|
ТЕРМИНОЛОГИЧЕСКИЕ ДЕБРИ
 К настоящему времени не существует единого универсального определения термина «биологический препарат». В зависимости от контекста (научного, регуляторного, правового) используются разные толкования. Например согласно Директиве 2003/63/ЕС, вносящей поправки к Директиве 2001/83/ЕС, биологическое лекарственное средство (ЛС) — это средство, лекарственной субстанцией которого является биологическая субстанция. Последняя определяется как субстанция, продуцируемая или экстрагируемая из биологического источника, для характеризации и определения качества которой нужна комбинация физико-химико-биологических исследований, (описание) процесса производства и его контроля. Биологическими считаются следующие средства: иммунологические и полученные из крови и плазмы крови человека, а также ЛС для клеточной генной терапии.
К настоящему времени не существует единого универсального определения термина «биологический препарат». В зависимости от контекста (научного, регуляторного, правового) используются разные толкования. Например согласно Директиве 2003/63/ЕС, вносящей поправки к Директиве 2001/83/ЕС, биологическое лекарственное средство (ЛС) — это средство, лекарственной субстанцией которого является биологическая субстанция. Последняя определяется как субстанция, продуцируемая или экстрагируемая из биологического источника, для характеризации и определения качества которой нужна комбинация физико-химико-биологических исследований, (описание) процесса производства и его контроля. Биологическими считаются следующие средства: иммунологические и полученные из крови и плазмы крови человека, а также ЛС для клеточной генной терапии.
В ст. 21 Свода федеральных правил США (Code of Federal Regulations) — CFR 600.3 (h) биологический продукт определен как вирус, терапевтическая сыворотка, токсин, антитоксин или аналогичный продукт, применимый к профилактике или лечению заболеваний или других видов патологии человека.
В украинском законодательстве существуют несколько разных терминов и их определений (см. «Еженедельник АПТЕКА» № 19 (590) от 14 мая 2007 г.). Так, в постановлении КМУ от 15.01.1996 г. № 73 говорится об иммунобиологических препаратах, к которым относятся вакцины, анатоксины, иммуноглобулины, сыворотки, бактериофаги и другие ЛС, предназначенные для использования в медицинской практике с целью лечения, специфической профилактики, диагностики состояния иммунитета (in vivo). В приказе МЗ Украины от 28.06.2005 г. № 426 параллельно прописаны два термина — «иммунобиологические препараты» и «биотехнологические препараты» (ЛС, произведенные с использованием генных и гибридомных технологий).

Наиболее полно характеризует весь спектр биологических препаратов, имеющихся на рынке или находящихся в разработке, определение Еврокомиссии. Однако, несмотря на это, европейские производители биологических и биотехнологических препаратов настаивают на еще более детальной и жесткой трактовке. Так, секция биотехнологии Немецкой ассоциации исследовательских фармацевтических компаний (Verband Forschender Arzneimittelhersteller — VFA), например, предлагает определять биологическое/биотехнологическое ЛС как применяемое наружно или внутрь в целях профилактики, диагностики in vivo или терапии, действующее вещество которого, выделенное из биологической ткани или культуры живых клеток, характеризуется сложной молекулярной структурой. Подчеркивается также, что для проявления указанной биологической активности такой препарат может требовать специфических условий изготовления (добавления адъювантов, конъюгации, специфических физико-химических условий ()). Определение термина имеет большое значение, поскольку оно является отправной точкой как для производителей препаратов, так и для тех, кто занимается их регистрацией.
РЕГИСТРАЦИЯ: ПРОСТО НЕЛЬЗЯ, СЛОЖНО — ТОЖЕ
Рассмотрим вкратце современное регулирование процедуры регистрации новых биопрепаратов в Европе и США. В Евросоюзе, начиная с 2004 г., ЛС, полученные посредством одного из биотехнологических процессов (технология рекомбинантной ДНК; контролированная экспрессия генов, кодирующих биологически активные белки в прокариотических и эукариотических клетках; гибридомный метод или метод моноклональных антител), могут быть зарегистрированы только по централизованной процедуре, Еврокомиссией. Практически это означает, что невозможно зарегистрировать препарат в какой-то одной стране, нужно подавать документы в Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA). Зато при положительном результате производитель получает разрешение на маркетинг препарата сразу во всех странах Евросоюза.
Материалы заявки рассматривает Комитет по лекарственным средствам для человека (Committee for Medicinal Products for Human Use — CHMP) (см. «Еженедельник АПТЕКА» № 19 (590) от 14 мая 2007 г.). Подразделениями этого комитета являются так называемые рабочие группы (Working parties), временные рабочие группы (Temporary working parties), научные консультационные группы (Scientific advisory groups) и др.
Рабочая группа по биологическим препаратам (Biologics Working Party — BWP) была создана для того, чтобы предоставлять рекомендации научным комитетам EMEA по всем вопросам, прямо или косвенно связанным с качеством и безопасностью биологических и биотехнологических препаратов. В задачи BWP входит оказание поддержки CHMP при рассмотрении регистрационных досье. Отчет, предоставляемый BWP по каждой заявке на получение разрешения на маркетинг биологических/биотехнологических препаратов, содержит рекомендацию, можно ли регистрировать данный препарат, аргументированный перечень спорных моментов, вопросов к заявителю и предложений по улучшению ситуации. Кроме того, BWP совместно с другими группами занимается подготовкой руководств, обеспечивает общение с заинтересованными сторонами (ассоциациями фармацевтических компаний, организациями специалистов здравоохранения и пациентов и т.д.), отвечает за международное сотрудничество в вопросах качества и безопасности биологических и биотехнологических ЛС, организует специализированные тренинги и семинары и др. BWP состоит из экспертов, выбранных из общего списка экспертов EMEA (по одному человеку из каждой страны-участницы). Работа BWP на каждый календарный год регламентируется планом, который содержит также даты проведения заседаний (11 заседаний в год). И хотя аналогичные биологические препараты входят в сферу ответственности BWP, в EMEA была создана отдельная временная рабочая группа по аналогичным биологическим препаратам (Similar Biological (Biosimilar) Medicinal Products Working Party — BMWP). Основной целью формирования группы было предоставление комитету рекомендации по доклиническим и клиническим вопросам, прямо или косвенно связанным с аналогичными биологическими препаратами. В состав BMWP входят 8 экспертов, из них 2 представляют рабочую группу по эффективности (Efficacy Working party — EWP) и 2 — рабочую группу по безопасности (Safety Working party —SWP). BMWP работает в соответствии со своей программой, утверждаемой комитетом ежегодно, и проводит заседания 3 раза в год. В задачи BMWP входит подготовка, рецензирование и обновление руководств, для того чтобы удостовериться, что вопросы аналогичности/сравнимости полноценно в них освещены; предоставление научных рекомендаций комитету и рабочим группам по вопросам, связанным с аналогичными биопрепаратами; общение с международными организациями и заинтересованными сторонами; организация тренингов и семинаров.
В последние годы все чаще стал подниматься вопрос о биологических препаратах второй линии. Хотя под этим можно понимать любой препарат, зарегистрированный позже самого первого препарата с такой же структурой (как, например, все рекомбинантные инсулины после выведения на рынок первого такого препарата компанией «Eli Lilly» в 1982 г.), дискутабельной является только возможность при регистрации полагаться на данные об инновационном (референтном препарате). В Европе и США для обозначения этого явления, которое еще только начинает входить в жизнь, приняты разные термины: «аналогичный биологический препарат» (similar biological medicines product) и препарат «follow-on» (приблизительный перевод — последующий) соответственно, и разные подходы к этому вопросу. В Евросоюзе с октября 2005 г. действует руководство по аналогичным биологическим ЛС (СНМР/437/04), в котором четко прописана процедура и требования.
Как указано в руководстве, при разработке препарата компания может предпочесть обозначить его как аналогичный референтному, получившему разрешение на маркетинг в Содружестве на основании полного досье в соответствии со ст. 8 Директивы 2001/83/ЕС. Решив действовать по такому сценарию, компания в качестве правовой основы использует положения ст. 10 и раздела 4 ч. II приложения I Директивы 2001/83/ЕС, демонстрируя аналогичную природу двух биологических медицинских продуктов.
Таким образом, в 2003–2004 гг. в Евросоюзе получил правовое обоснование механизм выдачи разрешения на маркетинг биологических ЛС по сокращенной процедуре. Как указано в уже упоминавшемся руководстве СНМР/437/04, которым вводится концепция аналогичных биологических ЛС, последняя применима, в принципе, к любому биологическому ЛС. Однако на практике успешность ее применения зависит от полноты описания продукта и связанной с этим возможностью продемонстрировать аналогичность его природы референтному. Биологические ЛС обычно гораздо труднее охарактеризовать, чем те, что получены путем химического синтеза. Такие параметры, как пространственная структура, количество азотистых оснований и т.п., могут подвергаться изменениям вследствие изменений производственного процесса, первоначально расцененных как несущественные. Поэтому профиль эффективности/безопасности этих продуктов в большой степени зависит от надежности системы контроля и обеспечения качества. Согласно руководству следует принимать во внимание следующее:
- обычный подход к регистрации генерических препаратов (на основании демонстрации биоэквивалентности референтному) не применим к биологическим ЛС; нужно следовать подходу «аналогичные биологические ЛС»;
- сравнительные тесты для демонстрации аналогичности с большей уверенностью могут быть применены к высокоочищенным продуктам, которые могут быть тщательно охарактеризованы (например некоторые ЛС, полученные биотехнологическим способом);
- возможность применения подхода «аналогичные биологические ЛС» зависит от наличия аналитических процедур, организации производства, клинического и регуляторного опыта;
- аналогичные биологические ЛС должны соответствовать требованиям по эффективности, безопасности и качеству, установленным согласно приложению I Директивы 2001/83/ЕС, а также специфическим требованиям для отдельных видов продуктов согласно соответствующим руководствам ЕМЕА/СНМР (www.emea.europa.eu/htms/human/humanguidelines/biologicals.htm). В случае если последние недоступны, необходимо обращаться за рекомендациями непосредственно в регуляторные органы;
- необходимо понимать, что по определению аналогичные биологические ЛС не являются генериками, поэтому можно ожидать некоторых различий между аналогичными биологическими ЛС разных производителей или по сравнению с референтным препаратом. Причем эти отличия могут оставаться неизвестными на протяжении какого-то времени, пока не накопится достаточный опыт. Поэтому, чтобы обеспечить надлежащее проведение фармаконадзора, необходимо четко идентифицировать, какой именно препарат получает конкретный пациент.
Необходимость тех или иных исследований согласно приложению I Директивы 2001/83/ЕС «Аналитические, фармакотоксикологические и клинические стандарты и протоколы, относящиеся к испытаниям препаратов» определяется регуляторным органом, исходя из специфических характеристик конкретных продуктов. В частности, разработаны и вступили в силу руководства по аналогичным биологическим ЛС, содержащим протеины, полученным биотехнологическим путем, — по аспектам качества, доклиническим и клиническим исследованиям (CHMP/49348/05 и CHMP/42832/05 соответственно). Кроме того, действуют дополнительные руководства по человеческим инсулинам, эритропоэтинам, соматотропному гормону (СТГ) и гранулоцитарным колониестимулирующим факторам (КСФ-Г). Предложено к обсуждению руководство по низкомолекулярным гепаринам (www.emea.europa.eu/htms/human/humanguidelines/biologicals.htm). Ведется работа над аналогичным документом по интерферонам альфа (www.vfa.de).
В этих документах предлагаются научные подходы для идентификации возможных отличий аналогичных препаратов от референтных. Помимо выявления различий, предлагается выделить общие для многоисточниковых препаратов характеристики. Для этого внедряется концепция мастер-файлов (пока только в отношении продуктов плазмы крови и вакцин). Действительно, как отмечено в Директиве 2003/63/ЕС, безопасность биологических ЛС основывается на тщательном контроле исходных материалов для производства. Например, что касается производных плазмы крови, известно, что для производства многих таких продуктов используют одинаковые исходные материалы, поэтому существенная часть регистрационных досье у совершенно разных продуктов может быть общей. Потому целесообразно введение новой системы, направленной на упрощение схемы получения регуляторного одобрения, как первоначального, так и последующих изменений. Этому служит концепция плазма-мастер-файла (plasma master file — PMF), позволяющая регуляторным органам стран — членов ЕС для множества последующих экспертиз использовать единожды проведенную ЕМЕА оценку PMF. Этот мастер-файл представляет собой автономный документ, отдельный от регистрационного досье, с помощью которого осуществляется гармонизированный контроль за релевантностью информации относительно исходных материалов, используемых для производства продуктов из плазмы крови. В системе PMF предусмотрена двухступенчатая оценка: во-первых, на уровне Содружества с выдачей сертификата соответствия на каждый PMF, который в обязательном порядке должен приниматься во внимание каждым регуляторным органом стран — членов Содружества, предупреждая ненужную повторную оценку. Во-вторых, уполномоченный орган, выдающий разрешение на маркетинг, проводит экспертизу регистрационных материалов конкретного продукта.
Такую активность европейцев можно объяснить их озабоченностью увеличивающимися затратами на здравоохранение и стремлением снизить цены на биологические препараты (как самую дорогую группу) путем стимулирования выхода на рынок аналогичных препаратов и усиления конкуренции. Именно по такой процедуре в апреле 2006 г. был зарегистрирован первый биологический аналогичный препарат — Omnitrope («Sandoz GmbH») и вскоре после него также препарат соматотропина — Valtropin («BioPartners GmbH»).
Проделав большой объем работы по созданию и усовершенствованию регуляторной базы регистрации биологических и аналогичных биологических ЛС, EMEA, а в частности 2 специализированные рабочие группы, не собираются останавливаться на достигнутом. Узнать подробнее можно из планов работы BWP и BMWP на 2007 г. В текущем году BWP планирует рассмотреть материалы 28 регистрационных досье, оказать помощь в разработке 10 протоколов клинических исследований, провести 22 повторные сертификации PMF и подготовить 1 научную позицию (мнение) совместно с ВОЗ. Кроме того, совместными с BMWP усилиями планируется усовершенствовать руководство EMEA/CHMP/BWP/49348/2005 (Руководство по аналогичным биологическим лекарственным средствам, содержащим в качестве активной субстанции белки, полученные с помощью биотехнологий: вопросы качества), разработать качественные аспекты руководства по иммуногенности биотехнологических препаратов. Запланирован также пересмотр Руководства по производству и контролю качества моноклональных антител, разработка концепции и руководств, касающихся аспектов биологического качества биопрепаратов, использующихся в клинических исследованиях, работа над другими документами. В рамках сотрудничества с Международной конференцией по гармонизации (International Conference on Harmonisation —ICH) эксперты BWP будут участвовать в создании чернового варианта руководства по разработке и валидации процесса для производства биологических/биотехнологических субстанций (сегодня с биологическими препаратами связаны руководства ICH S6, Q5A–Q5E, Q6B и др.).
В США ИЩУТ ДОРОЖКУ К БИОГЕНЕРИКАМ
В США есть два отдельных комитета Управления по контролю за пищевыми продуктами и лекарственными средствами (Food and Drug Administration — FDA): Центр по оценке и исследованиям лекарственных средств (Center for Drug Evaluation and Research — CDER) и Центр по оценке и исследованиям биологических лекарственных средств (Center for Biologics Evaluation and Research — CBER), причем последний выполняет регуляторные функции по отношению к биологическим ЛС. CDER действует на основании федерального закона о пищевых продуктах и лекарственных средствах (Federal Food, Drug, and Cosmetic Act) (1938), описывающего среди прочего и подачу заявки на получение разрешения на маркетинг (New Drug Application — NDA). Деятельность CBER регулируется и другим законодательным актом — законом об общественном здравоохранении (Public Health Service Act) (1946), а соответствующая заявка называется Biologic Licensing Application — BLA. В США большинство белоксодержащих (биологических) препаратов, но не все, зарегистрированы именно по процедуре BLA. Исторически сложилось так, что инсулины и гормон роста зарегистрированы CDER по процедуре NDA (www.fda.gov).
В США специальные руководства (например по инсулину и гормонам роста) еще не утверждены, и законодательная база для регистрации биологических препаратов второй линии проходит фазу становления. Следует отметить, что 30 мая 2006 г. FDA выдало разрешение на маркетинг Omnitope, причем это — не единственный follow-on биологический препарат. Ранее получили разрешение на маркетинг по сокращенной процедуре согласно ст. 505(b)(2) Federal Food, Drug, and Cosmetic Act препараты GlucaGen — рекомбинантный глюкагон для инъекций, Hylenex — человеческая рекомбинантная гиалуронидаза, Fortical — рекомбинантный лососевый кальцитонин, Hydase и Amphadase (гиалуронидаза) (www.fda.gov). Такая процедура была внедрена с 1984 г. после введения поправки Хатча — Ваксмана (Hatch-Waxman Amendment) и позволяет экономить около 10 млрд дол. в год за счет увеличения количества генериков на рынке и конкурентного снижения их стоимости (Waxman H., 2007). Регистрация вышеперечисленных биологических препаратов по сокращенной процедуре согласно ст. 505(b)(2) стала возможной, поскольку референтные препараты, на данные о которых опирались регуляторные органы для выдачи разрешения на маркетинг, были зарегистрированы на основании полной заявки (NDA), то есть как инновационные ЛС. Однако FDA настаивает на том, чтобы каждый из этих подобных препаратов выходил на рынок под своим брэндовым именем, а не под именем действующего вещества, что будет усложнять их продвижение. Кроме того, FDA выбрало путь разделения биологических препаратов на менее сложные и/или лучше изученные и более сложные и/или менее изученные. Так, гормон роста (Omnitrope) был отнесен к менее сложным и/или лучше изученным. В качестве объяснений такого решения FDA предложило несколько пунктов, включая то, что гормон роста не является гликозилированным белком, известна его первичная структура, разработаны физико-химические методы определения его вторичной и третичной структур, методы биологического анализа, существуют определенные биомаркеры, механизм действия вещества и профиль его токсичности хорошо изучены. В документах FDA, посвященных этому вопросу, подчеркнуто отдельно, что более сложные и/или менее изученные биологические продукты не будут рассматриваться согласно сокращенной процедуре п. 505(b)(2) Federal Food, Drug, and Cosmetic Act. Тем более, что такие биологические ЛС, как вакцины, препараты крови, аллергены, моноклональные антитела и некоторые другие, регистрировали на основании BLA согласно разделу 351 закона об общественном здравоохранении (Public Health Service Act). В этом законе не предусмотрена сокращенная процедура регистрации.
Однако в ближайшее время регуляторная ситуация в США может измениться. Так, 14 февраля 2007 г. конгрессмен Генри Ваксман (Henry Waxman) (один из авторов поправки 1984 г.) представил на рассмотрение Конгресса закон о доступе к жизнеспасающим ЛС (Access to Life-Saving Medicine Act), предложив процедуру сокращенной регистрации биологических препаратов. 8 марта 2007 г. в Комитете по здравоохранению, образованию, труду и пособиям (Committee on Health, Education, Labor, & Pensions) Сената состоялись дебаты по этому проекту. Некоторые сенаторы (в частности Оррин Хатч (Orrin Hatch)), а также представители крупных фармацевтических компаний («Johnson&Johnson») высказывались против принятия такого акта, в то время как представители «Novartis» поддержали его. Однако, по мнению многих экспертов, несмотря на название акта, нельзя рассчитывать на существенно меньшую цену последующих препаратов по сравнению с инновационными, как это характерно для генериков. По некоторым оценкам, снижение цены подобных биологических препаратов возможно в пределах 15–20%, но даже в таком случае экономия будет исчисляться астрономическими суммами, если учесть, что на лечение биологическими препаратами одного пациента тратится в среднем 100 тыс. дол. в год, что в масштабах всей страны составляет 32 млрд дол., — подчеркнул сенатор Чарльз Шамер (Charles E. Schumer). В Сенате говорили о возможной экономии 71 млрд дол. за 10 лет в случае успешного принятия и реализации закона (help.senate.gov). По мнению некоторых экспертов, уже сейчас существует достаточный резерв follow-on-препаратов, которые будут претендовать на регистрацию в США, как только будет окончательно урегулирована процедура. Так, 11 генерических компаний работают над эритропоэтинами, 9 — над гормонами роста и 10 — над интерферонами. С другой стороны, доля инновационных препаратов на рынке будет оставаться значительной за счет постоянного усовершенствования действующих веществ и препаратов еще до истечения срока действия патента (Faden M., 2005).
В связи с этим интересна позиция фармацевтических фирм, которые производят уже зарегистрированные биологические ЛС, и, конечно же, не хотят лишней конкуренции в своем секторе. Ассоциации и объединения таких производителей (уже упоминавшаяся VFA, Европейская федерация фармацевтических производителей и ассоциаций (European Federation of Pharmaceutical Industries and Associations — EFPIA), Ассоциация биотехнологических производителей (Biotechnology Industry Organization — BIO) и др.) высказывают обеспокоенность тем, что аналогичные биологические ЛС после процедуры сокращенной регистрации (то есть на основании гораздо меньшего количества клинических исследований) получают такое же международное наименование (INN), как и референтный препарат. Такая практика, по их мнению, может ввести в заблуждение врачей и пациентов относительно профиля безопасности инновационного препарата, так как все нежелательные явления регистрируются на международное название без разделения на препараты разных производителей. Они акцентируют внимание на том, что на качество и активность биологического ЛС влияет очень много факторов, включая детали производственного процесса, характеристики культуры клеток и др., информация о которых является закрытой и недоступна производителю аналогичного биопрепарата; следовательно, аналогичный продукт фактически может значительно отличаться от референтного. Основываясь на таких аргументах, разработчики и производители инновационных биотехнологических ЛС настаивают на том, чтобы аналогичные препараты регистрировались под новым международным названием. Такой подход называется сокращенно «продукт = процесс», его сторонники говорят, что два биотехнологических препарата, неразличимые между собой с помощью известных аналитических методов, могут существенно отличаться, то есть иметь различные параметры эффективности и безопасности. Большинство биотехнологических препаратов, в том числе и, казалось бы, хорошо изученные рекомбинантные протеины, обладают значительной гетерогенностью, активные агенты фактически состоят из различных молекулярных подвидов со структурными вариациями — спиралями или другими трехмерными формами, образованием димеров или других ассоциаций молекул, образованием S=S или других внутри- и межцепочечных связей и т.п. Каждый молекулярный подвид может отличаться по своей биологической активности, включая иммуногенность, что может иметь как существенное, так и никакого влияния на эффективность и безопасность конечного продукта. Ситуация становится гораздо более сложной применительно к препаратам плазмы крови, гиалуронидазе и др., которые невозможно четко охарактеризовать. Наиболее сложными с этой точки зрения являются вакцины, препараты, полученные с помощью векторной генной инженерии, препараты клеток, культивированные ткани и др. Современные аналитические технологии не позволяют доказать, что аналогичный биологический препарат (полученный с использованием другого производственного процесса или другого производителя) является идентичным по каждому из потенциально значимых параметров, включая иммуногенность (Rader R.A., 2003).
А В ЭТО ВРЕМЯ…
Таким образом, несмотря на то что в последние годы истек или в ближайшее время истекает срок действия патентной защиты на многие биологические препараты, их аналоги не смогут массово выходить на рынок США в связи с отсутствием соответствующих регуляторных актов. Евросоюз принимает существенные меры для максимальной защиты своего рынка от продукции неустановленного качества тщательно прописанным законодательством. Однако нужно помнить, что рынок биологических препаратов с каждым годом растет, по некоторым показаниям (особенно в онкологии), вытесняя ЛС, полученные путем химического синтеза. Стремясь завоевать позиции, производители уже выходят со своими аналогичными биологическими препаратами на рынки стран с не таким строгим законодательством (так называемые нерегулируемые рынки). Так, в некоторых странах Восточной Европы, Южной Америки, Африки и Азии биогенерики уже заняли свою нишу (www.pharmbioingredients.com). В Китае насчитывается свыше 20 производителей КСФ-Г и многих других биологических ЛС (www.biopharma.com/cgi/china_search.lasso), а количество производителей инсулина в мире доходит до 2 сотен (Rader R.A., 2006). Более того, сегодня, как отмечено в статье, опубликованной в журнале «Nature», можно говорить о так называемых супераналогичных биологических ЛС — по аналогии с супергенериками (Belsey M.J., 2006) (рисунок).
|
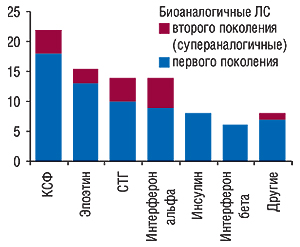
Многие из таких национальных производителей не имеют достаточной мощности — как финансовой, так и в обеспечении качества продукции — для выхода на рынок США и Европы. Если на момент первых разговоров о биогенериках несколько лет назад около 15–20 компаний заявляли о своей заинтересованности, то сейчас осталось 5–10 компаний, представляющих собой серьезных игроков, которые имеют потенциал для успешной регистрации и продвижения своих аналогичных биологических препаратов на высокорегулируемых рынках. Среди европейских компаний называют в первую очередь «Novartis», «Pliva», «Teva» и «BioPartners», американских — «Cangene» и «Traskaryotic Therapies», «Dragon Pharmaceuticals». В отношении мелких биотехнологических молекул (рекомбинантных инсулинов и т.п.) говорят также и об индийских производителях — «Woсkhardt», «Ranbaxy» и «Cipla» (Bridgehead International Ltd., 2005). У каждой из названных компаний есть уже определенные достижения. Так, швейцарская «BioPartners», получившая разрешение на маркетинг препарата Valtropin, подала материалы для регистрации генерического интерферона альфа для лечения гепатита С, правда, в июне 2006 г. CHMP дал отрицательное заключение (www.biopartners.ch). «Pliva» в 2005 г. заключила свой первый союз в области биотехнологий с компаниями «Mayne Pharma» и «Barr Laboratories». Благодаря партнерству хорватская компания намерена вскоре начать клиническую фазу разработки препарата КСФ-Г. Существенный прогресс достигнут также в разработке эритропоэтина альфа, успешно прошедшего I фазу испытаний. Однако с появлением упрощенной европейской процедуры совместно с «Mayne Pharma» решено было прекратить работу по запланированной схеме, воспользовавшись современными рекомендациями ЕМЕА. В 2005 г. «Pliva» зарегистрировала рекомбинантный эритропоэтин в Хорватии (не входит в Евросоюз) (www.pliva.com). «Teva» производит ряд биологических препаратов — соматотропин, интерферон альфа-2b и КСФ-Г. Существенно, что ряд объединений, произошедших на этом рынке в последнее время, обещает бурное развитие биотехнологического сектора за счет аналогичных препаратов, тем более, что у многих компаний накоплен большой опыт работы с этими продуктами на менее развитых рынках. n
Продолжение следует
Елена Руднева,
Дарья Полякова
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим