті Центру.
34. У випадку оновлення плану управління ризиками, якщо такі зміни зроблені внаслідок отримання нової інформації, що може призвести до значної зміни співвідношення користь/ризик, або при досягненні значних етапів (результатів) з фармаконагляду чи мінімізації ризику, заявник подає оновлений план управління ризиками до Центру не пізніше 30 календарних днів з моменту складання оновленої версії. При отриманні згоди від Центру заявник може подати тільки оновлені модулі, частини, розділи. Якщо необхідно, заявник подає на вимогу Центру оновлене резюме плану управління ризиками.
V.5 Вимоги до проведення післяреєстраційних досліджень з безпеки та ефективності лікарських засобів заявником
35. Післяреєстраційні неінтервенційні дослідження з безпеки та ефективності лікарських засобів (далі — дослідження з безпеки) здійснюються за ініціативою заявника або згідно із зобов’язаннями, встановленими МОЗ України та/або Центром. Дослідження з безпеки передбачають збір даних з безпеки, що надходять від пацієнтів чи медичних працівників.
36. Цей розділ застосовується без порушення законодавства, що гарантує здоров’я і права учасників досліджень з безпеки.
37. Дослідження з безпеки не проводиться, якщо факт його проведення сприяє промоції.
38. Центр може вимагати від заявника надати протокол і проміжні звіти про дослідження з безпеки.
39. Під час проведення дослідження з безпеки заявник здійснює моніторинг отриманих даних і аналізує їх вплив на співвідношення користь/ризик лікарського засобу.
Заявник подає до Центру всю нову інформацію, що може вплинути на співвідношення користь/ризик лікарського засобу. Ця вимога не скасовує необхідність подання результатів досліджень з безпеки у регулярних звітах.
40. Цей та наступні пункти цього розділу стосуються тільки досліджень з безпеки, що ініціюються, управляються заявником згідно із зобов’язаннями, встановленими МОЗ України та/або Центром. При цьому до проведення дослідження з безпеки заявник подає до Центру проект протоколу.
Протягом 60 днів з моменту отримання проекту протоколу Центр надає заявнику:
1) лист-повідомлення про затвердження або незатвердження проекту протоколу (у цьому разі Центр детально обґрунтовує несхвалення та зазначає, що проведення дослідження з безпеки сприяє промоції або не відповідає меті дослідження з безпеки);
2) лист-повідомлення про те, що дослідження є клінічним дослідженням та підпадає під сферу дії законодавства України щодо проведення клінічних досліджень.
41. Заявник може розпочинати проведення дослідження з безпеки лише після отримання письмового дозволу від Центру.
Після отримання листа-повідомлення про затвердження протоколу заявник може починати проведення цього дослідження з безпеки відповідно до затвердженого протоколу. При цьому, після початку проведення дослідження з безпеки, будь-які значні зміни до протоколу подаються на затвердження до Центру до їх впровадження. Центр проводить оцінку таких змін і поінформувати заявника про згоду або незгоду щодо їх впровадження.
42. Заявник упродовж 12 місяців з моменту закінчення збору даних:
1) подає до Центру заключний звіт про дослідження з безпеки українською мовою, якщо дослідження проводилось тільки на території України, або англійською мовою, якщо дослідження проводилось і в інших країнах. У цьому випадку заявник подає переклад українською мовою назви, резюме (abstract) протоколу дослідження з безпеки,
2) подає до Центру резюме заключного звіту про дослідження з безпеки українською мовою в електронному вигляді;
3) оцінює результати дослідження з безпеки, у разі необхідності, подає заяву на зміни до реєстраційних матеріалів протягом дії реєстраційного посвідчення у визначеному законодавством Порядку.
4) забезпечує зберігання в електронному вигляді та доступність для аудиту та перевірки аналітичного набору даних і статистичних програм, що використовувались для генерації даних, включених у заключний звіт про дослідження з безпеки;
5) забезпечує обробку та збереження усієї інформації з дослідження з безпеки, що дозволяє коректне звітування, інтерпретацію та перевірку цієї інформації і забезпечує захист конфіденційної інформації про учасників дослідження з безпеки;
6) складає протоколи, заключні звіти та резюме досліджень з безпеки відповідно до структури, наведеної у додатку 17 до цього Порядку;
7) публікує результати проведеного дослідження з безпеки у спеціалізованих медичних виданнях протягом року з дати завершення дослідження з безпеки.
43. На підставі результатів дослідження з безпеки Центр може прийняти рішення щодо реєстраційного посвідчення з обґрунтуванням причин. Центр повідомляє заявника про прийняте рішення. У випадку прийняття рішення щодо необхідності внесення змін заявник подає до Центру заяву на зміни до реєстраційних матеріалів протягом дії реєстраційного посвідчення у визначеному законодавством порядку, включаючи інструкцію для медичного застосування.
V.I ЗДІЙСНЕННЯ ФАРМАКОНАГЛЯДУ ЦЕНТРОМ
VІ.1. Загальні положення здійснення фармаконагляду Центром
1. Центр, для отримання інформації про побічні реакції, відсутність ефективності лікарських засобів, НППІ та інші проблеми з безпеки, пов’язані із застосуванням лікарських засобів, використовує будь-які методи збору інформації, включаючи метод спонтанних повідомлень, активний моніторинг, в тому числі активний моніторинг стаціонарів, рецептурний моніторинг, мета-аналіз, та залучає заявників, медичних працівників, фізичних осіб — підприємців, які здійснюють господарську діяльність з медичної практики, пацієнтів та/або їх представників, та організацій, що представляють інтереси пацієнтів.
2. Центр проводить аналіз інформації про побічні реакції, відсутність ефективності лікарських засобів, НППІ та інші проблеми, пов’язані із застосуванням лікарських засобів, отриманої з усіх доступних джерел інформації та усіма методами, що використовуються при здійсненні фармаконагляду.
3. Центр при проведенні аналізу інформації, зазначеної у карті-повідомленні, з метою з’ясування достовірності зазначених у ній даних про побічні реакції та/або відсутність ефективності лікарських засобів, НППІ чи інші проблеми, пов’язані із застосуванням лікарських засобів, має право запитувати та отримувати копії документів, включаючи первинну облікову медичну документацію, у медичних працівників, у закладів охорони здоров’я, у заявників, у пацієнтів та організацій, що надали таку інформацію.
4. Центр проводить інформаційну та просвітницьку роботу серед медичних працівників, пацієнтів та/або їх представників, організацій, що представляють інтереси пацієнтів та заявників з питань фармаконагляду із залученням співробітників із фармаконагляду в регіонах.
5. Центр уповноважує співробітників із фармаконагляду в регіонах організовувати та контролювати здійснення фармаконагляду в Україні за підтримки керівників усіх структурних підрозділів з питань охорони здоров’я, медичних працівників, фізичних осіб — підприємців, які здійснюють господарську діяльність з медичної практики, заявників, керівників та працівників Держлікслужби.
6. Центр проводить регулярний аудит власної системи фармаконагляду і перевірку системи фармаконагляду у заявника та оприлюднює її результати.
7. Центр веде публічний реєстр досліджень з безпеки лікарських засобів.
VІ.2. Порядок проведення Центром перевірки системи фармаконагляду заявників
8. Центр проводить перевірки системи фармаконагляду заявників для з’ясування питань щодо її наявності та функціонування.
9. Перевірки можуть бути:
планові:
якщо перевірка системи фармаконагляду заявника раніше не проводилася протягом 5 років після розміщення на фармацевтичному ринку України першого лікарського засобу заявника;
згідно з графіком проведення планових перевірок;
цільові:
з причин, що не мають відношення до питань, пов’язаних з безпекою лікарських засобів або недотримання встановлених вимог: зміни в організаційно-правовій формі підприємств; значна зміна власної системи фармаконагляду тощо;
з причин, що мають відношення до питань, пов’язаних з безпекою лікарських засобів або недотримання встановлених вимог: несвоєчасне подання повідомлень про випадки побічних реакцій та/або відсутності ефективності лікарського засобу; подання повідомлень не в повному обсязі; подання регулярних звітів, якість яких є невідповідною; невідповідність даних, зазначених у повідомленнях, даним з інших джерел інформації; зміни у співвідношенні користь/ризик; досвід та результати попередніх перевірок; оприлюднення інформації з фармаконагляду, яка є необ’єктивною чи вводить в оману громадськість, тощо.
10. Проведення перевірки передбачає:
1) початок не раніше ніж через 7 тижнів після надсилання попереднього повідомлення-запиту та узгодження із заявником початку її проведення, за винятком випадків, коли існує необхідність проведення перевірки без попередження або про проведення перевірки буде попереджено у термін, коротший ніж 7 тижнів. При цьому заявник надає Центру запитувані документи згідно з попереднім повідомленням-запитом не пізніше ніж за 14 календарних днів до початку проведення перевірки;
2) здійснення перевірки фахівцями з фармаконагляду Центру, які мають досвід роботи у сфері фармаконагляду та їх діяльність не призводить до конфлікту інтересів. У разі необхідності до участі у проведенні перевірки можуть залучатися інші фахівці (відповідно до особливостей застосування та безпеки лікарського засобу) за згодою. При проведенні перевірки обов’язковою умовою є присутність УЛОФ/КОВФ заявника, також можуть бути присутні інші представники заявника у разі необхідності (за згодою);
3) збереження фахівцями, які проводять перевірку, конфіденційної інформації, яку вони одержують під час її проведення, відповідно до вимог законодавства;
4) встановлення, у разі наявності, критичних, суттєвих або несуттєвих невідповідностей:
критичні невідповідності — це невідповідності, які унеможливлюють оцінку безпеки застосування та співвідношення користь/ризик лікарських засобів, що має негативні наслідки для здоров’я населення чи суспільного здоров’я. Критичні невідповідності включають у себе відсутність системи фармаконагляду або однієї чи декількох складових системи фармаконагляду;
суттєві невідповідності — це невідповідності, які можуть негативно вплинути на оцінку безпеки застосування та співвідношення користь/ризик лікарських засобів, що може мати негативні наслідки для здоров’я населення чи суспільного здоров’я. Суттєві невідповідності включають у себе, але не обмежуються: порушення вимог щодо кваліфікації, освіти та обов’язків УЛОФ/КОВФ; недоліки, виявлені у задокументованих процедурних процесах, пов’язаних із здійсненням фармаконагляду; недоліки ведення баз даних системи фармаконагляду; недоліки ведення системи управління ризиками; недоліки ведення документації стосовно фармаконагляду;
несуттєві невідповідності — це невідповідності, що не мають негативного впливу на оцінку безпеки застосування та співвідношення користь/ризик лікарських засобів, та не мають негативних наслідків для здоров’я населення чи суспільного здоров’я;
5) складання звіту за результатами перевірки, що підтверджує факт проведення перевірки та у якому зазначаються виявлені під час перевірки зауваження (за наявності), порушення/недоліки, невідповідності;
6) надсилання звіту заявнику в строк до 30 календарних днів після повного завершення перевірки;
7) надсилання заявником до Центру для погодження інформації щодо термінів коригувальних та запобіжних заходів для усунення заявником критичних та суттєвих невідповідностей та виявлених порушень (недоліків) під час проведеної перевірки; несуттєві невідповідності усуваються в робочому порядку.
11. Центр може провести цільову перевірку для підтвердження усунення невідповідностей.
12. У випадку, якщо протягом визначеного для усунення недоліків терміну, заявник не усуває критичні невідповідності, Центр подає до МОЗ України пропозиції про тимчасову заборону застосування лікарського(их) засобу(ів), шляхом припинення терміну дії реєстраційного посвідчення заявника у зв’язку з відсутністю у заявника системи фармаконагляду або її складових.
VІ.3. Вимоги до проведення аналізу інформації щодо безпеки та ефективності лікарських засобів
13. Центр систематизує та аналізує:
1) отриману інформацію про побічні реакції та/або відсутність ефективності лікарських засобів, НППІ, інших проблем з безпеки, пов’язаних із застосуванням лікарських засобів;
2) отриману інформацію про зареєстровані випадки захворювання на інфекційні хвороби, що керуються засобами специфічної імунопрофілактики у щеплених;
3) регулярні звіти;
4) плани управління ризиками.
Обробка персональних даних під час здійснення фармаконагляду здійснюється відповідно до вимог Закону України «Про захист персональних даних».
14. Центр проводить:
1) оцінку якості даних з безпеки та ефективності лікарських засобів;
2) зберігання даних з безпеки та ефективності лікарських засобів;
3) аналіз даних з безпеки та ефективності лікарських засобів;
4) оцінку причинно-наслідкового зв’язку, передбаченості/непередбаченості, ступеню серйозності;
5) аналіз інформації про зареєстровані випадки захворювання на інфекційні хвороби, що керуються засобами специфічної імунопрофілактики у щеплених;
6) виявлення/управління сигналом;
7) оцінку співвідношення користь/ризик;
8) оцінку ефективності заходів з мінімізації/усунення ризиків, вжитих заявником;
9) навчання з питань здійснення фармаконагляду;
10) післяреєстраційні неінтервенційні дослідження з безпеки та ефективності лікарського засобу (за необхідності).
15. Центр при проведенні аналізу даних з безпеки та ефективності лікарських засобів, оцінки причинно-наслідкового зв’язку між клінічними проявами будь-якої побічної реакції та/або відсутністю ефективності та застосуванням лікарського засобу, передбаченості/непередбаченості, ступеню серйозності, інформації про зареєстровані випадки захворювання на інфекційні хвороби, що керуються засобами специфічної імунопрофілактики у щеплених, а також оцінки співвідношення користь/ризик може залучати незалежних експертів та інших спеціалістів у разі необхідності (за згодою).
16. За результатами аналізу даних з безпеки та ефективності лікарських засобів, Центр подає пропозиції до МОЗ України:
1) щодо прийняття рішень стосовно повної або тимчасової заборони медичного застосування:
у разі виявлення раніше невідомих небезпечних властивостей лікарського засобу, що призвели або можуть призвести до тяжких наслідків для здоров’я населення чи суспільного здоров’я;
у випадках, коли ризик від застосування лікарського засобу перевищує користь;
у разі невиконання заявником умов видачі реєстраційного посвідчення;
у разі невиконання взятих заявником на себе гарантійних зобов’язань.
2) обмеження застосування лікарського засобу.
17. За результатами аналізу даних з безпеки та ефективності лікарських засобів, Центр рекомендує заявнику:
1) внести уточнення, доповнення або зміни до інструкції для медичного застосування з наданням вмотивованих висновків та рекомендацій;
2) проведення досліджень з безпеки та ефективності;
3) розроблення плану управління ризиками.
18. Висновки Центру щодо подальшого медичного застосування лікарського(их) засобу(ів) можуть бути оскаржені згідно з вимогами чинного законодавства України.
| Начальник Управління фармацевтичної діяльності та якості фармацевтичної продукції |
Т. Лясковський |
Повідомлення заповнюється та надається за місцезнаходженням: Державне підприємство «Державний експертний центр Міністерства охорони здоров’я України», Департамент післяреєстраційного нагляду, вул. Ушинського, 40, м. Київ, 03151; тел/факс: +38 044 4984358; e-mail: [email protected]; електронна форма повідомлення розміщена на http://www.dec.gov.ua
ВИМОГИ ДО заповнення
карти-повідомлення для надання пацієнтом та/або його представником, організацією, що представляє інтереси пацієнтів, інформації про побічну реакцію та/або відсутність ефективності лікарського засобу та несприятливу подію після імунізації
1. Інформація про пацієнта: зазначається прізвище, ім’я та по батькові пацієнта, у якого спостерігалась побічна реакція/відсутність ефективності лікарського засобу, несприятлива подія після імунізації його адреса та телефон.
2. Інформація про підозрюваний лікарський засіб: зазначається торгівельне найменування, форма випуску, виробник
3. Інформація про призначення підозрюваного лікарського засобу: відмічається відповідна позиція
4. Опис проявів побічної реакції, несприятливої події після імунізації або зазначення про відсутність ефективності: детально описується побічна реакція, несприятлива подія після імунізації уключаючи безпосередній прояв побічної реакції, несприятливої події після імунізації а також короткий опис усієї клінічної інформації, що може стосуватися виявленої побічної реакції, несприятливої події після імунізації або надається інформація щодо відсутності ефективності.
5. Інформація про повідомника: зазначається прізвище, ім’я та по батькові, адреса та телефон того, хто подає це повідомлення.
6. Інформація про лікаря, заклад охорони здоров’я та місце проживання пацієнта, у якого спостерігалась побічна реакція, несприятлива подія після імунізації або відсутність ефективності: зазначається прізвище, ім’я та по батькові лікуючого лікаря, адреса, телефон та назва закладу охорони здоров’я, де працює лікуючий лікар; місце проживання пацієнта, у якого спостерігалась побічна реакція, несприятлива подія після імунізації або відсутність ефективності лікарського засобу.
ВИМОГИ ДО заповнення
Звіту про випадки побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подій після імунізації у закладах охорони здоров’я
_____________________________________________________за 20__ рік
I. Таблиця 1000 заповнюється відповідальною особою структурного підрозділу з питань охорони здоров’я обласних, Київської міської державних адміністрацій. До звіту вносяться випадки виникнення побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подійпісля імунізації у закладах охорони здоров’я усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування у такому порядку: до кожної графи таблиці вносяться дані стосовно усіх виявлених випадків побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подійпісля імунізації у закладах охорони здоров’я області (міста) в послідовному порядку по закладах (обласного значення, міського, районного значення). Кожен наступний випадок вноситься у наступний рядок таблиці.
1. Порядковий номер.
2. П. І. Б.: прізвище, ім’я, по батькові пацієнта зазначаються ініціалами.
3. Стать: жінка або чоловік.
Якщо повідомлення стосується лікарського засобу, який приймала вагітна жінка, а побічна реакція та/або відсутність ефективності, виникла у плода, то всі дані (за винятком побічної реакції) надаються про матір із зазначенням триместру вагітності.
4. Вік: для пацієнтів віком від 3 років і старше зазначаються роки; для пацієнтів молодше 3 років — місяці; для пацієнтів віком до місяця — дні.
5. Номер історії хвороби або амбулаторної карти.
6. Підозрюваний лікарський засіб: торгівельне найменування, форма випуску, виробник (повне найменування), країна.
7. Опис проявів побічної реакції підозрюваного лікарського засобу, зазначення про відсутність ефективності, опис несприятливої події після імунізації: зазначаються негативні клінічні прояви, пов’язані з переважаючим або комбінованим впливом на функції органів травлення, шкіри, центральної нервової системи, серцево-судинної системи, системи дихання, сечовивідної, імунної та інших систем, що виникли унаслідок застосування підозрюваного лікарського засобу або комбінацій лікарських засобів, вакцин, туберкуліну і призвели до відповідного порушення життєдіяльності організму. У випадку відсутності ефективності при зазначається: відсутність ефективності. У випадку несприятливої події після імунізації надається опис несприятливої події після імунізації.
8. Основні клінічний та супутній діагнози (із зазначенням шифру за МКХ-10): зазначаються основний клінічний та супутній діагнози пацієнта, у якого спостерігалася побічна реакція, відсутність ефективності, НППІ із зазначенням шифру МКХ-10.
ІІ. Таблиця 1001 заповнюється відповідальною особою структурного підрозділу з питань охорони здоров’я обласних, Київської міської державних адміністрацій. До звіту вносяться випадки побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подійпісля імунізації усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування у такому порядку: до граф таблиці вносяться абсолютні показники стосовно даного регіону (міста).
1. Кількість закладів охорони здоров’я: зазначається кількість усіх діючих закладів охорони здоров’я усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування, що розташовані на території адміністративно-територіальної одиниці (області, міста).
2. Кількість закладів охорони здоров’я, що подавали карти-повідомлення про побічну реакцію та/або відсутність ефективності лікарських засобів та несприятливу подіюпісля імунізації (далі — карти-повідомлення): зазначається кількість усіх діючих закладів охорони здоров’я усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування, що розташовані на території адміністративно-територіальної одиниці (області, міста), які протягом звітного року надавали карти повідомлення про випадки побічних реакцій та/або відсутності ефективності лікарських засобів та несприятливі події після імунізації.
3. Кількість лікарів (за винятком тих, які не займаються лікарсько-профілактичною діяльністю (далі — ЛП): зазначається загальна кількість лікарів, за винятком тих, які не займаються ЛП діяльністю (патологоанатоми, лікарі-лаборанти, лікарі-статистики тощо).
4. Кількість населення (середньорічна): зазначається загальна середньорічна кількість населення в адміністративно-територіальній одиниці (області, місті) у звітному році (за даними статистичних управлінь).
5. Із них діти (0 — 18 років): зазначається середньорічна кількість дитячого населення (0 — 18 років включно) із загальної кількості населення в адміністративно-територіальній одиниці, вказаної у графі 4.
6. Кількість карт-повідомлень: зазначається загальна кількість карт-повідомлень, що були надіслані з даної адміністративно-територіальної одиниці (області, міста). Цей показник повинен співпадати з загальною кількістю карт-повідомлень зазначеною в таблиці 1000.
ВИМОГИ ДО заповнення
Звіту про випадки побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подій після імунізації у закладах охорони здоров’я
_________________________________________за 20__ рік
I. Таблиця 1000 заповнюється відповідальною особою з питань фармаконагляду закладу охорони здоров’я усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування (далі — ЗОЗ). До звітупро випадки побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подійпісля імунізаціїу закладах охорони здоров’я у такому порядку: до кожної графи таблиці вносяться дані стосовно усіх виявлених випадків побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подійпісля імунізаціїу закладі охорони здоров’я області (міста) в послідовному порядку. Кожен наступний випадок вноситься у наступний рядок таблиці.
1. Порядковий номер.
2. П. І. Б.: прізвище, ім’я, по батькові пацієнта зазначаються ініціалами.
3. Стать: жінка або чоловік.
Якщо повідомлення стосується лікарського засобу, який приймала вагітна жінка, а побічна реакція та/або відсутність ефективності виникла у плода, то всі дані (за винятком побічної реакції) надаються про матір із зазначенням триместру вагітності.
4. Вік: для пацієнтів віком від 3 років і старше зазначаються роки; для пацієнтів молодше 3 років — місяці; для пацієнтів віком до місяця — дні.
5. Номер історії хвороби або амбулаторної карти.
6. Підозрюваний лікарський засіб: торгівельне найменування, його форма випуску, виробник (повне найменування), країна.
7. Опис проявів побічної реакції підозрюваного лікарського засобу, зазначення про відсутність ефективності, опис несприятливої події після імунізації: зазначаються негативні клінічні прояви, пов’язані з переважаючим або комбінованим впливом на функції органів травлення, шкіри, центральної нервової системи, серцево-судинної системи, системи дихання, сечовивідної, імунної та інших систем, що виникли унаслідок застосування підозрюваного лікарського засобу або комбінацій лікарських засобів і призвели до відповідного порушення життєдіяльності організму. У випадку відсутності ефективності при зазначається: відсутність ефективності. У випадку несприятливої події після імунізації надається опис несприятливої події після імунізації.
8. Основні клінічний та супутній діагнози (із зазначенням шифру за МКХ-10): зазначаються основний клінічний та супутній діагнози пацієнта, у якого спостерігалася побічна реакція, відсутність ефективності, НППІ із зазначенням шифру МКХ-10.
II. Таблиця 1001 відповідальною особою закладу охорони здоров’я усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування. До звіту вносяться випадки побічних реакцій та/або відсутності ефективності лікарських засобів, несприятливих подійпісля імунізації усіх рівнів надання медичної допомоги незалежно від форм власності та відомчого підпорядкування у такому порядку: залишається пустою.
3-й ЕТАП: Алгоритм визначення причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну
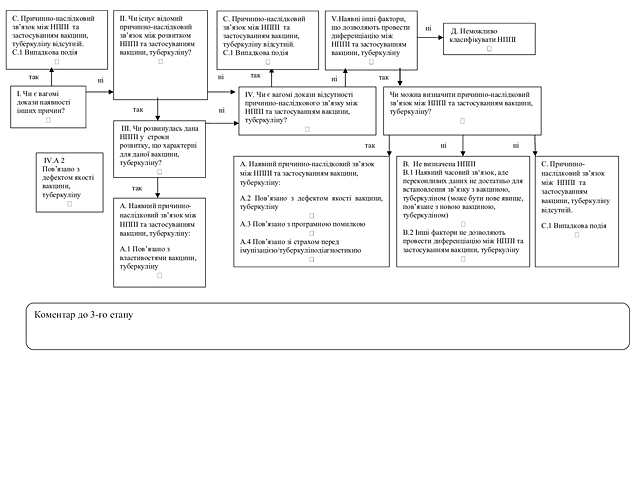
4-ЕТАП Категорії причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну згідно алгоритму визначення (3-го етапу)
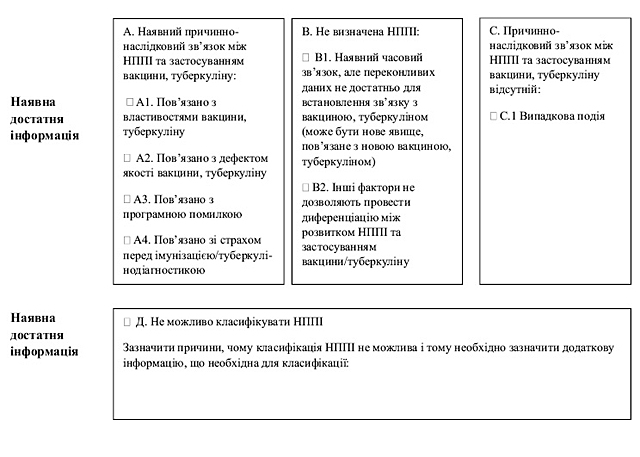
Коротке обґрунтування визначення категорії причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну:
На підставі доступних даних регіональна/центральна група оперативного реагування
(необхідне підкреслити)
прийшла до висновку, що категорію причинно-наслідкового зв’язку визначено як ____________________________________________________________________________________________,
оскільки ____________________________________________________________________________________
Голова групи: ________________________ Підпис _______________
Дата: ________________
ВИМОГИ ДО ЗАПОВНЕННЯ
Протоколу розслідування серйозних та/або групових несприятливих подій після імунізації (НППІ) після застосування вакцини, туберкуліну та визначення причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну
1-й ЕТАП: Збір інформації, що стосується розвитку серйозних та/або групових НППІ після застосування вакцини, туберкуліну
Розділ І Загальна інформація
1. Назва організації (закладу охорони здоров’я), де проводилося щеплення/туберкулінодіагностика: зазначити повністю назвуорганізації, де була проведена імунізація/туберкулінодіагностика.
2. Категорія імунізації або туберкулінодіагностики: У графі зазначається категорія імунізації або туберкулінодіагностики згідно наведеного переліку.
3. Адреса місця проведення імунізації/туберкулінодіагностики: зазначити повністю адресу місця проведення імунізації /туберкулінодіагностики.
4. ПІБ особи, яка надає протокол: зазначити прізвище, ім’я, по батькові особи, яка надає цей протокол, її посаду, місце роботи, робочий телефон (з кодом) та/або, мобільний телефон, e-mail, дату розслідування та заповнення протоколу із зазначенням, який він згідно наведеного переліку.
Інформація про особу, якій застосовували вакцину/туберкулін :
5. ПІБ особи: зазначити прізвище, ім’я, по батькові особи, якій застосувалась вакцина або туберкулін, із зазначенням статі, дати народження та повної адреси згідно наведеного переліку.
Заповнюється окрема форма протоколу для кожного випадку групової НППІ.
6. Торгівельна назва вакцин(и)/розчинника або туберкуліну, що отримані особою раніше: зазначити дату, час імунізації або туберкулінодіагностики, дозу, номер серії та дату закінчення терміну придатності вакцини або туберкуліну, місце введення вакцини/туберкуліну згідно наведеного переліку, дату першого/основного симптому із зазначенням дня/місяця/року, час першого симптому (годин/хвилин), дату госпіталізації (день/місяць/ рік), дату першого звернення до закладу охорони здоров’я (день/ місяць/рік), стан на дату розслідування згідно наведеного переліку (у разі смерті, дата та час смерті). У разі, якщо проведений розтин необхідно зазначити дату (день/місяць/рік) та додати коментарі щодо результатів розтину. У разі, якщо розтин непроведений необхідно зазначити заплановану дату (день/місяць/рік) та час (години/хвилини).
Розділ ІІ Наявна інформація про особу, якій застосовували вакцину/туберкулін, до проведення імунізації/туберкулінодіагностики
1. Наявність подібного НППІ у минулому незалежно від щеплення/туберкулінодіагностики: зазначається згідно наведеного переліку результату. Якщо результат «так» необхідно надати коментар.
2. Наявність подібного НППІ після попередньої(х) щеплення(ь)/туберкулінодіагностики: зазначається згідно наведеного переліку результату. Якщо результат «так» необхідно надати коментар.
3. Обтяжений алергологічний анамнез на вакцину/туберкулін, лікарський засіб або харчовий продукт: зазначається згідно наведеного переліку результату. Якщо результат «так» необхідно надати коментар.
4. Обтяжений родинний або сімейний алергологічний анамнез (наявність хвороб, що мають відношення до НППІ): зазначається згідно наведеного переліку результату. Якщо результат «так» необхідно надати коментар.
5. Госпіталізація в анамнезі за останні 30 днів із зазначенням причини: зазначається згідно наведеного переліку результату. Якщо результат «так» необхідно надати коментар.
6. Чи застосовує особа, якій застосовували вакцину/туберкулін, інші лікарські засоби у даний час? Зазначається згідно наведеного переліку результату. Якщо «так», вказується торгівельна назва лікарського засобу, показання, доза та дата початку та дата закінчення прийому.
7. Чи передувала будь-яка хвороба розвитку НППІ (30 днів)/ наявність генетичних порушень: Зазначається згідно наведеного переліку результату. Якщо результат «так» необхідно надати коментар.
8. Для дорослих жінок, яким застосовували вакцину необхідно зазначити: інформацію щодо вагітності згідно наведеного переліку. Якщо «так» вказується тиждень вагітності.
Також, зазначається інформація щодо годування груддю згідно наведеного переліку.
9. Для дітей до 1 року, яким застосовували вакцину, необхідно зазначити: від яких пологів дитина народилися, які були пологи згідно наведеного переліку. Також зазначити вагу при народженні у грамах, стан новонародженого за шкалою Апгар.
Розділ ІІІ Детальна інформація щодо серйозного та/або групового випадку НППІ після застосування вакцини, туберкуліну
1. Джерело інформації: зазначається згідно наведеного переліку;
2. ПІБ, яка першою оглядала/лікувала особу, якій проведено імунізацію/ туберкулінодіагностику: зазначити прізвище, ім’я, по батькові особи, яка першою оглядала/лікувала особу, якій проведено імунізацію/ туберкулінодіагностику;
3. ПІБ інших осіб, які консультували та надавали медичну допомогу особі, якій проведено імунізацію/ туберкулінодіагнстику: зазначити прізвище, ім’я, по батькові осіб, які консультували та надавали медичну допомогу особі, якій проведено імунізацію/ туберкулінодіагностику. Якщо є наявна інформація щодо серйозного, групового випадку НППІ з іншого джерела її необхідно зазначити.
4. Ознаки та симптоми НППІ у хронологічному порядку після імунізації/туберкулінодіагностики: зазначити детально у хронологічному порядку всі ознаки та симптоми даної НППІ.
5. ПІБ та контактна інформація про особу, яка надавала детальну інформацію: зазначити прізвище, ім’я, по батькові особи, яка надавала детальну інформацію, її посаду, місце роботи, робочий телефон (з кодом) та/або, мобільний телефон, e-mail та дату (день/місяць/рік), час (години/хвилини) заповнення.
6. Документи, що містять документальне підтвердження стану пацієнта: зазначається згідно наведеного переліку.
7. Попередній/заключний діагноз: зазначити читко попередній або заключний діагноз на підставі документів, що містять документальне підтвердження стану пацієнта згідно наведеного переліку.
Розділ ІV Додаткова інформація про вакцину/туберкулін у випадку серйозного та/або групової НППІ
1. Кількість осіб, яким було проведено щеплення та/або туберкулінодіагностика по організації (закладу охорони здоров’я) області/місту/району: зазначається торгівельна назва вакцини/туберкуліну та кількість введених доз/туберкулінових проб.
2. Номер дози вакцини/туберкуліну у особи, у якої зареєстровано серйозний та/або груповий випадок НППІ: зазначається інформація згідно наведеного переліку. У разі використання вакцини з багатодозових флаконів зазначити номер за порядком щодо даного пацієнта. У разі застосування туберкуліну зазначити номер за порядком щодо даного пацієнта.
3. Чи була допущена помилка при застосуванні вакцини, туберкуліну в результаті недотримання рекомендацій щодо їх використання (наприклад, імунізація та/або туберкулінодіагностика після закінчення терміну придатності вакцини, туберкуліну, не дотримання протипоказань тощо)? Зазначається згідно наведеного переліку. У разі якщо вибрано «так» необхідно надати роз’яснення.
4. Чи була вакцина, туберкулін введені з порушенням умов стерильності? Зазначається згідно наведеного переліку. У разі якщо вибрано «так» необхідно надати роз’яснення.
5. При застосуванні вакцини, туберкуліну їх фізичний стан (колір, помутніння, присутність сторонніх речовин та ін.) не відповідали нормі? Зазначається згідно наведеного переліку. У разі якщо вибрано «так» необхідно надати роз’яснення.
6. Чи була допущена особою, яка проводила імунізацію/ туберкулінодіагностику, помилка при:
а) розведенні/ приготуванні вакцини (застосовувався інший лікарський засіб або розчинник, порушена процедура змішування, заповнення шприца тощо);
б) застосуванні туберкуліну (застосовувався інший лікарський засіб, заповнення шприца тощо).
Зазначається згідно наведеного переліку. У разі якщо вибрано «так» необхідно надати роз’яснення.
7. Чи була допущена помилка при введенні/ застосуванні вакцини, туберкуліну (невірно вибрана доза або шлях введення; невірне місце введення, невірний розмір голки тощо)? Зазначається згідно наведеного переліку. У разі якщо вибрано «так» необхідно надати роз’яснення.
8. Кількість осіб, яким було проведено імунізацію із даного флакону/ампули з вакциною або кількість осіб, яким була проведена туберкулінодіагностика із даного флакону/ампули у конкретній організації (заклади охорони здоров’я) де проводилося імунізація/туберкулінодіагностика. Зазначається кількість щеплених із даного флакону/ампули з вакциною або кількість проведеної туберкулінодіагностики із даного флакону/ампули у конкретній організації (заклади охорони здоров’я) де проводилося імунізація/туберкулінодіагностика.
9. Кількість осіб, яким було проведено щеплення даною вакциною або кількість осіб, яким була проведена туберкулінодіагностика даним туберкуліном у конкретній організації (заклади охорони здоров’я) протягом цього дня: зазначається яким було проведено щеплення даною вакциною або кількість осіб, яким була проведена туберкулінодіагностика даним туберкуліном у конкретній організації (заклади охорони здоров’я) протягом цього дня.
10. Кількість осіб, яким було проведено щеплення даною серією вакцини або кількість осіб, яким була проведена туберкулінодіагностика даною серією туберкуліну у конкретній організації (закладі охорони здоров’я): Зазначається кількість осіб, яким було проведено щеплення даною серією вакцини або кількість осіб, яким була проведена туберкулінодіагностика даною серією туберкуліну у конкретній організації (закладі охорони здоров’я протягом цього дня.
11. Цей випадок НППІ відноситься до групової НППІ. Зазначається згідно наведеного переліку. Якщо «так», зазначити скільки інших групових НППІ виявлено.
12. Чи в усіх групових НППІ особам було проведено імунізацію/туберкулінодіагностику із того самого флакону/ампули вакцини/туберкуліну? Зазначається згідно наведеного переліку. Якщо «ні», зазначте серії та кількістьфлаконів/ампул, що застосовувалися у груповому випадку НППІ при проведенні щеплення/туберкулінодіагностики.
Розділ V Інформація про організації (заклади охорони здоров’я), де проводилась імунізація/туберкулінодіагностика
1. Шприці та голки, що застосуються: чи застосовувалися для імунізації/туберкулінодіагностики самоблокуючі шприці (для уникнення повторного використання)? Якщо «ні», зазначте тип шприців, що використовуються згідно наведеного переліку.
Зазначте основні результати/додаткові спостереження та коментарі:
2. Відновлення (заповнюється, якщо застосовувався розчинник):
процедура відновлення
– один й той же шприц застосовувався для флаконів з тією ж самою вакциною? Зазначається статус згідно наведеного переліку;
– один й той же шприц застосовувався для розведення різних вакцин? Зазначається статус згідно наведеного переліку;
– окремий шприц застосовувався для розведення окремого флакону з вакциною? Зазначається статус згідно наведеного переліку;
– окремий шприц застосовувався для розведення для кожної вакцинації? Зазначається статус згідно наведеного переліку.
3. Чи застосовувалися вакцина та розчинник, що рекомендований виробником? Зазначається статус згідно наведеного переліку.
У разі наявності зазначаються особливі результати/додаткові спостереження та коментарі до цього розділу.
Розділ VI «Холодовий ланцюг» при зберіганні та транспортуванні вакцини/туберкуліну
1. Зберігання вакцини/туберкуліну у кабінеті щеплень:
– чи контролюється температурний режим у холодильнику, де зберігається вакцина/туберкулін? Зазначається згідно наведеного переліку. Якщо «так», зазначити згідно наведеного переліку чи були будь-які відхилення від діапазону 2-8°С після того, як вакцина/туберкулін була поміщена у холодильник та надати детальну інформацію про температурний контроль (моніторинг) окремо;
– чи застосовується окремий холодильник для зберігання вакцин/туберкуліну, розчинників та шприців? Зазначається згідно наведеного переліку;
– чи були наявні у холодильнику (чи морозильнику холодильника) інші предмети окрім вакцин/туберкуліну, розчинників, шприців, голок? Зазначається згідно наведеного переліку;
– чи були наявні у холодильнику розведені вакцини, що частково використані? Зазначається згідно наведеного переліку;
– чи були наявні у холодильнику вакцини/туберкулін з терміном придатності, що закінчився, без маркування, заморожені, зі зміненим кольором квадрату термоіндикатору тощо. Зазначається згідно наведеного переліку;
– чи присутні у холодильнику непридатні розчинники з терміном придатності, що закінчився, не рекомендовані виробником/заявником, флакони/ампули з мікротрищінами, забруднені флакони/ампули. Зазначається згідно наведеного переліку.
У разі наявності особливих результатів/додаткових спостережень надаються коментарі.
2. Транспортування вакцини/туберкуліну:
– зазначити в чому транспортувалась вакцина/туберкулін до кабінету щеплень? Зазначається інформація в чому транспортувалась вакцина/туберкулін до кабінету щеплень;
– вакцина/туберкулін була/був доставлена(ий) в той же день коли проводилося імунізація/туберкулінодіагностика? Зазначається згідно наведеного переліку.
– чи застосовувався при транспортуванні конденсований хладоелемент? Зазначається згідно наведеного переліку.
Розділ VII Розслідування серед населення
1.Чи були повідомлення про подібні клінічні прояви протягом часу, коли виникла дана НППІ, у цій місцевості? Зазначається згідно наведеного переліку.
Якщо «так», зазначте та опишіть (врахувати фоновий рівень захворюваності по області/місту, де зареєстрована дана НППІ
2. Зазначте скільки випадків НППІ з числа: щеплених, нещеплених або невідомо: Зазначається кількість випадків НППІ у щеплених, нещеплених або у осіб з невідомим щеплювальним анамнезом.
У разі наявності коментарів надаються коментарі.
Розділ VIII Інші результати/спостереження/коментарі
У разі наявності результатів/спостережень/коментарів надаються.
2-й ЕТАП: Збір інформації для встановлення причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну
2-ЕТАП
І. Чи є вагомі докази наявності інших причин?
1. Чи підтверджують результати клінічного або лабораторного обстеження особи, якій застосувалась(ися) вакцина(и)/туберкулін наявність іншої причини у виникненні серйозної та/або групової НППІ? у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язково надати обґрунтування. При відповіді на питання необхідно врахувати дані клінічного огляду, результати лабораторних досліджень, що допоможе провести диференціацію інших станів, захворювань або вроджених вад, що могли викликати дану НППІ.
ІІ. Чи існує відомий причинно-наслідковий зв’язок між розвитком НППІ та застосуванням вакцини/туберкуліну?
ІІ.1 Вакцина(и), туберкулін (підкреслити потрібне)
1. Чи існують у літературі (науковій) дані про те, що дана вакцина/туберкулін може викликати зареєстровану НППІ навіть при правильному застосуванні згідно інструкції для медичного застосування?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно врахувати дані літератури (наукової), щодо даної НППІ, причиною якої є властивості конкретної вакцини, що застосовувалась при умові дотримання вимог щодо її введення.
2. Чи підтверджують результати спеціального(их) лабораторного(их) дослідження(ь) причинно-наслідковий зв’язок між даною НППІ та вакциною(ами), що застосовувалась(ись)?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно врахувати дані спеціальних лабораторних досліджень для конкретної вакцини. Наприклад, дослідження спинномозкової рідини для ідентифікації вакцинного вірусу, та встановлення що дана НППІ у вигляді асептичного менінгіту виникла внаслідок застосування вакцини, що містила штам Urabe.
ІІ.2 Програмна помилка при щепленні/туберкулінодіагностики
3. Чи була допущена особою, яка проводила імунізацію/туберкулінодіагностику, помилка при застосуванні вакцини/ туберкуліну в результаті недотримання рекомендацій щодо їх використання (наприклад, імунізація та/або туберкулінодіагностика після закінчення терміну придатності вакцини/туберкуліну, не дотриманні протипоказань тощо)?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно з’ясувати застосовувалась вакцина(и), туберкулін у відповідності до інструкції для медичного застосування щодо показань, протипоказань, доз, схеми, умов зберігання, тощо. Вакцини різних виробників можуть мати різні специфікації, невідповідність яких може призвести до розвитку даної НППІ.
4. Чи була вакцина, туберкулін введені з порушенням умов стерильності?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосоване. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на наявність або відсутність симптомів, що характерні для синдрому токсичного шоку (блювота, діарея, ціаноз, підвищення температури) та строки їх появи (протягом декількох годин), а також місцева чутливість та інфільтрація тканин.
5. При застосуванні вакцини, туберкуліну їх фізичний стан (колір, помутніння, присутність чужорідних речовин тощо.) не відповідали нормі?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на наявність або відсутність незвичайного кольору, помутніння або присутність сторонніх часток, що підтверджує порушення вмісту вакцини та може бути причиною даної НППІ.
6. Чи була допущена особою, яка проводила імунізацію/туберкулінодіагностику помилка при:
– розведенні/ приготуванні вакцини (застосовувався інший лікарський засіб або розчинник, порушена процедура змішування, заповнення шприца тощо);
– застосуванні туберкуліну (застосовувався інший лікарський засіб, заповнення шприца тощо) : у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на застосування відповідного до вакцини розчинника, правильності заповнення шприца.
7. Чи була допущена помилка відповідальною особою при транспортуванні/зберіганні вакцини, туберкуліну (недотримання температурних умов при транспортуванні, зберіганні, тощо): у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на дотримання відповідних температурних умов при транспортуванні/зберіганні вакцини, не виконання яких може призводити до загальних та місцевих реакцій, неефективності вакцинації.
8. Чи була допущена помилка особою, яка проводила імунізацію/туберкулінодіагностику, при введенні/застосуванні вакцини, туберкуліну (невірно вибрана доза або шлях введення; невірне місце введення, невірний розмір голки тощо)?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу чи відповідають вікова доза, місце та шлях введення вимогам інструкції для медичного застосування конкретної вакцини.
ІІ.3 Страх перед імунізацією/туберкулінодіагностикою
9. Чи міг страх при імунізації/туберкулінодіагностиці викликати дану НППІ (вагусна судинна реакція, гіпервентиляційний синдром або розлад, пов’язаний зі стресом)?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно врахувати вік дитини (характерна подія для підлітків), де була проведена імунізація (дошкільний дитячий заклад або школа).
ІІІ Строки розвитку даної НППІ з урахуванням типу вакцини, що застосовувалась, туберкуліну
1. Чи розвинулась дана НППІ у строки розвитку, що характерні для вакцини/ туберкуліну?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на строки появи даної НППІ після застосування конкретної вакцини/туберкуліну.
IV. Чи є вагомі докази відсутності причинно-наслідкового зв’язку?
Чи є вагомі докази відсутності причинно-наслідкового зв’язку?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на наявність факторів, що підтверджують відсутність причинно-наслідкового Зв’язку між даною НППІ та вакциною, що застосовувалась.
V. Інші фактори, що дозволяють провести диференціацію між НППІ та застосуванням вакцини/туберкуліну
1. Чи могла дана НППІ статися незалежно від щеплення/ туберкулінодіагностики (врахувати фоновий рівень захворюваності по області/місту, де зареєстрована дана НППІ)?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно враховувати фонові рівні соматичних та інфекційних захворювань для різних вікових груп населення регіону, що дозволить порівняти частоту розвитку даної НППІ серед щеплених та нещеплених осіб одної вікової групи, що мешкають в одному регіоні.
2. Чи могла дана НППІ бути проявом іншої патології?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно враховувати інші стани здоров’я, які могли спровокувати виникнення даної НППІ.
3. Чи мала місце подібна НППІ після введення будь-якої попередньої дози аналогічної вакцини/туберкуліну?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно враховувати попередній щеплювальний анамнез, який повинен бути детально проаналізований.
4. Чи мав місце вплив потенційних факторів ризику або токсинів до виникнення даної НППІ?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на те, чи не передувало щепленню хірургічне втручання, проведення хіміотерапії, застосування будь-яких лікарських засобів.
5. Чи передувало даній НППІ захворювання?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно звернути увагу на те, чи не передувало щепленню будь-яке захворювання, розвиток НППІ у післявакцинальному періоді є причиною цього захворювання.
6. Чи спостерігався подібний стан в минулому незалежно від щеплення/ туберкулінодіагностики?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно з’ясувати, чи виникала подібна НППІ у щепленої особи та його родині у минулому незалежно від імунізації.
7. Чи приймала особа, якій проведено щеплення/ туберкулінодіагностику будь-який лікарський засіб перед щепленням/туберкулінодіагностикою?: у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. При відповіді на питання необхідно з’ясувати, чи не приймала щеплена особа у день вакцинації будь-який лікарський засіб тому, що дана НППІ може бути як наслідком застосування лікарського засобу так і вакцини.
8. Чи існує біологічна ймовірність того, що вакцина могла викликати дану НППІ? у відповідному віконечку символом () відмічається відповідь на зазначене питання — так, ні, невідомо (недостатньо інформації), не застосовано. У разі позитивної відповіді на питання у розділі «Примітка» обов’язкове обґрунтування. Біологічна ймовірність, як додатковий уточнюючий фактор може використовуватися у випадку, коли результаті лабораторних досліджень або симптоми даної НППІ подібні і відповідають природньому перебігу інфекції.
3-й ЕТАП: Алгоритм визначення причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну
На підставі отриманих відповідей 1-го та 2-го етапівПротоколу розслідування серйозних та/або групових несприятливих подій після імунізації (НППІ) після застосування вакцини, туберкуліну та визначення причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну та керуючись стрілковими показчиками для встановлення причинно-наслідкового зв’язку символом () ставляться позначки у відповідних віконичках. Алгоритм надає можливість у логічній послідовності зробити відповідні висновки щодо наявності або відсутності причинно-наслідкового зв’язку між НППІ та застосуванням вакцини/туберкуліну або дане НППІ неможливо класифікувати.
В окремому віконечку «коментар до 3 етапу» надаються обґрунтування щодо причинно-наслідкового зв’язкуміж серйозними та/або груповими НППІ та застосуванням вакцини/ туберкуліну.
4-ЕТАП Категорії причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну згідно алгоритму визначення (3-го етапу)
Категорія причинно-наслідкового зв’язку між серйозними та/або груповими НППІ та застосуванням вакцини, туберкуліну: визначається згідно алгоритму визначення (3-го етапу) та наведеного переліку з урахуванням обґрунтування щодо причинно-наслідкового зв’язкуміж серйозними та/або груповими НППІ та застосуванням вакцини/туберкуліну.
Вимоги до заповнення
Карти-повідомлення про побічну реакцію, відсутність ефективності лікарського засобу, вакцини та туберкуліну (ЛЗ) та несприятливу подію після імунізації (НППІ)
I розділ. Інформація про пацієнта
У графах зазначаються:
Ініціали пацієнта. Прізвище, ім’я та по батькові пацієнта зазначаються першими літерами: якщо повідомлення стосується лікарського засобу, який приймала вагітна жінка, а побічна реакція виникла у плода, то всі дані (за винятком побічної реакції) надаються про матір.
Номер історії хвороби/амбулаторної карти. Указуються номер історії хвороби чи амбулаторної карти пацієнта.
Дата народження/вік. Зазначається день, місяць та рік народження пацієнта. Для пацієнтів віком від 3 років та старше — зазначаються роки (наприклад, 4 роки); для пацієнтів до 3 років — місяці (наприклад, 24 місяці); для пацієнтів віком до місяця — дні (наприклад, 5 днів).
Стать. Позначається так: Ж або Ч. Якщо повідомлення стосується лікарського засобу, який приймала вагітна жінка, а побічна реакція виникла у плода, то всі дані (за винятком побічної реакції) надаються про матір із зазначенням триместру вагітності.
Зріст. Зазначається зріст пацієнта у см.
II розділ. Підозрювані ПР/ВЕ/НППІ
У графах зазначаються:
Підозрювана ПР/НППІ (Опишіть кожен клінічний прояв ПР/НППІ із зазначенням дати та часу початку, закінчення та наслідку) /Зазначення ВЕ. Описується кожна ПР/НППІ із зазначенням дати та часу початку і закінчення та наслідку ПР/ВЕ/НППІ. Початок ПР/ВЕ/НППІ лікарського засобу. Зазначається день, місяць, рік та час виникнення ПР/ВЕ/НППІ. Повідомлення щодо вроджених аномалій плода супроводжується датою народження дитини або терміну вагітності.
Наслідок ПР/ВЕ/НППІ. Зазначаються відповідні позиції.
Корекція ПР/ВЕ/НППІ. Зазначаються відповідні позиції.
Чи вважаєте Ви ці прояви ПР/НППІ серйозними? (стосується загалом випадку ПР/НППІ) Зазначаються відповідні позиції. У разі групової НППІ Карти-повідомлення про ПР чи ВЕ лікарського засобу та НППІ заповнюються на кожного пацієнта, у якого зареєстрована дана НППІ та якому була проведена імунізація/туберкулінодіагностика.
III розділ. Інформація про підозрюваний ЛЗ.
У графах зазначаються:
Підозрюваний лікарський засіб (торгівельне найменування, форма випуску, виробник). Указуються торгівельне найменування підозрюваного лікарського засобу, який підозрюється у причетності до виникнення побічної реакції/відсутності ефективності/НППІ, його форма випуску, виробник.
Номер серії. Зазначається номер серії підозрюваного лікарського засобу.
Покази. Зазначаються покази для призначення підозрюваного лікарського засобу за можливості по МКХ-10.
Сила дії. Указується вміст діючої(их) речовини (речовин) у кількісному вираженні на одиницю дози, або одиницю об’єму, або одиницю маси відповідно до лікарської форми підозрюваного лікарського засобу.
Разова доза. Указується разова доза підозрюваного лікарського засобу.
Кратність приймання. Указується кратність приймання підозрюваного лікарського засобу.
Спосіб введення. Указується спосіб введення підозрюваного лікарського засобу.
Дата та час початку терапії. Вказується день, місяць, рік та час призначення підозрюваного лікарського засобу.
Дата та час закінчення терапії. Указується день, місяць, рік та час закінчення застосування підозрюваного лікарського засобу.
Заходи, що вживались стосовно підозрюваного ЛЗ для корекції ПР/ВЕ/НППІ Зазначаються відповідні позиції.
Розділ ІІІ а. Додаткова інформація у випадку НППІ на вакцини або туберкулін
Категорія імунізації або туберкулінодіагностики. У графі зазначається категорія імунізації або туберкулінодіагностики згідно наведеного переліку.
Категорія НППІ. У графі зазначається категорія НППІ згідно наведеного переліку.
Номер дози (для вакцини). У графі зазначається номер дози вакцинального комплексу згідно наведеного переліку.
Місце уведення вакцини/ туберкуліну. У графі зазначається місце уведення вакцини/ туберкулінузгідно наведеного переліку.
Спосіб уведення вакцини/ туберкуліну. У графі зазначається спосіб уведення вакцини/ туберкуліну згідно наведеного переліку.
Дані анамнезу життя особи, якій було проведено щеплення/туберкулінодіагностику (щеплювальнийанамнез, наявність реакцій на попередні введення вакцин, туберкуліну, наявність гострого або загострення хронічного захворювання протягом 1–1,5 місяців до проведення імунізації/туберкулінодіагностики, застосування імуносупресивної терапії протягом 1 місяця та препаратів крові протягом 3-х місяців до проведення імунізації тощо). Зазначається інформація щодо щеплювального анамнезу, наявності реакції на попередні введення вакцин, туберкуліну, наявності гострого або загострення хронічного захворювання протягом 1–1,5 місяців до проведення імунізації/туберкулінодіагностики, застосування імуносупресивної терапії протягом 1 місяця та препаратів крові протягом 3-х місяців до проведення імунізації/туберкулінодіагностики тощо.
IV розділ. Інформація про супутні ЛЗ (за виключенням препаратів, які застосовувалися для корекції наслідків ПР/НППІ/ВЕ).
У графах зазначаються:
Супутні ЛЗ (торгівельне найменування, форма випуску). Указується торгівельне найменування супутніх лікарських засобів, які призначались, їх форма випуску, виробник, номер серії.
Покази. Зазначаються покази для призначення супутніх лікарських засобів за можливості по МКХ-10.
Сила дії. Указується вміст діючої(их) речовини (речовин) у кількісному вираженні на одиницю дози, або одиницю об’єму, або одиницю маси відповідно до лікарської форми супутніх лікарських засобів.
Разова доза. Указується разова доза підозрюваного лікарського засобу.
Кратність приймання. Указується кратність приймання підозрюваного лікарського засобу.
Спосіб введення. Указується спосіб введення супутніх лікарських засобів.
Дата початку терапії. Указується день, місяць та рік призначення супутніх лікарських засобів.
Дата закінчення терапії. Указується день, місяць та рік закінчення застосування супутніх лікарських засобів.
Інша важлива інформація (супутні діагнози, дані лабораторно-інструментальних досліджень, алергоанамнез, вагітність із зазначенням: терміну вагітності, способу зачаття, результату вагітності, якщо вагітність завершилась, то — дати пологів, типу пологів тощо). Зазначаються дані, які можуть впливати на прояв побічної реакції/відсутність ефективності, але безпосередньо з ним не пов’язані.
V розділ. Інформація про повідомника.
У графі зазначається прізвище, ім’я, по батькові повідомника, спеціальність, організація (заклад охорони здоров’я), поштова адреса організації, електронна адреса, телефон, дата заповнення.
VІ розділ. Інформація про медичного/фармацевтичного спеціаліста (якщо не повідомник).
У графі зазначається прізвище, ім’я, по батькові медичного/фармацевтичного спеціаліста, спеціальність, організація (заклад охорони здоров’я), поштова адреса організації, електронна адреса, телефон, дата заповнення.
Додаток 10
до Порядку здійснення фармаконагляду
ПОЛОЖЕННЯ
про регіональні та центральну групи оперативного реагування
1. Це Положення визначає порядок створення, склад та завдання регіональних та центральної груп оперативного реагування (далі — група оперативного реагування) на несприятливі події після імунізації (далі — НППІ).
2. Регіональна група оперативного реагування створюється при структурних підрозділах з питань охорони здоров’я обласних, Київської міської державних адміністрацій.
3. Центральна група оперативного реагування створюється при Міністерстві охорони здоров’я України.
4. Групу оперативного реагування очолює голова, який має спеціальність лікар-педіатр або лікар-терапевт.
5. Група оперативного реагування залучається до дій головою групи оперативного реагування.
6. Група оперативного реагування у своїй діяльності керується цим Положенням та іншими чинними нормативно-правовими актами у сфері охорони здоров’я.
7. Склад регіональної групи оперативного реагування затверджується наказом структурних підрозділів з питань охорони здоров’я обласних, Київської міської державних адміністрацій, центральної — наказом МОЗ України.
До груп оперативного реагування входять фахівці за спеціальністю: невролог (дитячий невролог), інфекціоніст (дитячий інфекціоніст), алерголог (дитячий алерголог), імунолог (дитячий імунолог), анестезіолог (дитячий анестезіолог), епідеміолог, педіатр/терапевт (в залежності від спеціальності голови групи), судово-медичний експерт, патологоанатом.
8. Регіональна група оперативного реагування:
1) отримує карти-повідомлення про усі випадки НППІ, що сталися у регіоні;
2) здійснює аналіз карт-повідомлень про усі випадки НППІ, що сталися у регіоні, на предмет визначення категорії серйозності;
3) направляє усі отримані карти-повідомлення про випадки НППІ до Центру;
4) проводить розслідування усіх серйозних та групових НППІ та встановлює причинно-наслідковий зв’язок між НППІ та застосуванням вакцини, туберкуліну;
5) за результатами розслідування заповнює Протокол розслідування та встановлення причинно-наслідкового зв’язку серйозних та групових НППІ (далі — протокол розслідування НППІ) та надає його до Центру разом із копією відповідної карти-повідомлення про випадок НППІ;
6) голова регіональної групи оперативного реагування надає копії карт-повідомлень про випадки НППІ головному штатному/позаштатному спеціалісту структурного підрозділу з питань охорони здоров’я обласних, Київської міської державних адміністрацій за спеціальністю «терапія».
9. Центральна група оперативного реагування залучається до дій протягом 48 годин після отримання від Центру інформації, що зазначена у підпункту 4 пункту 2 розділу ІІІ до цього Порядку.
10. За результатами аналізу Центральна група надає до МОЗ України та/або заявника (за необхідністю) рекомендації щодо прийняття відповідних рішень.
11. Копії протоколів засідання Центральної групи надаються до Центру.
12. Центральна група оперативного реагування, у разі необхідності, надає консультативну допомогу у разі інших проблем, виявлених при застосуванні вакцин, туберкуліну, що пов’язані з безпекою та ефективністю.
ВИМОГИ ДО СКЛАДАННЯ
зведених даних про випадки побічних реакцій після застосування вакцин, туберкуліну
Зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну заповнюється відповідальною особою в такому порядку:
у заголовку зведених даних про випадки побічних реакцій після застосування вакцин, туберкуліну зазначається заклад охорони здоров’я, структурний підрозділ з питань охорони здоров’я та звітний період;
2) торгове найменування вакцини та туберкуліну;
3) найменування підприємства-виробника, країна;
4) серія вакцини та туберкуліну. Уважно вказується серія із усіма наявними літерами (латиною або кирилицею) та цифрами зазначеними на упаковці вакцини або анатоксину чи алергену туберкульозного. Наприклад: AD12CN234DE — правильний варіант заповнення серії, AД12CН234ДE — неправильний варіант заповнення серії;
5) кількість фактично введених доз відповідної серії вакцини, туберкуліну;
6) кількість осіб, яким була проведена імунізація або туберкулінодіагностика конкретною серією вакцини, туберкуліну;
7) інформація про побічні реакції,згідно з переліком клінічних проявів побічних реакцій, що відповідно зазначений у додатку до цього Порядку. Якщо клінічні прояви побічної реакції не зазначені в переліку, але зазначені в інструкції для медичного застосування відповідної вакцини, туберкуліну, ця побічна реакція зазначається у вільному рядку, без зазначення коду;
8) у випадку відсутності за звітний період побічних реакцій у відповідній графі узагальненого звіту про випадки побічних реакцій після застосування вакцин, туберкуліну має бути зазначено «0».
9) Зведені дані про випадки побічних реакцій після застосування вакцин, туберкуліну надаються до:
– Державного підприємства «Державний експертний центр Міністерств охорони України» у паперовому вигляді разом із супровідним листом на адресу: Державне підприємство «Державний експертний центр Міністерства охорони здоров’я України», Департамент післяреєстраційного нагляду, вул. Ушинського, 40, м. Київ, 03151; тел/факс: +38 044 4984358; e-mail; у електронному вигляді на електронну адресу: [email protected]
– відповідного структурного підрозділу з питань охорони здоров’я у паперовому вигляді разом із супровідним листом на адресу відповідного структурного підрозділу з питань охорони здоров’я. У електронному вигляді на електронну адресу відповідного структурного підрозділу з питань охорони здоров’я.
Додаток 13
до Порядку здійснення фармаконагляду
СТРУКТУРА
майстер-файлу системи фармаконагляду заявника
Інформація в майстер-файлі системи фармаконагляду (далі — МФСФ) повинна бути представлена згідно структури та змісту додатків. Додаток Е не може бути перейменований в додаток D у випадку, коли додаток про комп’ютеризовані системи і бази даних не використовується, додаток D має бути позначений як «невикористаний» у змісті до МФСФ для того, щоб одержувачі МФСФ були впевнені, що такий додаток відсутній не в результаті помилки.
Титульна сторінка
Представлення системи фармаконагляду
1. Інформація про уповноважену особу з фармаконагляду
2. Інформація про організаційну структуру заявника/власника реєстраційного посвідчення
3. Інформація про джерела даних з безпеки
4. Інформація про комп’ютеризованих системах і базах даних
5. Інформація про процеси фармаконагляду
6. Інформація про продуктивність системи фармаконагляду
7. Інформація про систему якості в фармаконагляду
Додатки.
Вимоги до заповнення
майстер-файлу системи фармаконагляду заявника
Титульна сторінка повинна містити інформацію про:
унікальний номер;
найменування заявника/власника реєстраційного посвідчення;
найменування заявника/власника реєстраційного посвідчення уповноваженої особи, відповідальної за описану систему фармаконагляду (якщо відрізняється), а також відповідну назву третьої сторони (якщо є) уповноваженої особи, відповідальної за описану систему фармаконагляду;
назви інших зацікавлених заявників/власників реєстраційних посвідчень (у разі спільного використання системи фармаконагляду);
список МФСФ заявника/власника реєстраційного посвідчення (для лікарських засобів, що мають різні системи фармаконагляду);
дату складання/останнього оновлення.
Представлення системи фармаконагляду
Надаються посилання на нормативні документи, на підставі яких розроблена і функціонує система фармаконагляду, надається стислий опис/представлення власника реєстраційного посвідчення/заявника.
Розділ 1. Інформація про уповноважену особу з фармаконагляду
Надається інформація, що включає, але не обмежується: інформація про УОВФ, включаючи контактну інформацію про неї, дані про кваліфікацію і досвід роботи; резюме з ключовою інформацією про роль УОВФ; переліком обов’язків для гарантування того, що УОВФ має достатні повноваження щодо створення системи фармаконагляду, підтримки та покращення її функціонування, у тому числі в Україні; відомостями про резервні механізми, що будуть задіяні у випадку відсутності УОВФ; інформацією про КОФ, у тому числі в Україні, якщо вона відмінна від УОВФ, включаючи контактну інформацію про неї, дані про кваліфікацію і досвід роботи; інформацією щодо виконання обов’язків УОВФ/КОФ, у разі його відсутності (хвороба, відпустка).
Розділ 2. Інформація про організаційну структуру заявника/ власника реєстраційного посвідчення
Надається опис організаційної структури власника реєстраційного посвідчення/заявника, у тому числі у розрізі здійснення фармаконагляду. Опис повинен містити інформацію щодо залучених компаній, головних підрозділів з фармаконагляду та взаємозв’язків між організаціями і підрозділами, що мають відношення до виконання зобов’язань з фармаконагляду. В опис необхідно включати третіх осіб, залучених до діяльності, пов’язаної із здійсненням фармаконагляду. Зокрема, МФСФ повинен описувати:
а) організаційну структуру власника(ів) реєстраційного посвідчення із зазначенням позиції УОВФ в організації;
б) місце(я), де проводиться діяльність з фармаконагляду, що включає збір повідомлень про випадки побічних реакцій, їх оцінку, введення у базу даних з безпеки, генерування регулярно оновлюваних звітів з безпеки, виявлення та аналіз сигналів, підготовку та виконання плану управління ризиками, а також клінічними та післяреєстраційними дослідженнями, внесення змін до реєстраційних матеріалів, що стосуються безпеки лікарського(их) засобу(ів) та/або системи фармаконагляду.
Опис повинен містити інформацію щодо делегованої діяльності, залучених компаній, головних підрозділів з фармаконагляду та взаємозв’язків між організаціями і підрозділами, що мають відношення до виконання зобов’язань з фармаконагляду. Така інформація може бути представлена у вигляді списку/таблиці із зазначенням залучених сторін, їх обов’язків та відповідних лікарських засобів і країн. Список повинен бути організований відповідно до постачальників послуг (наприклад, медична інформація, аудитори, організатори програм підтримки пацієнтів, управління даними досліджень і т.д.), комерційних угод (дистриб’ютори, партнери, спільний маркетинг тощо) та інших технічних послуг (хостинг комп’ютерних систем і т.д.). У додатках надають список договорів, а самі договори повинні бути доступні на запит або під час проведення інспекцій і аудитів.
Розділ 3. Інформація про джерела даних з безпеки
Надається опис основних підрозділів зі збору даних з безпеки, що повинен включати інформацію про всі відповідальні сторони на глобальному рівні для організованого та спонтанного збору даних з безпеки лікарських засобів. Джерела даних з безпеки повинні включати дані, що надходять з будь-яких досліджень, реєстрів, спостережень чи програм підтримки, що фінансуються власником реєстраційного посвідчення, за допомогою яких можна збирати повідомлення про побічні реакції. Перелік повинен бути вичерпним та включати усі поточні дослідження/програми, а також дослідження/програми, завершені протягом останніх двох років, і може міститися в додатках або надаватися окремо.
Розділ 4. Інформація про комп’ютеризовані системи і бази даних
Надається опис розташування, функціональності та відповідальність за комп’ютеризовані системи і бази даних, що використовуються для отримання, обробки, запису та надання інформації з безпеки, а також оцінка придатності таких систем і баз даних для виконання фармаконагляду заявником. Якщо використовуються декілька комп’ютеризованих систем/баз даних, їх придатність для діяльності з фармаконагляду повинна бути описана таким чином, щоб надати чітке уявлення про ступінь комп’ютеризації в рамках системи фармаконагляду. Також повинен бути описаний валідаційний статус баз даних; інформація про процедури контролю внесення змін, характер тестування, процедури резервування та електронні сховища даних, важливих для відповідності фармаконагляду повинна бути включена в узагальнену інформацію, повинен бути описаний характер доступної документації. Для систем на основі паперових носіїв (коли електронна система може використовуватися тільки для подачі термінових повідомлень про побічні реакції) повинні бути описані організація та ведення даних і механізми забезпечення цілісності та доступності даних про безпеку, зокрема, узагальнення інформації про побічні реакції лікарських засобів.
Розділ 5. Інформація про процеси фармаконагляду
Надається опис процесів, обробки даних і записів для здійснення фармаконагляду, що охоплює наступні аспекти (але може не обмежуватись):
а) безперервний моніторинг профілю користь/ризик лікарського засобу(ів), результати оцінки, процес прийняття рішень для вжиття відповідних заходів; генерація сигналу, його виявлення і оцінка, а також ряд письмових процедур та інструкцій, що стосуються вихідних даних бази даних з безпеки, взаємодії з клінічними департаментами і т.д.;
б) систему(и) управління ризиками і моніторинг результатів заходів з мінімізації ризиків; до даної діяльності можуть бути залучені декілька підрозділів, і їх взаємодія повинна бути регламентована в письмових процедурах або угодах;
в) збір повідомлень про індивідуальні випадки, пов’язані з безпекою, узагальнення даних повідомлень, подальше відстеження (follow-up), оцінка та звітування; процедури, що при цьому застосовуються, повинні уточнювати діяльність на центральному та національному рівнях;
г) планування термінів генерації регулярно оновлюваних звітів з безпеки, їх підготовка та подання (див. модуль VII);
д) інформування з питань безпеки споживачів, спеціалістів системи охорони здоров’я і уповноваженого органу;
ж) внесення змін з безпеки в інструкцію для медичного застосування; процедури повинні охоплювати як внутрішню, так і зовнішню комунікацію.
Опис повинен супроводжуватися переліком процесів щодо контролю відповідності, а також взаємодії з іншими функціями. Взаємодія з іншими функціями включає (однак не обмежується) функціональні обов’язки та відповідальність УОВФ/КОФ, відповіді на запити уповноваженого органу про надання інформації, літературний пошук, контроль змін в базі даних з безпеки, угоди про обмін даними з безпеки, архівування даних з безпеки, аудит фармаконагляду, контроль якості і навчання персоналу. Цей перелік може бути розміщений у додатках, і повинен містити реєстраційний номер процедурного документу, заголовок, дату набуття чинності і тип документу (для усіх стандартних операційних процедур, робочих інструкцій, інструкцій і т.д.). Повинні бути чітко ідентифіковані процедури, що стосуються постачальників послуг та інших третіх сторін. До переліку не вимагається включати документи, що стосуються специфічних національних процедур, однак їх перелік може вимагатися на рівні країни. Якщо жодна або лише деякі країни використовують спеціальні місцеві процедури, це має бути зазначено (і вказано назви відповідних країн).
Розділ 6. Інформація про продуктивність системи фармаконагляду
Надається опис методів моніторингу продуктивності системи фармаконагляду власника реєстраційного посвідчення/заявника, що застосовуються. Необхідно описати і пояснити цільові показники продуктивності системи фармаконагляду. Перелік показників продуктивності потрібно надати у додатку до МФСФ разом з фактичними результатами оцінки такої ефективності. Така інформація може бути надана у вигляді цифрових даних/графіків, що ілюструють та підтверджують продуктивність системи фармаконагляду. МФСФ повинен включати опис методів моніторингу, що застосовуються, і містити щонайменше:
а) опис процедури оцінки коректності подання повідомлень про індивідуальні випадки, пов’язані з безпекою. У додатку повинні бути надані цифрові дані/графіки, що ілюструють та підтверджують дотримання термінів та своєчасність подання повідомлень про індивідуальні випадки, пов’язані з безпекою, у передньому році;
б) загальні відомості про подання регулярно оновлюваних звітів з безпеки до регуляторних органів (додаток повинен містити останні дані, що використовувалися власником реєстраційного посвідчення/заявником для оцінки відповідності термінів подання таких звітів);
в) короткий опис методів, що використовуються для забезпечення своєчасності подання заяв на зміни з безпеки у порівнянні з внутрішніми граничними строками та строками, передбаченими вимогами законодавства, а також дату та опис необхідних змін з безпеки, які були виявлені, але заяви на зміни ще не були подані;
г) у відповідних випадках, — узагальнені дані щодо дотримання зобов’язань виконання плану управління ризиками або інших зобов’язань чи умов, що стосуються фармаконагляду, дотримання яких було умовою видачі реєстраційного посвідчення.
Розділ 7. Інформація про систему якості в фармаконагляду
Надається опис управління системою якості потрібно представити в рамках структури організації і застосування системи якості до фармаконагляду. Такий опис повинен включати інформацію про:
а) контроль документації та записів: потрібно надати опис механізмів архівування електронних та/або друкованих версій МФСФ, повідомлень про індивідуальні випадки, пов’язані з безпекою, регулярно оновлюваних звітів з безпеки, планів управління ризиками, інших записів і документів, що мають відношення до фармаконагляду;
б) процедурні документи: потрібно надати загальний опис документів, що використовуються у фармаконагляді (стандарти, операційні процедури, робочі інструкції тощо), доступність різних документів на глобальному, регіональному або локальному рівні в рамках організації, а також методів контролю їх доступності, впровадження та супроводу; дані щодо системи ведення документації, що використовуються для відповідних процесуальних документів під контролем третіх осіб.
в) навчання: потрібно надати опис управління ресурсами для виконання діяльності з фармаконагляду (організаційна структура із зазначенням кількості співробітників (повної зайнятості), залучених до діяльності з фармаконагляду, що можуть бути надані в розділі, який описує організаційну структуру організації); короткий опис концепції навчання персоналу (не тільки співробітники підрозділів з фармаконагляду, а й будь-яких працівників, які можуть отримувати повідомлення про індивідуальні випадки, пов’язані з безпекою), в тому числі посилання на розміщення навчальних файлів.
г) аудити: потрібно надати інформацію про аудити забезпечення якості у системі фармаконагляду. У додатку необхідно надати опис підходів, що використовуються для планування аудитів системи фармаконагляду, механізмів звітності і строків, а також поточний перелік запланованих і проведених аудитів, що стосуються системи фармаконагляду. Цей перелік повинен містити дату проведення аудиту, дату подання звіту, мету і стан здійснення аудитів постачальників послуг, специфічних видів діяльності з фармаконагляду або підрозділів, що здійснюють діяльність з фармаконагляду, та їх взаємодію з іншими підрозділами організації, що мають відношення до виконання зобов’язань з фармаконагляду, і повинен охоплювати 5-річний період.
МФСФ повинен також містити коротку інформацію про всі аудити, в результаті яких було виявлено значимі результати, із наданням короткого опису коригуючих та/або запобіжних заходів щодо значимих результатів аудиту, дат їх виявлення та очікуваних дат їх усунення з перехресним посиланням на звіт з аудиту та задокументований план(и) коригуючих та запобіжних заходів.
У переліку проведених аудитів, що надається у додатках, повинні бути позначені ті, інформація про які міститься у МФСФ. Коротка інформація про аудити, в результаті яких було виявлено критичні дані, та короткий опис коригуючих і запобіжних заходів повинні міститися в МФСФ до тих пір, поки коригуючі та/або запобіжні заходи не будуть вжиті в повному обсязі. Тобто ця інформація видаляється лише після того, як було продемонстровано досягнуті результати вжитих коригуючих дій та/або підтвердження незалежною стороною істотного покращення системи. Доповнення, зміни або видалення інформації про аудити в МФСФ повинні реєструватися в журналі.
В якості засобу управління системою фармаконагляду та підстави для проведення аудиту, МФСФ повинен також описувати процес обліку, управління та усунення виявлених відхилень в системі якості. МФСФ повинен також документувати відхилення від процедур фармаконагляду, їх вплив і управління ними до моменту їх вирішення. Ці відхилення можна документувати у вигляді списку з посиланням на звіт про відхилення, його дату та процедуру.
Додатки до Майстер-файлу системи фармаконагляду
Інформація у додатках повинна бути представлена відповідно до розділів Майстер-файлу системи фармаконагляду, в яких йде посилання на певний додаток, із наданням інформації, що представлена нижче, але не обмежуючись:
Додаток А до розділу 1. Інформація про уповноважену особу з фармаконагляду;
Додаток В до розділу 2. Інформація про організаційну структуру заявника/власника реєстраційного посвідчення;
Додаток С до розділу 3. Інформація про джерела даних з безпеки;
Додаток D до розділу 4. Інформація про комп’ютеризованих системах і базах даних;
Додаток Е до розділу 5. Інформація про процеси фармаконагляду: переліки процедурних документів;
Додаток F до розділу 6. Інформація про продуктивність системи фармаконагляду: переліки показників ефективності, поточні результати оцінки ефективності по відношенню до показників;
Додаток G до розділу 7. Інформація про систему якості в фармаконагляду: графік аудитів, перелік проведених та завершених аудитів, інформація щодо застосованих заходів;
Додаток H до представлення системи фармаконагляду: перелік лікарських засобів, що охоплені цією системою фармаконагляду;
Додаток I. Контроль документації та записів: журнал коригування; документація щодо історії змін змісту, відповідно проіндексовані, та опис таких змін.
Додаток 14
до Порядку здійснення фармаконагляду
СТРУКТУРА
регулярно оновлюваного звіту з безпеки лікарського засобу
Частина І. Титульна сторінка з підписом
Частина II. Основні положення
Частина III. Зміст регулярно оновлюваного звіту з безпеки
1. Вступ
2. Міжнародний реєстраційний статус
3. Заходи з безпеки, вжиті протягом звітного періоду
4. Зміни у довідковій інформації з безпеки
5. Оцінка експозиції та схем застосування
5.1. Кумулятивна експозиція учасників клінічних випробувань
5.2. Кумулятивна та інтервальна експозиція пацієнтів у післяреєстраційному періоді
6. Дані зведених таблиць
6.1. Довідкова інформація
6.2. Кумулятивні зведені таблиці про серйозні побічні явища, що виявлені під час проведення клінічних випробувань
6.3. Кумулятивні та інтервальні зведені таблиці згідно даних з безпеки, отриманих у післяреєстраційному періоді
7. Резюме значимих результатів клінічних випробувань протягом звітного періоду
7.1. Завершені клінічні випробування
7.2. Поточні клінічні випробування
7.3. Тривале спостереження
7.4. Інше терапевтичне застосування лікарського засобу
7.5. Нові дані з безпеки, пов’язані з фіксованим комбінованим лікуванням
8. Результати неінтервенційних досліджень
9. Інформація, отримана з інших джерел та під час проведення інших клінічних випробувань
10. Неклінічні дані
11. Дані з літературних джерел
12. Інші регулярно оновлювані звіти з безпеки
13. Відсутність ефективності в контрольованих клінічних дослідженнях
14. Інформація, отримана в останній момент
15. Огляд сигналів: нові, поточні та закриті
16. Оцінка сигналів та ризиків
16.1. Резюме проблем безпеки
16.2. Оцінка сигналу
16.3. Оцінка ризиків та нової інформації
16.4. Характеристика ризиків
16.5. Ефективність заходів з мінімізації ризиків (якщо застосовано)
17. Оцінка користі
17.1. Важлива базова інформація з ефективності
17.2. Нова виявлена інформація з ефективності
17.3. Характеристика користі
18. Інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань
18.1. Контекст співвідношення користь/ризик — медична необхідність та важливі альтернативи
18.2. Оцінка аналізу співвідношення користь/ризик
19. Висновки та заходи
20. Додатки до регулярно оновлюваного звіту з безпеки
Вимоги до заповнення регулярно оновлюваного звіту з безпеки лікарського засобу
Частина І. Титульна сторінка
Титульна сторінка повинна містити назву лікарського засобу(ів)1та активної субстанції, міжнародну дату народження лікарського засобу, звітний період, дату складання звіту, інформацію про заявника та твердження про конфіденційність інформації, що представлена у регулярно оновлюваному звіті з безпеки (далі — регулярний звіт).
На титульній сторінці повинен стояти підпис.
__________________
1 Для регулярно оновлюваних звітів з безпеки, що включають декілька лікарських засобів, вакцин, туберкуліну, з практичних причин цю інформацію можна надавати на супровідній до титульної сторінці регулярно оновлюваного звіту з безпеки (див. Додаток ІІ)
Частина II. Основні положення
Надається стисле резюме змісту та найбільш важливої інформації, що представлена у регулярному звіті:
вступ і звітний період;
лікарський засіб(и), хіміко-терапевтична або фармакологічна група (и), механізм(и) дії, показання, форми випуску, доза(и) та спосіб(и) застосування;
узагальнена оцінка кумулятивного впливу лікарського засобу, на учасників клінічних випробувань;
узагальнена оцінка кумулятивного та інтервального впливу лікарського засобу на пацієнтів у післяреєстраціному періоді;
кількість країн, в яких лікарський засіб зареєстровано;
резюме загальної оцінки аналізу співвідношення користь/ризик (на основі даних, представлених у підрозділі 18.2 «Оцінка аналізу співвідношення користь/ризик» регулярного звіту);
вжиті та пропоновані заходи з питань безпеки (наприклад, значущі зміни до інформації про лікарський засіб чи інші заходи з мінімізації ризиків)
висновки.
Частина III. Зміст регулярного звіту
Розділ 1. Вступ
Надається короткий опис лікарського засобу (ів) для того, щоб регулярний був «автономним» документом, однак також відображав зв’язок з попередніми регулярним звітом та інші обставини. Вступ повинен містити наступну інформацію:
міжнародна дата народження лікарського засобу та звітний період;
лікарський(і) засіб(и) хіміко-терапевтична або фармакологічна(і) група(и), механізм(и) дії, затверджене(і) показання, форма(и) випуску, доза(и) та спосіб(оби) застосування;
короткий опис пролікованих та досліджуваних популяцій
Розділ 2. Міжнародний реєстраційний статус
Надається короткий опис із зазначенням наступної інформації: дата першої реєстрації у світі, показання, зареєстрована доза(и), де зареєстрований лікарський засіб.
Розділ 3. Заходи з безпеки, прийняті протягом звітного періоду
Надається опис значущих заходів з безпеки, що були запроваджені у світі протягом звітного періоду власником реєстраційного посвідчення, спонсорами клінічних досліджень, етичними комітетами чи уповноваженими органами незалежно від того стосувалися вони застосування лікарського засобу при проведенні досліджень чи у післяреєстраційний період, та мали значний вплив на співвідношення користь/ризик зареєстрованого лікарського засобу та/або вплив на проведення окремого клінічного дослідження(ь) або на загальну програму клінічних досліджень.
Якщо відомо, слід надати обґрунтування кожного заходу та за необхідності включити до регулярного звіту будь-яку додаткову важливу інформацію. У цьому розділі слід також надати в узагальненому вигляді відповідні оновлення до попередніх заходів з питань безпеки. Наприклад: заходи, пов’язані із застосуванням лікарського засобу з дослідницькою метою (відмова у реєстрації клінічного випробування з етичних причин або з міркувань безпеки; часткове2 або повне припинення клінічного випробування або дострокове припинення поточного клінічного випробування з причин безпеки або відсутності ефективності; відкликання досліджуваного лікарського засобу або компаратора; відмова у реєстрації для досліджуваних показань, включаючи добровільне відкликання заяви на реєстрацію; заходи з управління ризиками); заходи, пов’язані з досвідом знаходження на ринку (відмова у перереєстрації чи не подання заяви на перереєстрацію; відкликання або призупинення дії реєстраційного посвідчення; заходи, вжиті через проблеми, пов’язані з якістю; призупинення постачання лікарського засобу власником реєстраційного посвідчення; заходи з управління ризиками)
_____________
2 Часткове припинення може включати декілька заходів (наприклад, припинення досліджень повторних доз, але продовження досліджень однієї дози; припинення досліджень за одним показанням, але продовження за іншим та/або припинення застосування певної схеми прийому у дослідженні).
Розділ 4. Зміни у довідковій інформації з безпеки
Повинні бути перераховані будь-які значні зміни до довідкової інформації з безпеки у звітний період. Такі зміни можуть стосуватися протипоказань, особливостей застосування, застережень, серйозних побічних реакцій, взаємодій, важливих результатів поточних чи завершених клінічних випробувань або значущих результатів неклінічних досліджень (наприклад, дослідження канцерогенності). Специфічна інформація стосовно таких змін повинна надаватися у відповідних розділах регулярного звіту.
Розділ 5. Оцінка експозиції та схем застосування
Надається точна оцінка популяцій, які піддавалися впливу лікарського засобу, включаючи обсяги продажів та кількість призначень. Така оцінка повинна супроводжуватися якісним та кількісним аналізом фактичного застосування лікарського засобу, що повинен, коли доцільно, вказувати наскільки фактичне застосування відрізняється від показаного застосування на підставі всіх даних, доступних власнику реєстраційного посвідчення, включаючи результати обсерваційних досліджень чи досліджень застосування лікарського засобу. Потрібно надати короткий опис методу(ів), що використовувались для підрахунку експозиції суб’єктів/пацієнтів та із зазначенням обмежень такого методу. Для підрахунку експозиції суб’єктів/пацієнтів у всіх регулярних звітах для одного й того ж лікарського засобу слід використовувати порівняні методи. Якщо з об’єктивних причин метод був змінений, обидва методи та розрахунки слід надавати у регулярних звітах, із зазначенням такої зміни та будь-якої важливої відмінності між результатами при використанні обох методів. Інформація повинна бути надана із зазначенням.
Підрозділ 5.1. Кумулятивна експозиція учасників клінічних випробувань
Цей підрозділ регулярного звіту повинен містити наступну інформацію про суб’єкти клінічних випробувань, спонсорованих власником реєстраційного посвідчення, представлену за можливості у вигляді таблиці:
сукупна кількість суб’єктів поточних та завершених клінічних випробувань, які піддавалися впливу досліджуваного лікарського засобу, плацебо та/або компаратор(и), починаючи з міжнародної дати народження лікарського засобу, що знаходився в стадії розробки;
детальні дані про експозицію суб’єктів клінічних випробувань (наприклад, дані згруповані за віком, статтю та расовою приналежністю для всієї програми випробування);
якщо можливо, в таблицях повинні бути зазначені суттєві розбіжності у випробуваннях у дозах, способах застосування чи популяціях пацієнтів; можна також використовувати окремі таблиці;
якщо клінічні випробування проводилися або проводяться у спеціальних популяціях (наприклад, вагітні жінки, пацієнти з нирковою, печінковою або серцевою недостатністю; або пацієнти з відповідним генетичним поліморфізмом), необхідно надати дані про їх експозицію;
якщо існують значні розбіжності у тривалості експозиції між суб’єктами, рандомізованими до досліджуваного лікарського засобу чи компаратора, або у випадку невідповідності у тривалості експозиції у різних клінічних випробуваннях, може бути корисним надати інформацію про експозицію препарату у суб’єкто-часі (суб’єкто-днях, — місяцях або — роках);
експозиція досліджуваного лікарського засобу у здорових добровольців може бути менш важливою для оцінки загального профілю безпеки, залежно від типу побічної реакції, зокрема, коли суб’єкти застосовують одну дозу. Такі дані можуть бути представлені окремо з відповідним поясненням;
якщо серйозні побічні явища, виявлені під час клінічних випробувань представлені у зведених таблицях за показаннями, експозиція пацієнтів повинна бути також представлена за показаннями, якщо можливо;
для окремих особливо важливих досліджень, демографічні характеристики повинні надаватися окремо.
Підрозділ 5.2. Кумулятивна та інтервальна експозиція пацієнтів у післяреєстраційному періоді
Післяреєстраційна (неклінічні дослідження) експозиція.
Якщо у післяреєстраційному періоді лікарський засіб застосовувався у спеціальних популяціях.
Інше застосування лікарського засобу у післяреєстраційному періоді.
Розділ 6. Дані, представлені у зведених таблицях
Необхідно надати дані з безпеки у вигляді зведених таблиць серйозних побічних явищ з клінічних досліджень, спонтанних повідомлень про серйозні та несерйозні реакції з післяреєстраційного досвіду застосування препарату (включаючи повідомлення від працівників з медичною та фармацевтичною освітою, споживачів, з наукової літератури, повідомлення від уповноважених органів (на міжнародному рівні) та серйозних реакцій з неінтревенційних досліджень та інших неінтервенційних джерел інформації про побічні реакції. При кодуванні термінів побічних явищ/реакцій з використанням медичного словника для регуляторної діяльності (Medical Dictionary for Regulatory Activities (MedDRA) у зведених таблицях необхідно представляти дані на рівні термінів переважного застосування (preferred term (PT) та класів систем і органів (system organ class (SOC). Серйозність побічних явищ/реакцій у зведених таблицях повинна відповідати серйозності явищ/реакцій у повідомленнях про індивідуальні випадки побічних реакцій, що визначається із застосуванням критеріїв, наведених в ICH-E2А3. Якщо серйозні та несерйозні явища/реакції відносяться до одного й того ж випадку, у зведених таблицях необхідно надати інформацію про серйозність кожної реакції. Не можна змінювати серйозність побічного явища/реакції спеціально для підготовки регулярного звіту.
_______________
3 ICH тема E2А. Управління даними з клінічної безпеки: визначення та стандарти для термінового повідомлення.
Підрозділ 6.1. Довідкова інформація
Необхідно вказати версію(ї) довідкової інформації (словника), що використовувався для кодування побічних явищ/реакцій.
Підрозділ 6.2. Кумулятивні зведені таблиці серйозних побічних явищ, виявлених під час клінічних випробувань
Потрібно надати основні дані для додатку, у якому надаються зведені таблиці серйозних побічних реакцій з клінічних досліджень власника реєстраційного посвідчення, починаючи з міжнародної дати розробки до дати закриття бази даних поточного регулярного звіту. Власник реєстраційного посвідчення повинен надавати пояснення з приводу будь-якого невключення даних у таблиці (наприклад, дані клінічного дослідження можуть бути недоступними для препаратів, присутніх на ринку впродовж багатьох років). Таблиця(і) повинна бути структурованою за MedDRA на рівні SOC (впорядкованою згідно з міжнародними правилами) як для досліджуваного препарату, так і для груп(и) компаратора (активні компаратори, плацебо), що використовувалися у програмі клінічних досліджень. Дані можуть бути зібрані з усієї програми. Якщо доцільно та можливо, дані можуть бути розподілені по дослідженнях, показаннях, способах застосування чи за іншими параметрами. Цей підрозділ не повинен використовуватися для надання аналізу чи висновків на підставі серйозності побічних явищ.
При підготовці цього підрозділу потрібно враховувати наступне:
оцінка причинно-наслідкового зв’язку зазвичай корисна для оцінки індивідуальних випадків рідкісних побічних реакцій. Ця оцінка має меншу цінність для аналізу узагальнених даних, коли стає можливим групове порівняння частоти випадків. Тому у зведені таблиці потрібно включати усі серйозні побічні явища, а не лише серйозні побічні реакції досліджуваного препарату, компараторів та плацебо. Може бути доцільним надавати частоту відповідно до доз;
загалом, таблиці серйозних побічних явищ з клінічних досліджень повинні містити лише терміни, що використовувалися для оцінки випадку як серйозного, а несерйозні побічні явища повинні бути включені у звіти з дослідження;
таблиці повинні містити дані сліпих та відкритих клінічних досліджень. Інформація про серйозні побічні явища можуть походити із завершених досліджень та з індивідуальних випадків, що були виведені з сліпого дослідження з міркувань безпеки (наприклад, термінове повідомлення). Спонсори клінічних досліджень та власники реєстраційних посвідчень не повинні розкривати дані сліпих досліджень з метою підготовки регулярного звіту;
деякі побічні явища можуть бути виключені зі зведених таблиць клінічних досліджень, однак у звіті такі виключення необхідно обґрунтувати. Наприклад, побічні явища, які були визначені у протоколі як такі, що не підлягають (exempt) спеціальному збору та внесенню до бази даних з безпеки оскільки їх розвиток передбачався у популяції пацієнтів, а також ті, що представляють кінцеві точки клінічного дослідження (наприклад, повідомлення про смерть при дослідженні препарату для лікування застійної серцевої недостатності, коли смерть незалежно від її причини є первинною кінцевою точкою, чи прогресування захворювання у дослідженнях лікування раку).
Підрозділ 6.3. Кумулятивні та інтервальні зведені таблиці з джерел післяреєстраційних даних
Потрібно надати основні дані для додатку, у якому надаються кумулятивні та інтервальні зведені таблиці побічних реакцій, зібраних за період від міжнародної дати народження лікарського засобу до дати закриття бази даних поточного регулярного звіту. Ці побічні реакції, отримані зі спонтанних повідомлень, що включають повідомлення від працівників з медичною та фармацевтичною освітою, споживачів, з наукової літератури, від уповноважених органів (на глобальному рівні), а також обов’язкові повідомлення з неінтревенційних досліджень4. Дані щодо серйозних та несерйозних реакцій зі спонтанних джерел, а також серйозних побічних реакцій з неінтервенційних досліджень та інших неінтервенційних джерел, слід надавати в одній таблиці з інтервальними та кумулятивними поряд. Таблицю слід структурувати згідно з MedDRA на рівні SOC (впорядкованою згідно з міжнародними правилами). Дані зі специфічних проблем безпеки можуть бути представлені у додаткових таблицях з розподілом за показаннями, способом застосування препарату чи іншими параметрами. У цьому підрозділі регулярного звіту не потрібно надавати аналіз чи висновки на підставі даних зведених таблиць.
________________
4 ICH-E2D Управління даними з безпеки у післяреєстраційному періоді: визначення та стандарти для термінового повідомлення.
Розділ 7. Резюме значимих результатів клінічних досліджень протягом звітного періоду
Потрібно надати стислий опис клінічно важливих результатів з ефективності та безпеки лікарського засобу, отриманих за звітний період із спонсорованих власником реєстраційного посвідчення клінічних досліджень, з джерел, зазначених у підрозділах нижче. Якщо можливо і доцільно, дані слід розподілити за статтю та віком (особливо діти у порівнянні з дорослими), показанням, дозою та регіоном.
Сигнали, що походять з джерел клінічних досліджень, слід звести у таблиці в розділі 15 регулярного звіту («Огляд сигналів: нові, поточні чи закриті»). Оцінка сигналів, незалежно від того це спростований сигнал, потенційний або виявлений ризик, що були закриті за звітний період, повинні бути надані у розділі 16.2 регулярного звіту («Оцінка сигналу»). Нова інформація стосовно уже відомих потенційних чи виявлених ризиків, що не підпадає під поняття нового виявленого сигналу, повинна оцінюватися і характеризуватися у розділах 16.3 («Оцінка ризиків та нової інформації») і 16.4 («Характеристика ризиків»), відповідно.
Результати клінічних досліджень, що не спонсорувалися власником реєстраційного посвідчення повинні описуватися у відповідних розділах регулярного звіту.
Якщо інформація з клінічних досліджень про відсутність ефективності для лікування не загрозливих життю захворювань відповідно до зареєстрованих показань впливає на оцінку співвідношення користь/ризик, її необхідно коротко описати у цьому розділі.
Інформація щодо відсутності ефективності лікарського засобу, отримана із клінічних досліджень, у яких препарат використовувався для лікування та профілактики серйозних або загрозливих життю захворювань, повинна узагальнюватися у р. 13 (Відсутність ефективності у контрольованих клінічних дослідженнях).
Інформація з інших клінічних досліджень повинна бути включена у підрозділ 9.1 регулярного звіту.
Крім того, власник реєстраційного посвідчення повинен надати у додатку перелік спонсорованих ним за звітний період завершених та поточних післяреєстраційних інтервенційних досліджень, основною метою яких було виявити, характеризувати чи кількісно визначити загрозу безпеки або підтвердити профіль безпеки лікарського засобу інтервенційних досліджень.
Перелік повинен містити наступну інформацію по кожному дослідженню:
ідентифікатор дослідження (наприклад, номер протоколу чи інший ідентифікатор);
назва дослідження (абревіатурна назва дослідження, якщо застосовується);
вид дослідження (наприклад, рандомізоване клінічне дослідження, когорт не дослідження, дослідження випадок-контроль);
досліджувана популяція, включаючи країну та інші відповідні характеристики популяції (наприклад, педіатрична популяція, або дослідження пацієнтів з нирковою недостатністю);
дата початку (визначена власником реєстраційного посвідчення) і планована дата завершення;
статус: поточне (клінічне дослідження почалося) чи завершене (завершено написання звіту клінічного дослідження).
Підрозділ 7.1. Завершені клінічні дослідження
Потрібно надати короткий опис клінічно важливих результатів з ефективності та безпеки, отриманих з клінічних досліджень, завершених у звітний період. Ця інформація може бути представлена у вигляді опису чи синопсису. Він може включати інформацію, яка підтверджує або спростовує попередні виявлені проблеми безпеки, а також докази нових сигналів з безпеки.
Підрозділ 7.2. Поточні клінічні дослідження
Якщо власнику реєстраційного посвідчення відомі клінічно важливі результати ефективності та безпеки, виявлені у поточних клінічних дослідженнях (наприклад, при проміжному аналізі даних з безпеки або внаслідок виведення з сліпого дослідження пацієнтів з побічними явищами), їх необхідно коротко узагальнити у цьому підрозділі. Це може бути інформація, що підтверджує чи спростовує попередні ідентифіковані проблеми безпеки, а також підтверджує нові сигнали з безпеки.
Підрозділ 7.3. Тривале спостереження
Потрібно представити відому інформацію про тривале спостереження за суб’єктами клінічних досліджень, зокрема, високотехнологічних препаратів (наприклад, продуктів генної інженерії, клітинної терапії та тканинного інжинірингу).
Підрозділ 7.4. Інше терапевтичне застосування лікарського засобу
Повинна бути викладена клінічно важлива інформація з безпеки, виявлена в інших програмах, що проводилися власником реєстраційного посвідчення за спеціальним протоколом з обов’язковим поданням повідомлень про побічні реакції (наприклад, програми розширеного доступу, програми застосування незареєстрованого лікарського засобу при тяжких патологіях, програми застосування певними пацієнтами та інший організований збір даних).
Підрозділ 7.5. Нові дані з безпеки, пов’язані з фіксованим комбінованим лікуванням
Необхідно враховувати наступне:
якщо діюча речовина, що є предметом регулярного звіту, також зареєстрована чи розробляється як компонент фіксованого комбінованого препарату чи схеми комбінованої терапії, необхідно резюмувати важливі результати з безпеки комбінованої терапії;
якщо сам препарат, що є предметом регулярного звіту, є фіксованою комбінацією, необхідно узагальнити важливу інформацію з безпеки окремих його компонентів, як вже зареєстрованих, або так і тих, що знаходяться у розробці.
Дані, специфічні для комбінації можуть надаватися в окремому(их) розділі(ах) регулярного звіту по одному чи по всіх його компонентах.
Розділ 8. Результати неінтервенційних досліджень
Слід узагальнити відповідну інформацію з безпеки чи інформацію, що має потенційний вплив на оцінку співвідношення користь/ризик, отриману за звітний період із спонсорованих власником реєстраційного посвідчення неінтервенційних досліджень (наприклад, обсерваційні дослідження, епідеміологічні дослідження, реєстри та програми активного фармаконагляду). До даного розділу також слід включати відповідну інформацію з досліджень використання препаратів, якщо вона властива багатьом регіонам. Власник реєстраційного посвідчення повинен надати додаток з переліком спонсорованих ним неінтервенційних досліджень, головною метою яких було ідентифікувати, характеризувати або кількісно визначити загрозу безпеки, підтвердити профіль безпеки лікарського засобу або оцінити ефективність заходів з управління ризиками, що були завершені або продовжувалися протягом звітного періоду.
Заключні звіти досліджень, завершених у звітний період, для досліджень, зазначених у пункті вище, слід також включити до регіонального додатку регулярного звіту(див. розділ VII.B.5.20. та розділ VII.С.5.4.). У цьому пункті можна також надавати підсумкову інформацію, що ґрунтується на оцінці сукупних даних, згенерованих з програм підтримки пацієнтів, якщо вони не надавалися в інших розділах регулярного звіту. Що стосується інших інформаційних джерел, то власник реєстраційного посвідчення повинен надавати сигнали і ризики, виявлені з таких джерел, у відповідних розділах регулярного звіту.
Розділ 9. Інформація з інших клінічних досліджень та джерел
Підрозділ 9.1 Інші клінічні випробування
Потрібно узагальнити інформацію, що стосується оцінки співвідношення користь/ризик лікарського засобу, доступну з інших джерел клінічних випробувань/джерел дослідження, що стала доступною власнику реєстраційного посвідчення під час звітного періоду (наприклад, результати узагальненого або мета-аналізу рандомізованих клінічних випробувань, інформація з безпеки, надана партнерами або ініціаторами досліджень).
Підрозділ 9.2 Помилки, пов’язані з лікарським засобом
Потрібно узагальнити інформацію про помилки, пов’язані з лікарським засобом, і потенційні помилки, навіть якщо вони не пов’язані з розвитком небажаних ефектів. Потенційні помилки, пов’язані з лікарським засобом — це розпізнавання обставин, що можуть призводити до помилок, пов’язаних з лікарським засобом і можуть включати пацієнта, а можуть і не включати. Помилки, пов’язані з лікарським засобом, можуть виникати на будь-якому етапі процесу медичного застосування і можуть включати пацієнтів, споживачів, працівників з медичною чи фармацевтичною освітою Ця інформація може стосуватися інтерпретації даних з безпеки чи загальної оцінки співвідношення користь/ризик лікарського засобу.
Розділ 10. Неклінічні дані
Потрібно узагальнити основні результати з безпеки з неклінічних in vivo та in vitro досліджень (наприклад, дослідження канцерогенності, репродуктивності та імунотоксичності), поточних чи завершених у звітний період. Результати з досліджень, що проводилися з метою вивчення специфічних проблем безпеки, повинні включатися в регулярному звіті, незалежно від наслідків. Наслідки цих результатів повинні обговорюватися у відповідних розділах регулярного звіту.
Розділ 11 Література
Потрібно відобразити резюме нових та важливих даних з безпеки, що стосуються лікарського засобу, опублікованих в спеціальній медичній літературі або доступних у вигляді неопублікованих рукописів, про які стало відомо власнику реєстраційного посвідчення у звітний період.
Літературний пошук для регулярного звіту були повинен бути ширшим, ніж у випадку пошуку випадків побічних реакцій, оскільки він повинен також включати дані з безпеки з досліджень для груп пацієнтів та для інших препаратів, що містять однакову діючу речовину. Наприклад, інформація з безпеки, що повинна бути включена до регулярного звіту, але яку можна не знайти шляхом літературного пошуку випадків побічних реакцій:
наслідки вагітності (включаючи її завершення) без несприятливих наслідків;
застосування у педіатричних популяціях;
застосування незареєстрованого препарату у тяжко хворих пацієнтів, чи застосування поіменованими пацієнтами;
відсутність ефективності;
безсимптомне передозування, зловживання або неправильне застосування;
лікопов’язана помилка без виникнення побічних подій;
важливі неклінічні результати з безпеки.
Якщо доцільно, слід розглянути інформацію про інші діючі речовини того ж класу.
Розділ 12. Інші регулярні звіти
Заповнюється тільки для фіксованих комбінованих препаратів чи лікарських засобів з багатьма показаннями та/або формами випуску у випадках, коли за погодженням з уповноваженим органом готується декілька регулярних звітів, тоді у цьому розділі також необхідно зазначити значні результати з інших регулярних звітів, якщо вони не представлені в інших розділах звіту. Керуючись контрактними домовленостями, власник реєстраційного посвідчення повинен узагальнити важливі дані з регулярних звітів, надані у звітний період іншими сторонами (наприклад, спонсорами або іншими контрактними партнерами), якщо такі є доступними.
Розділ 13. Відсутність ефективності в контрольованих клінічних дослідженнях
Потрібно резюмувати дані клінічних досліджень, що вказують на відсутність ефективності, чи відсутність ефективності, пов’язану із обраним видом терапії, для препаратів, призначених для лікування і профілактики серйозних чи загрозливих життю захворювань, що можуть відображати значний ризик для лікованої популяції.
Розділ 14 Інформація, отримана в останній момент
Необхідно узагальнити потенційно важливі дані з безпеки, ефекту та ефективності препарату, про які йому стало відомо після дати закриття бази даних, але під час підготовки регулярного звіту. Наприклад, клінічно значимі нові публікації, важливі дані спостережень, клінічні токсикологічні дані та будь-які заходи, вжиті власником реєстраційного посвідчення, комітетом з моніторингу даних або уповноваженим органом (на міжнародному рівні) з міркувань безпеки. Нові повідомлення про випадки побічних реакцій не слід рутинно включати у регулярний звіт, крім важливих репрезентативних випадків (тобто, перший приклад важливого явища) або даних щодо важливих сигналів з безпеки коли вони можуть надати додаткову інформацію для оцінки проблем безпеки, що вже представлені у регулярному звіті. Потрібно також включати усі значні зміни до довідкової інформації лікарського засобу (наприклад, нова побічна реакція, застереження або протипоказання), внесені у цей період.
Представлені дані потрібно також враховувати при оцінці ризиків та нової інформації.
Розділ 15 Огляд сигналів: нові, поточні та закриті
Надається загальний огляд сигналів, що були закриті (тобто, їх оцінка завершена) протягом звітного періоду, а також поточних сигналів, оцінка яких ще продовжувалася на момент завершення звітного періоду. У регулярному звіті сигнал повинен включатися одразу як тільки він пройшов початковий скринінг чи був класифікований, і власник реєстраційного посвідчення вирішив проводити його подальшу оцінку. Сигнали можуть бути якісними чи кількісним. Джерелами сигналів можуть бути інформаційні запити чи розслідування питання безпеки уповноваженими органами в усьому світі. Рішення стосовно класифікації цих сигналів і висновки з їх оцінки вимагають медичного судження і наукової інтерпретації доступних даних. Новим сигналом вважається сигнал, виявлений у звітний період. Якщо у звітний період стає відомою нова клінічно значима інформація стосовно раніше закритого сигналу, вона буде новим сигналом на підставі того, що новий аспект попередньо спростованого сигналу або визнаного ризику потребує подальших кроків для його верифікації. Залежно від стадії оцінки сигналу на момент завершення звітного періоду регулярного звіту, новий сигнал може бути класифікований як закритий чи поточний.
У цьому розділі або у додатку надаються у вигляді таблиці усі сигнали поточні чи закриті на момент завершення звітного періоду. Ця таблиця повинна включати наступну інформацію: короткий опис сигналу; дата коли стало відомо про сигнал; статус сигналу на момент завершення звітного періоду (закритий чи поточний); дата закриття сигналу, якщо сигнал закритий; джерело сигналу; короткий опис ключових даних; плани щодо подальшої оцінки; і вжиті чи заплановані заходи. Не потрібно надавати детальну оцінку закритих сигналів.
Оцінку нової інформації, пов’язаної з будь-якими раніше відомими виявленими і потенційними ризиками, яка не вважається новим сигналом, слід надавати у підрозділі 16.3 регулярного звіту («Оцінка ризиків та нової інформації»).
Якщо уповноважений орган (на міжнародному рівні) вимагав, щоб специфічне питання, що не вважається сигналом, моніторувалося та про нього звітували у регулярний звіт, необхідно узагальнити результати аналізу у цьому розділі якщо вони негативні. У випадку, якщо специфічне питання стає сигналом, воно повинно бути включене у таблицю сигналів і проаналізоване у підрозділі 16.2 («Оцінка сигналів»).
Потрібно також включати усі значні зміни до довідкової інформації лікарського засобу (наприклад, нова побічна реакція, застереження або протипоказання), внесені у цей період (див. розділ 4). Представлені у цьому розділі дані потрібно також враховувати при оцінці ризиків та нової інформації (див. підрозділ 16.3).
Розділ 16 Оцінка сигналів та ризиків
Необхідно надати: короткий опис того, що було відомо про важливі виявлені і потенційні ризики і відсутню інформацію на початок звітного інтервалу, що охоплює регулярний звіт; оцінку усіх сигналів закритих протягом звітного періоду; оцінку нової інформації стосовно раніше ідентифікованих і потенційних ризиків; оновлену характеристику важливих потенційних і ідентифікованих ризиків; резюме ефективності заходів з мінімізації ризику в будь-якій країні чи регіоні, що можуть корисними в інших країнах чи регіонах. Інформація у наведених нижче підрозділах не повинна дублювати інформацію, представлену у попередніх розділах, а повинна надавати інтерпретацію та критичну оцінку інформації з метою характеристики профілю ризиків, що були оцінені як важливі. Немає необхідності включати описи випадків у розділи регулярного звіту для оцінки ризиків. Проте, якщо вони є частиною наукового аналізу сигналу або ризику, необхідно надати клінічну оцінку важливих або ілюстративних випадків.
Підрозділ 16.1 Резюме проблем безпеки
Надається резюме важливих питань з безпеки, відомих на початку звітного періоду, щодо яких можна зробити оцінку нової інформації. Для лікарських засобів з існуючою специфікацією з безпеки цей розділ буде або таким як резюме специфікацій з безпеки дійсного на початок звітного періоду регулярного звіту, або походитиме з нього. Слід надати інформацію про важливі виявлені та потенційні ризики, та важливу відсутню інформацію.
Для лікарських засобів без специфікації з безпеки необхідно надати інформацію про важливі виявлені та потенційні ризики і важливу відсутню інформацію, пов’язані з їх застосуванням, на підставі до- та післяреєстраційного досвіду.
Підрозділ 16.2 Оцінка сигналу
Необхідно узагальнити результати оцінки усіх сигналів з безпеки, закритих у звітний період, незалежно від того, чи були вони класифіковані як важливі чи ні. Сигнал з безпеки після його оцінки може бути закритий або тому, що він спростований, або тому, що визначений як потенційний чи ідентифікований ризик. Необхідно включити наступні дві головні категорії сигналів та надати достатньо детальний опис оцінки кожного сигналу з обох категорій закритих сигналів:
1) сигнали, які після їх оцінки були спростовані як помилкові на підставі наукової оцінки наявної на даний момент інформації;
2) сигнали, які після оцінки були класифіковані як потенційний або виявлений ризик, включаючи відсутність ефективності.
Якщо для закритих сигналів обох категорій використовуються різні оцінки, вони можуть бути представлені у наступному порядку:
закриті та спростовані сигнал;
закриті сигнали, класифіковані як важливі потенційні ризики;
закриті сигнали, класифіковані як важливі виявлені ризики;
закриті сигнали, що є потенційними ризиками, не класифіковані як важливі;
закриті сигнали, що є виявленими ризиками, не класифіковані як важливі.
Де доцільно, оцінку закритих сигналів можна представити за показанням або популяцією.
Описи оцінки сигналів можуть бути включені у цей підрозділ регулярного звіту або у додаток до регулярного звіту. Кожна оцінка повинна містити наступну інформацію, залежно від ситуації:
джерело або тригер сигналу;
основні дані, важливі для оцінки;
метод(и) оцінки, включаючи джерела даних, критерії пошуку (якщо можливо, з застосуванням спеціальних MedDRA термінів (наприклад, на рівні PT, HLT, SOC і т.п.) або перегляд стандартизованих запитів (SMQs) MedDRA) та аналітичні підходи;
результати — резюме та критичний аналіз даних, що розглядаються при оцінці сигналу; можуть містити опис серії випадків або окремого випадку (наприклад, випадок детально документованого агранулоцитозу або синдрому Стівенса-Джонсона), якщо вони є частиною оцінки;
обговорення;
висновок.
Необхідно надати оцінку та висновки власника реєстраційного посвідчення щодо спростованих сигналів.
Підрозділ 16.3. Оцінка ризиків та нової інформації
Необхідно надати критичну оцінку новій інформації, пов’язаній з раніше визнаними ризиками, що не була включена у підрозділ 16.2.
Нову інформацію, що є сигналом для раніше визнаного ризику або для раніше спростованого сигналу, слід представити у таблиці сигналів і оцінити у підрозділі 16.2, якщо сигнал був також закритий протягом звітного періоду.
Слід включити оновлену інформацію, що стосується раніше визнаного ризику, і не становить сигнал. Нову інформацію можна представити у такому вигляді:
1. Нова інформація щодо важливих потенційних ризиків.
2. Нова інформація щодо важливих виявлених ризиків.
3. Нова інформація щодо інших потенційних ризиків, що не класифіковані як важливі.
4. Нова інформація щодо інших виявлених ризиків, що не класифіковані як важливі.
5. Оновлені дані щодо важливої відсутньої інформації.
При оцінці нової інформації основна увага повинна приділятися новій інформації, що стала відомою у звітний період, така оцінка складатиме основу для оновленої характеристики важливих потенційних та виявлених ризиків у підрозділі 16.4. Рівень деталізації оцінки, включеної у цей підрозділ, має бути пропорційним наявним доказам ризику і його медичній важливості та впливу на охорону здоров’я.
Оцінка нової інформації та оновленої відсутньої інформації може входити до цього підрозділу або надаватися у додатку. Кожна оцінка повинна включала наступну відповідну інформацію:
джерело нової інформації;
основні дані, важливі для оцінки;
метод(и) оцінки, включаючи джерела даних, критерії пошуку та аналітичні підходи;
результати — резюме та критичний аналіз даних, що розглядаються при оцінці ризику;
обговорення;
висновок, в тому числі й про те впливає чи ні дана оцінка на оновлення характеристик будь-яких важливих та виявлених ризиків, зазначених у підрозділі 16.4 («Характеристика ризиків»).
Слід критично оцінити будь-яку нову інформацію щодо популяцій, які застосовують лікарський засіб, або даних, згенерованих для аналізу раніше відсутньої інформації. Слід також описати наявні невирішені проблеми та непевності.
Підрозділ 16.4 Характеристика ризиків
Слід на основі узагальнених даних, не обмежених звітним періодом, охарактеризувати важливі виявлені та потенційні ризики та описати важливу відсутню інформацію.
Якщо відсутня інформація становить важливий ризик, то вона повинна розглядатися як проблема безпеки. Необхідно також проаналізувати обмеження баз даних з безпеки (стосовно кількості досліджуваних пацієнтів, кумулятивної експозиції, тривалого застосування і т.п.).
У регулярних звітах, що складаються для лікарських засобів з кількома показаннями, формами випуску, способами застосування, коли можуть мати місце значні розбіжності у виявлених та потенційних ризиках, може бути доцільним представити ризики за показаннями, формами випуску або способами застосування.
Підрозділ 16.5 Ефективність заходів з мінімізації ризиків (якщо застосовувалися)
Слід надати результати оцінки ефективності заходів з мінімізації ризиків, що стосуються оцінки користь/ризик, узагальнити усю доступну за звітний період інформацію щодо результативності та/або обмежень специфічних заходів з мінімізації важливих виявлених ризиків у будь-якій країні чи регіоні.
Якщо доречно і можливо, інформацію можна узагальнити по регіонах.
Якщо вимагається надання результатів оцінок ефективності заходів з мінімізації ризиків у звітний період, що стосуються окремого регіону, вони повинні надатися у регіональному додатку до регулярного звіту.
Розділ 17 Оцінка користі
Підрозділ 17.1. Важлива базова інформація з ефективності лікарського засобу
Узагальнюється інформація з ефекту й ефективності лікарського засобу, відома на початку звітного періоду, що становить базис для оцінки користі. Ця інформація повинна відповідати зареєстрованими показаннями лікарського засобу, що містяться у довідковій інформації про лікарський засіб.
Для лікарських засобів з різними показаннями, популяціями та/або способами застосування, якщо доцільно, користь лікарського засобу слід характеризувати окремо за цими факторами. Якщо не було значних змін у профілях користі та ризику лікарського засобу у звітний період, резюме має бути коротким; переважно це має бути зміст довідкової інформації або має входити до показань, що перераховуються у вступній частині регулярного звіту. Рівень деталізації інформації, що надається у цьому підрозділі регулярного звіту, повинен бути достатнім для підтвердження характеристики користі у підрозділі регулярного звіту («Характеристика користі») і оцінки співвідношення користь/ризик у розділі 18 («Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань»).
Підрозділ 17.2 Нова виявлена інформація з ефективності
Слід представити додаткову інформацію з ефекту та ефективності для зареєстрованих показань, яка з’явилася протягом звітного періоду. Також слід надати нову інформацію з ефекту і ефективності для зареєстрованих показань щодо умов фактичного (практичного) застосування, якщо вона доступна. Не слід надавати нову інформацію з ефекту і ефективності при застосуванні за незареєстрованими показаннями, крім випадків, коли вона є важливою для оцінки співвідношення користь/ризик препарату за зареєстрованими показаннями. Слід також представити нову інформацію щодо показань, зареєстрованих у звітний період. Рівень деталізації інформації, що надається у цьому підрозділі регулярного звіту, повинен бути достатнім для підтвердження характеристики користі і оцінки співвідношення користь/ризик.
Особливу увагу слід приділити вакцинам, антиінфекційним та іншим лікарським засобам, для яких зміни терапевтичних умов можуть з часом вплинути на їх ефект/ефективність
Підрозділ 17.3. Характеристика користі
Надається сукупна базова та нова інформація з користі для зареєстрованих показань, що стала доступною у звітний період. Рівень деталізації інформації повинен бути достатнім для забезпечення оцінки співвідношення користь/ризик. Якщо немає нових важливих даних з користі, слід надати характеристику інформації, представленої у підрозділі 17.1 регулярного звіту. Якщо є нова позитивна інформація з користі але немає значних змін у профілі ризику препарату у цей звітний період, сукупна базова та нова інформація, повинна надаватися досить стисло.
У цьому підрозділі слід надати коротку, але критичну оцінку сильних сторін та обмежень доказів ефекту та ефективності препарату.
Розділ 18 Інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань
Надається загальна оцінка користі та ризику лікарського засобу при його застосуванні у клінічній практиці. При цьому не слід дублювати інформацію з користі та ризику, представлену в розділах 16.2 («Оцінка сигналу»), 16.3 («Оцінка ризиків та нової інформації») та 17.3 («Характеристика користі»), а надати критичний аналіз та узагальнену ключову інформацію з користі та ризику із зазначених розділів.
Підрозділ 18.1. Контекст користь/ризик — медична необхідність та важливі альтернативи
Надається короткий опис медичної потреби в лікарському засобі за зареєстрованими показаннями та резюме альтернатив (медичних, хірургічних або інших, включаючи відсутність лікування).
18.2. Оцінка аналізу користь/ризик
Слід оцінювати та представляти профіль користь/ризик за кожним показанням окремо. Якщо в межах одного показання існують важливі розбіжності у профілі користь/ризик серед різних популяцій, оцінку користь/ризик слід представити для кожної популяції, якщо це можливо.
Оцінку користь/ризик слід представити і проаналізувати таким чином, щоб забезпечити порівняння користі і ризиків. При оцінці необхідно зазначити ключові переваги та ризики Необхідно узагальнити ключову інформацію з користі та ризиків, представлену у попередніх розділах/підрозділах. Слід врахувати обставини застосування лікарського засобу. Розглянути величину ефекту, всі елементи користі. Слід розглянути клінічну значимість ризику та умови його виявлення.
Необхідно надати чітке пояснення методології та обґрунтування, що використовувались для оцінки користь/ризик.
У випадку, якщо існує важлива нова інформація або вимагається надання спеціального регулярному звіті, детальний аналіз користь/ризик повинен ґрунтуватися на кумулятивних даних. І навпаки, якщо протягом звітного періоду отримано небагато нової інформації, першочергова увага при оцінці користь/ризик може приділятися оцінці оновлених даних з безпеки за звітний період.
Розділ 19 Висновки та заходи
Слід зробити висновки щодо будь-якої нової інформації, отриманої протягом звітного періоду, відносно загальної оцінки користь/ризик препарату для кожного зареєстрованого показання, а також відповідних підгруп пацієнтів. Слід оцінити необхідність внесення змін до довідкової інформації і запропонувати зміни, якщо вони необхідні. У разі доцільності слід включити попередні рекомендації щодо оптимізації чи подальшої оцінки балансу користь/ризик для подальшого їх обговорення з відповідним уповноваженим органом. Також можуть бути включені пропозиції щодо додаткових заходів з мінімізації ризиків. Якщо для лікарського засобу розроблений план з фармаконагляду чи план управління ризиками, необхідно також обговорити пропозиції щодо них. Спираючись на оцінку зведених даних з безпеки та аналіз співвідношення користь/ризик, надається висновок щодо необхідності змін та/або заходів, включаючи зміни до затвердженої короткої характеристики лікарського засобу.
20. Додатки до регулярного звіту
1. Довідкова інформація (див. розділ 4).
2. Зведені таблиці серйозних побічних явищ з клінічних досліджень; зведені та проміжні таблиці серйозних і несерйозних побічних реакцій з післяреєстраційних джерел даних.
3. Зведена таблиця сигналів з безпеки (якщо не включена до змісту відповідного розділу регулярного звіту)5.
4. Перелік всіх інтервенційних і неінтервенційних досліджень, спонсорованих власником реєстраційного посвідчення, головною метою яких є ідентифікація, характеристика чи кількісне визначення загрози безпеці або підтвердження профілю безпеки лікарського засобу.
5. Перелік джерел інформації, що використовувалися для підготовки регулярного звіту (за бажанням заявника).
6. Регіональний/локальний додаток.
__________________
5 Зведену таблицю сигналів з безпеки рекомендується включати у змісту відповідного розділу регулярного звіту.
до Порядку здійснення фармаконагляду
СТРУКТУРА
протоколу, заключного звіту та резюме післяреєстраційного дослідження з безпеки
Структура протоколу
1. Назва: інформативна назва, що містить загальновживаний термін дизайну дослідження, і досліджуваний лікарський засіб, діючу речовину або АТС код; і підзаголовок з ідентифікатором версії та датою останньої версії.
2. Власник реєстраційного посвідчення.
3. Відповідальні сторони включно з переліком усіх партнерських установ та відповідних баз дослідження.
4. Резюме: резюме протоколу дослідження, що містить наступні підрозділи:
4.1. Заголовок з підзаголовками, включаючи версію та дату протоколу, а також прізвище та назва організації головного автора.
4.2. Обґрунтування та історія питання.
4.3. Предмет дослідження та цілі.
4.4. Дизайн дослідження.
4.5. Досліджувана популяція.
4.6. Змінні.
4.7. Джерела даних.
4.8. Масштаб дослідження.
4.9. Аналіз даних.
4.10. Основні етапи.
5. Зміни та оновлення: будь-яка значна зміна та оновлення до протоколу дослідження після початку збору даних, включаючи обґрунтування кожної зміни або оновлення, дату кожної зміни та посилання на розділ протоколу, до якого було внесено зміну.
6. Основні етапи: таблиця із запланованими датами для наступних основних етапів:
6.1. Початок збору даних.
6.2. Завершення збору даних.
6.3. Звіт(и) про хід дослідження.
6.4. Проміжний звіт(и) результатів дослідження, якщо застосовно.
6.5. Заключний звіт за результатами дослідження.
7. Обґрунтування та історія питання: короткий опис загроз(и) безпеці, профілю безпеки або заходів з управління ризиками, які зумовили вимогу дослідження у якості зобов’язання.
8. Предмет та цілі дослідження у відповідності з рішенням компетентного органу, що призначив дослідження у якості зобов’язання.
9. Методи дослідження: опис методів дослідження, включаючи:
9.1. Дизайн дослідження.
9.2. Параметри дослідження: досліджувана популяція щодо осіб, місця, періоду часу та критеріїв відбору, включаючи обґрунтування будь-яких критеріїв включення та виключення до вибірки та їх вплив на кількість осіб, доступних для аналізу. Якщо проводиться відбір з визначеної популяції, має бути наданий опис визначеної популяції та детальний опис методики формування вибірки. Якщо дизайн дослідження — систематичний огляд або мета-аналіз, повинні бути надані пояснення критеріїв вибору та правомірності досліджень.
9.3. Змінні.
9.4. Джерела даних: стратегії та джерела даних для визначення експозиції, результатів та всіх інших змінних, пов’язаних з цілями дослідження. Якщо дослідження буде використовувати існуюче джерело даних, таке як електрона медична документація, слід зазначити будь-яку інформацію щодо валідності записів та кодування даних. У випадку систематичного огляду або мета-аналізу слід описати стратегію та процедури пошуку, а також будь-які методи для підтвердження даних, отриманих від дослідників.
9.5. Масштаб дослідження: будь-який запланований масштаб дослідження, точність оцінок дослідження та будь-який підрахунок розміру вибірки, який як мінімум здатен виявити попередньо визначений ризик з попередньо заданою статистичною точністю.
9.6. Управління даними.
9.7. Аналіз даних.
9.8. Контроль якості.
9.9. Обмеження методів дослідження.
10. Захист досліджуваних: заходи з безпеки з метою дотримання національних вимог щодо гарантування захисту прав учасників неінтервенційних післяреєстраційних досліджень безпеки.
11. Обробка та звітування про побічні явища/ побічні реакції та інші медично важливі явища під час проведення дослідження.
12. Плани щодо розповсюдження та комунікації результатів дослідження.
13. Посилання.
Структура резюме заключного звіту
1. Заголовок та підзаголовки, включаючи дату резюме та прізвище і місце роботи головного автора.
2. Ключові слова (не більше п’яти ключових слів, що дають основну характеристику дослідження).
3. Обґрунтування та історія питання.
4. Предмет дослідження та цілі.
5. Дизайн дослідження.
6. Параметри дослідження.
7. Суб’єкти та масштаб дослідження, включаючи вибулих учасників.
8. Змінні та джерела даних.
9. Результати.
10. Обговорення (включаючи, якщо можливо, оцінку впливу результатів дослідження на співвідношення ризик-користь препарату).
11. Власник реєстраційного посвідчення.
12. Прізвища та місця роботи основних дослідників.
Структура заключного звіту
1. Назва: назва, що містить загальновживаний термін дизайну дослідження; підзаголовки з датою заключного звіту, прізвищем та місцем роботи головного автора.
2. Резюме: окреме стисле резюме у форматі, представлене розділі 2 цього додатку.
3. Власник реєстраційного посвідчення: назва та адреса власника реєстраційного посвідчення.
4. Дослідники: прізвища, посади, вчені ступені, адреси та місця роботи усіх основних дослідників і співдослідників, перелік усіх партнерських установ та відповідних дослідницьких баз.
5. Основні етапи: заплановані та фактичні дати для наступних основних етапів:
5.1. Початок збору даних (запланована і фактична дати).
5.2. Завершення збору даних (запланована і фактична дати).
5.3. Звіт(и) про хід дослідження.
5.4. Проміжний звіт(и) результатів дослідження, якщо необхідно.
5.5. Заключний звіт про результати дослідження.
5.6. Будь-які інші важливі етапи, що стосуються дослідження, включаючи дату реєстрації протоколу у електронному реєстрі досліджень.
6. Обґрунтування та історія питання: опис проблем безпеки, що призвели до ініціювання дослідження, та критичний аналіз важливих опублікованих та неопублікованих відповідних даних і відсутньої інформації, яку дослідження призначене заповнити.
7. Предмет дослідження та цілі: предмет та цілі дослідження, включаючи будь-які попередньо висунуті гіпотези, як зазначено у протоколі дослідження.
8. Зміни та оновлення до протоколу: перелік будь-яких значних змін та оновлень до початкового протоколу дослідження після початку збору даних, включаючи обґрунтування кожної зміни або оновлення.
9. Методи дослідження:
9.1. Дизайн дослідження: основні елементи дизайну дослідження та обґрунтування його вибору.
9.2. Параметри дослідження: місце проведення та відповідні дати дослідження, включаючи періоди набору пацієнтів, спостереження та збору даних. У випадку, якщо за дизайном дослідження є систематичним оглядом або мета-аналізом, характеристики дослідження використовуються як критерії відбору, з обґрунтуванням.
9.3. Суб’єкти дослідження: будь-яка первинна популяція та критерії відбору суб’єктів у дослідження. Необхідно надати джерела та методи відбору учасників, включаючи, якщо необхідно, методи підтвердження випадку, а також кількість та причини вибулих учасників дослідження.
9.4.Змінні: усі результати, експозиції, прогностичні параметри, потенційні систематичні помилки та модифікатори ефекту, включаючи робочі визначення та діагностичні критерії, якщо вони застосовуються.
9.5. Джерела даних та вимірювання: для кожної змінної, що представляє інтерес, джерела даних та детальний опис методів оцінки та вимірювання. Якщо дослідження використовує існуюче джерело даних, таке як електронна медична документація, слід повідомляти будь-яку інформацію щодо достовірності записів та кодування даних. У випадку систематичного огляду або мета-аналізу потрібно надати опис усіх інформаційних джерел, стратегії пошуку, методів відбору досліджень, методів добування даних та будь-яких процесів для отримання або підтвердження даних від дослідників.
9.6. Систематичні помилки.
9.7. Масштаб дослідження: масштаб дослідження, обґрунтування будь-якого підрахунку розміру вибірки та будь-якого методу досягнення запланованого розміру дослідження.
9.8. Перетворення даних: перетворення, підрахунок або оперування даними, включаючи, яким чином оброблялись кількісні дані при аналізі, яке групування було вибрано та чому.
9.9. Статистичні методи: опис:
– основних підсумкових показників;
– статистичних методів, що застосовувалися;
– будь-яких методів, що використовувалися для вивчення підгруп та взаємодій;
– яким чином вирішувалась проблема відсутніх даних;
– будь-яких аналізів чутливості;
– будь-яких змін до плану аналізу даних, включеного до протоколу дослідження з обґрунтуванням змін.
9.10. Контроль якості: механізми гарантії якості та цілісності даних.
10. Результати: складається з наступних підрозділів:
10.1. Учасники: кількість суб’єктів на кожному етапі дослідження; у випадку систематичного огляду або мета-аналізу — кількість відібраних досліджень, оцінених на придатність та включених у дослідження, із зазначенням причин виключення на кожному етапі.
10.2. Описові дані: характеристика учасників дослідження, інформація щодо експозиції та потенційних систематичних помилок та кількість учасників з відсутніми даними. У випадку систематичного огляду або мета-аналізу, характеристика кожного дослідження, з якого були взяті дані.
10.3. Дані про результати: кількість учасників по категоріях основних результатів.
10.4. Основні результати: нескореговані та, якщо необхідно, скореговані показники та їх точність. Якщо доцільно, потрібно розрахункові показники відносного ризику трансформувати в абсолютний ризик за значимий період часу.
10.5. Інші розрахунки.
10.6. Побічні явища та побічні реакції.
11. Обговорення:
11.1. Основні результати: основні результати з посиланням на цілі дослідження, попереднє дослідження на підтримку та на противагу результатам завершеного післяреєстраційного дослідження безпеки та, у відповідних випадках, вплив результатів на співвідношення ризик-користь лікарського засобу.
11.2. Обмеження: обмеження дослідження, враховуючи обставини, які могли вплинути на якість або цілісність даних, обмеження підходу і методів дослідження, джерела потенційних систематичних помилок та неточностей і оцінка явищ. Необхідно описати тенденцію та масштаб потенційних систематичних помилок.
11.3. Інтерпретація: інтерпретація результатів з урахуванням цілей, обмежень, складності аналізу, результатів подібних досліджень та інших відповідних доказів.
11.4. Узагальнення.
12. Посилання.
ВИМОГИ
до визначення та перегляду Періодичності подання регулярно оновлюваних звітів з безпеки лікарських засобів за міжнародною непатентованою назвою активного фармацевтичного інгредієнта або комбінацій активних фармацевтичних інгредієнтів
1. Періодичність подання регулярно оновлюваних звітів з безпеки лікарських засобів за міжнародною непатентованою назвою активного фармацевтичного інгредієнта або комбінацій активних фармацевтичних інгредієнтів (далі — Періодичність подання регулярних звітів за МНН АФІ або комбінацій АФІ) складається і переглядається Державним підприємством «Державний експертний центр Міністерства охорони здоров’я України» (далі — Центр).
2. Періодичність подання регулярних звітів за МНН АФІ або комбінацій АФІ оприлюднюється на веб-сайті Центру.
3. Періодичність подання регулярних звітів за МНН АФІ або комбінацій АФІ містить таку інформацію:
1) міжнародна непатентована назва (далі — МНН) активного фармацевтичного інгредієнта (далі — АФІ) або комбінацій АФІ;
2) частота подання регулярних звітів;
3) кінцева дата для включення даних до регулярного звіту за певний період;
4) кінцева дата подання регулярних звітів;
5) дата публікації.
4. Критерії визначення Періодичності подання регулярних звітів за МНН АФІ чи комбінації АФІ:
1) інформація щодо ризиків чи користі, що може вплинути на здоров’я та життя пацієнтів;
2) новий лікарський засіб, для якого на даний момент існує обмежена інформація з безпеки (включаючи до- та післяреєстраційний досвід);
3) значні зміни лікарського засобу (наприклад, зареєстровано нове показання, нова форма випуску чи спосіб застосування, що розширюють популяцію пацієнтів, які застосовують лікарський засіб);
4) вразливі/недостатньо вивчені популяції пацієнтів, відсутня інформація (наприклад, діти, вагітні жінки), коли такі популяції пацієнтів ймовірно будуть застосовувати лікарський засіб у післяреєстраційному періоді;
5) сигнал про/або потенційна загроза неправильного застосування, лікопов’язаної помилки, ризик передозування або залежності від лікарського засобу;
6) наявність даних з безпеки та застосування лікарського засобу;
7) лікарські засоби, що підлягають додатковому моніторингу.
5. Критерії перегляду Періодичності подання регулярних звітів за МНН АФІ або комбінацій АФІ:
1) поява/виявлення нової інформації, що може вплинути на співвідношення користь/ризик АФІ чи комбінації АФІ та потенційно на здоров’я населення чи суспільне здоров’я;
2) будь-яка зміна критеріїв, що використовується для визначення періодичності подання регулярних звітів;
3) на вимогу заявника з причин, що стосуються здоров’я населення чи суспільного здоров’я; з метою уникнення дублювання оцінки; з метою досягнення міжнародної гармонізації;
4) новий зареєстрований АФІ.
6. У разі необхідності, заявники подають заяви на відповідні зміни до реєстраційних матеріалів протягом дії реєстраційного посвідчення у визначеному законодавством порядку, крім випадків, коли реєстраційне посвідчення містить пряме посилання на Періодичність подання регулярних звітів за МНН АФІ або комбінацій АФІ.
* — Для лікарських засобів з АФІ або комбінаціями АФІ не зазначених у цьому Додатку, строки подання регулярних звітів за МНН АФІ або комбінацій АФІ повинні відповідати стандартному графіку подання регулярних звітів для кожного лікарського засобу, починаючи з його міжнародної дати народження.
** — Для лікарських засобів, які були зареєстровані в Україні до 2012 р., періодичність надання регулярних звітів — згідно з цим Додатком, для лікарських засобів, які були зареєстровані в Україні як у першій країні світу, після 2012 р., періодичність надання регулярних звітів відповідно до стандартного графіка для кожного лікарського засобу, але не рідше, ніж зазначено у цьому Додатку.
АНАЛІЗ РЕГУЛЯТОРНОГО ВПЛИВУ
до проекту наказу Міністерства охорони здоров’я України «Про внесення змін до деяких наказів Міністерства охорони здоров’я України»
I. Визначення проблеми
Відповідно до законів України «Про лікарські засоби», «Про захист населення від інфекційних хвороб», «Про забезпечення санітарного та епідемічного благополуччя населення», «Про загальнодержавну програму адаптації законодавства України до законодавства Європейського Союзу», Порядку державної реєстрації (перереєстрації) лікарських засобів і Розмірів збору за державну реєстрацію (перереєстрацію) лікарських засобів, затвердженого постановою Кабінету Міністрів України від 26 травня 2005 року № 376, суб’єкт господарювання, що здійснює державну реєстрацію лікарського засобу (далі — заявник), несе відповідальність за ефективність, безпеку та якість зареєстрованого лікарського засобу, а управління у сфері створення, виробництва, контролю якості та реалізації лікарських засобів щодо безпеки та ефективності дозволених до застосування лікарських засобів здійснює центральний орган виконавчої влади, що забезпечує формування державної політики у сфері охорони здоров’я, оскільки на момент видачі реєстраційного посвідчення інформація про безпеку лікарського засобу досить обмежена. Це пов’язано з багатьма чинниками, зокрема, з відносно невеликою кількістю учасників клінічних випробувань, популяція яких обмежена за віком, статтю, етнічною приналежністю, супутніми захворюваннями, а також з обмеженим досвідом застосування супутніх лікарських засобів, обмеженими умовами застосування, порівняно нетривалим періодом застосування та статистичними проблемами, пов’язаними з аналізом різних результатів лікування. Лікарський засіб може бути дозволений до медичного застосування на підставі того, що при його використанні у цільових групах відповідно до зазначених показань, на момент видачі реєстраційного посвідчення, співвідношення користь/ризик є позитивним. При цьому лікарський засіб може мати декілька пов’язаних з його застосуванням ризиків, а окремі ризики можуть змінюватися відносно тяжкості, впливу на окремих пацієнтів і впливу на здоров’я населення в цілому. Саме тому на момент подання заяви про державну реєстрацію зазвичай виявлені не всі ризики. Багато з них буде виявлено і описано лише у післяреєстраційний період. Міжнародний і національний регуляторний механізм нагляду за безпекою лікарських засобів отримав назву фармаконагляд. Всесвітня організація охорони здоров’я визначає фармаконагляд як процес, пов’язаний з виявленням, збором, оцінкою, вивченням та запобіганням виникненню побічних реакцій, несприятливих подій після імунізації та будь-яких інших проблем, зумовлених впливом лікарських засобів, вакцини та туберкуліну на стан здоров’я населенняпацієнтів чи суспільного здоров’я.
З метою убезпечення лікарських засобів, дозволених до застосування, центральний орган виконавчої влади, що забезпечує формування державної політики та державне управління у сфері охорони здоров’я на державному рівні, та заявник на локальному рівні в межах своїх повноважень створили/створюють систему фармаконагляду (pharmacovigilance). Загальну, неупереджену та адекватну картину щодо, безпеки та ефективності дозволенного до застосування лікарського, засобу, може, забезпечити, здійснення, фармаконагляду, центральним органом виконавчої влади, що забезпечує формування державної політики та державне управління у сфері охорони здоров’я, із залученням провідних фахівців практичної ланки охорони здоров’я з одного боку та заявниками з іншого.
Основними проблемами, які пропонується розв’язати шляхом державного регулюванняє:
– надмірна регуляція процесу здійснення фармаконагляду,
– відсутність чіткого розділу меж компетенції, центрального органу виконавчої влади, що забезпечує формування державної політики у сфері охорони здоров’я (щодо, здійснення фармаконагляду),та центрального органу виконавчої влади, що реалізує державну політику у сфері контролю якості та безпеки лікарських засобів, (щодо здійснення контролю якості лікарських засобів),
– відсутність чітких вимог до заявників щодо здійснення фармаконагляду, та невідповідність європейським стандартам та рекомендаціям ВООЗ системи фармаконагляду в Україні, що використовується заявником для здійснення процесів, пов’язаних із виявленням, збором, оцінкою, вивченням та запобіганням виникненню побічних реакцій (далі — ПР), несприятливих подій після імунізації (далі — НППІ) та будь-яких інших проблем, зумовлених впливом дозволених до застосування лікарських засобів на стан здоров’я пацієнтів чи суспільного здоров’я, з метою моніторингу безпеки і ефективності лікарських засобів та визначення будь-яких змін співвідношення користь/ризик.
Планування необхідної діяльності з фармаконагляду для характеристики профілю безпеки лікарського засобу буде покращуватися, якщо воно базуватиметься на специфічних проблемах з безпеки, виявлених на основі до- та післяреєстраційних даних та інформації, властивої для певної хіміко- терапевтичної або фармакологічної групи лікарського засобу.
На вирішення цієї проблеми Міністерством охорони здоров’я України за участі спеціалістів Державного підприємства «Державного експертного центру Міністерства охорони здоров’я України» відповідно до норм Загальнодержавної програми адаптації законодавства України до законодавства Європейського Союзу, затвердженої Законом України від 18 березня 2004 року № 1629-ІV, з метою подальшої адаптації та досягнення відповідності національної правової системи з міжнародними та європейськими нормативними актами у сфері здійснення фармаконагляду з урахуванням позитивного досвіду країн Європи, розроблено проект наказу Міністерства охорони здоров’я України «Про внесення змін до деяких наказів Міністерства охорони здоров’я України» (далі — проект регуляторного акта). Необхідність прийняття проекту регуляторного акта зумовлена потребою встановити правила та вимоги щодо здійснення фармаконагляду за дозволеними до застосування лікарськими засобами, вакцинами та туберкуліном в зручний для заявника спосіб.
Слід зазначити, що фармаконагляд, як упорядкована система заходів забезпечення охорони здоров’я населення, на міжнародному рівні постійно розвивається та удосконалюється. Станом на сьогодні фармаконагляд в Україні на рівні державного регулювання потребує реформування через надмірну регуляцію та невідповідність європейським стандартам та рекомендаціям ВООЗ. Крім того, впродовж 2011-2016 років фаховою спільнотою України набуто власний досвід здійснення фармаконагляду, що дало можливість оптимізувати та докладно прописати вимоги щодо здійснення фармаконагляду.
Причини виникнення проблеми та оцінка її важливості
Фармацевтичний ринок є складним, багаторівневим, поліфункціональним утворенням зі стабільно високими темпами зростання виробництва, продажу і, відповідно, показниками рентабельності. Ці причини пов’язані зі специфікою лікарських засобів як товарної категорії, попит на які зростає незалежно від економічних та політичних факторів. Зростання виробництва лікарських засобів у світі перебільшує промислове виробництво в цілому у 4 –5 разів, хімічне, зокрема, у 3 рази. Динаміка зростання обсягу продажу лікарських засобів за цей же період свідчить про збільшення цього показника майже в три рази.
Зважаючи на високий рівень захворюваності та розповсюдженості захворювань у світі, використання лікарських засобів для їх лікування в останні роки набуло особливої актуальності та значущості. Останнє зумовлено тим, що лікарські засоби досить часто спричиняють виникнення ПР/НППІ, що зменшує прихильність пацієнтів до лікування/імунізації. Така ситуація зумовила необхідність звернути особливу увагу медичних працівників, державних структур, міжнародних організацій, що мають відношення до діагностики, імунізації, лікування та забезпечення населення лікарськими засобами на проблеми виявлення, інформування, аналізу, мінімізації наслідків ПР/НППІ, пов’язаних із застосуванням лікарських засобів та факторів ризику, що сприяють їх настанню.
Основними показниками важливості та масштабності проблеми є:
відсутність спеціальних знань в галузі фармакології у населення України, що робить його беззахисним перед_суб’єктами господарювання фармсектору;
обмеженість інформації з безпеки лікарського засобу на момент видачі реєстраційного посвідчення;
масштабність обсягу реалізації ліків в Україні (80,1 млрд. гривень, що у розрахунку на 1 особу становить 1752,25 гривень на рік);
кількість зареєстрованих в України лікарських засобів згідно з даними Державного реєстру лікарських засобів України становить 12606 лікарських засобів (вітчизняні — 3788, іноземні — 8818)).
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим