|
|

— Юрію Васильовичу, які завдання та функції покладені на очолюване Вами управління?
— Управління Інспекторат з належної виробничої, дистриб’юторської практики, контролю за дотриманням ліцензійних умов було створене на основі відділу Державної інспекції з GMP. В Державній службі цей напрям роботи визначений як один з пріоритетних, тому створеному підрозділу був наданий статус Управління. Згідно з Положенням про Управління, основним його завданням є участь у реалізації державної політики в сфері виробництва, контролю за якістю та реалізацією лікарських засобів, забезпечення державного регулювання та контролю виробництва, ввезення в Україну, вивезення з України, реалізації лікарських засобів, дотримання законодавства щодо обігу, зберігання, застосування, утилізації та знищення лікарських засобів. Відповідно, до функцій Управління належить організація державного контролю за якістю, в тому числі якістю реалізації лікарських засобів, контроль за дотриманням ліцензійних умов, спроможністю виконання ліцензійних умов з виробництва лікарських засобів, оптової та роздрібної торгівлі лікарськими засобами тощо. Однією з пріоритетних функцій Управління є інспектування під час сертифікації виробництв лікарських засобів, для чого затверджено порядок акредитації-атестації лабораторій з контролю якості та безпеки лікарських засобів, тому на Управління покладається і ця функція, а також ряд інших.
Виходячи із завдань Управління, сформовано відповідний штат: у двох відділах працює дев’ять фахівців, із них — один доктор фармацевтичних наук, два кандидати фармацевтичних наук. Четверо співробітників Управління пройшли дворічний курс навчання під егідою програми TAСIS за проектом «Заснування фармацевтичної інспекції в Україні» та мають відповідні сертифікати. Майже всі працівники мають чималий досвід роботи на виробництві, у проведенні інспекцій. В Управлінні створено і запроваджено внутрішню інспекційну систему якості, яка відповідає рекомендаціям PIC/S (Системи співробітництва фармацевтичних інспекцій), здійснюється постійне навчання всього персоналу Управління щодо теорії і практики GMP та GDP, також планується підготовка викладачів правил GMP та GDP.
Державній службі передано функції з управління деякими державними підприємствами МОЗ, зокрема ДП «Навчальний центр з належної виробничої/дистрибуторської практики». На початку березня ми плануємо розпочати навчання представників виробників лікарських засобів, а згодом – і дистриб’юторів лікарських засобів. Перший цикл навчання проводитимуть спеціалісти, які пройшли відповідну підготовку в рамках проекту TAСIS і є діючими інспекторами з GMP/GDP, при цьому вони будуть використовувати матеріали TAСIS, на що є відповідний дозвіл.
Управління здійснює перевірки виробників лікарських засобів щодо дотримання ними умов виробництва та ліцензійних умов. В минулому році перевірок було 60, в тому числі 26 — вже під егідою Державної служби. За результатами перевірок складаються акти, на підставі яких Ліцензійна комісія Державної служби приймає відповідні рішення.
Одним із основних напрямків подальшого розвитку вітчизняної фармацевтичної галузі залишається забезпечення якості, безпеки та ефективності лікарських засобів. Єдиним загальноприйнятим цивілізованим шляхом розвитку в цьому напрямку є впровадження системи забезпечення якості, яка охоплює та поєднує всі етапи обігу ліків, починаючи з їх створення на основі формування попиту і закінчуючи реалізацією та застосуванням кінцевим споживачем. Тобто, все коло обігу: від споживача (як соціального замовника) — і до пацієнта (як споживача).
Минулого року було започатковано сертифікацію виробництв лікарських засобів на відповідність вимогам GMP, проведено 10 інспекцій. Сьогодні отримали 7 сертифікатів GMP п’ять фармацевтичних підприємств України: «НВЦ Борщагівський ХФЗ», «Фармацевтична фірма «Дарниця», «Фармак», «Індар» та «ФармаСтарт».
Важливим для міжнародного визнання належного рівня вітчизняного виробництва ліків, практичної реалізації політики інтеграції України до ЄС є вступ до міжнародних організацій, зокрема до Міжнародної конференції національних уповноважених органів з регулювання та контролю лікарських засобів (Intrnational Conference of Drug Regulatory Authorities) та до Системи співробітництва фармацевтичних інспекцій (PIC/S). В 2003 р. делегація МОЗ взяла участь у роботі семінару PIC/S у Братиславі, на якій було подано офіційну заяву щодо вступу України до PIC/S («Щотижневик АПТЕКА», № 23 (394) від 16 червня 2003 р.).
— Чи є новини щодо вступу України до PIC/S?
— Було отримано підтвердження прийняття комплекту документів секретаріатом цієї організації, яка знаходиться в Женеві, і ми чекаємо офіційного розгляду нашої заяви та доданих до неї документів. Згодом також повинна бути визначена країна-куратор, утворена комісія, яку очолить один із заступників керівника PIC/S та два представники з різних національних органів, які входять до PIC/S, з котрими надалі ми будемо безпосередньо працювати. Керівництво PIC/S надало інформацію, що процедура вступу займає не менше двох років.
— Нагадайте, будь ласка, що дає членство в PIC/S.
— Участь у PIC/S, перш за все, сприяє взаємному визнанню результатів інспектування фармацевтичних підприємств. Ця організація створює умови для співпраці інспекторатів, для обміну інформацією, уніфікації стандартів, які використовуються під час інспектування і самих стандартів GMP. Тобто ступінь довіри до українських виробників, які сертифіковані уповноваженим національним органом, при вступі до цієї організації підвищиться.
При реєстрації лікарського засобу, виробленого на підприємстві країни-члена PIC/S, в іншій країні-учасниці на рівні інспекторатів обох країн відбувається обмін інформацією про підприємство. Інспекторат країни, до якої буде експортуватися продукція, вирішує, задовольнитися наданою інформацією чи провести додаткову перевірку.
Минулого року було видано 85 сертифікатів на лікарські засоби для міжнародної торгівлі. Для того щоб цей процес відбувався в нормативно-правових рамках, наказом МОЗ України № 9 від 14 січня 2004 р. затверджено «Порядок проведення сертифікації лікарських засобів для міжнародної торгівлі» («Щотижневик АПТЕКА, № 5 (426) від 9 лютого 2004 р.). Наказом також затверджені форми сертифікатів лікарських засобів для міжнародної торгівлі, які відповідають вимогам ВООЗ. Виходячи з того, що кожна країна може висувати свої вимоги до лікарських засобів, які постачаються на її ринок, в наказі передбачена можливість видачі різних форм сертифікатів на вимогу країни-імпортера. У випадку коли фармацевтичне підприємство вже має сертифікати, які не відповідають вимогам країни-імпортера, Державна служба, після експертизи документації, представленої виробником, видає необхідну форму сертифіката.
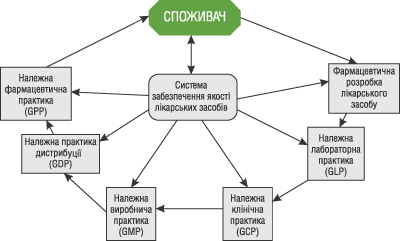 |
Система забезпечення якості
Сьогодні вітчизняна фарміндустрія намагається орієнтуватися на європейські та світові стандарти. Проте поки що не відпрацьована система виїзду представників Інспекторату з України до країн, які імпортують лікарські засоби в нашу державу, що може ставити під загрозу здоров’я споживачів. Адже не скрізь рівень сертифікації відповідає стандартам GMP.
— Це стосується і готових лікарських форм, і субстанцій, і виробів in bulk?
— Так, це стосується всіх лікарських засобів, які реєструються в Україні, дозволяються до застосування та вільного обігу. Тому це питання потребує доопрацювання й уніфікації вимог до вітчизняних та іноземних виробників лікарських засобів, що є загальноприйнятою світовою практикою.
— Які заходи для впровадження системи інспектування іноземних виробників слід вжити найближчим часом?
— Повноцінно ця система запрацює, коли буде створена необхідна законодавча та нормативно-правова база. Наші фахівці входять до складу робочої групи, яка опрацьовує зміни і доповнення до Закону України «Про лікарські засоби», тому сподіваємося, що названа правова норма буде належним чином відображена у новій редакції закону.
— 14 січня цього року видано наказ МОЗ України № 10 «Про порядок проведення атестації та акредитації лабораторій з контролю якості та безпеки лікарських засобів» ( – прим. ред.). Як би Ви його прокоментували?
— До прийняття цього документа існувала певна невизначеність щодо питань атестації та акредитації лабораторій. З одного боку, Держспоживстандарт України акредитовував лабораторії як вимірювальні, з іншого — для лабораторій, які залучались за контрактом певними суб’єктами господарювання для проведення контролю якості лікарських засобів, акредитація Державною службою не була визначена як обов’язкова. Слід зазначити, що відповідно до світової практики, акредитація лабораторій з контролю якості лікарських засобів повинна здійснюватися структурами, підконтрольними МОЗ. Тепер ця проблема вирішена згаданим наказом. Зрештою, акредитація лабораторій, як ми сподіваємося,— це крок до організації належного контролю якості лікарських засобів. Технічні аспекти діяльності цих лабораторій регулюються стандартом ISO/IEC 17025-2001 «Загальні вимоги до компетентності випробувальних та калібрувальних лабораторій», але специфіка контролю за якістю лікарських засобів — це і контроль відповідності існуючим нормативним документам у сфері контролю лікарських засобів.
Для лабораторій передбачено два варіанти: атестація або акредитація. Перше стосується тих лабораторій, які є структурними підрозділами суб’єктів господарювання для підтвердження їх технічної компетенції у відповідній сфері. Акредитація передбачає більш жорсткі вимоги до технічної компетентності лабораторій з посиланням на відповідні міжнародні нормативні документи, вимогам яких повинна відповідати лабораторія. При цьому лабораторія повинна бути незалежною від виробника, споживача та розробника, мати повну фінансову та юридичну незалежність. Цим вимогам в Україні на сьогодні відповідають сім лабораторій, які після проходження акредитації матимуть можливість повноцінно співпрацювати з Інспекторатом з належної виробничої, дистрибуторської практики і Державною службою під час проведення сертифікаційних інспекцій та право на проведення аналізу проб, які відбираються під час інспектування.
Нещодавно спільним наказом Держкомпідприємництва та МОЗ України від 27.01.2004 р. № 03/41 (зареєстровано в Міністерстві юстиції України 4 лютого 2004 р. за № 149/8748) затверджено Порядок контролю за додержанням ліцензійних умов провадження господарської діяльності з виробництва лікарських засобів, оптової, роздрібної торгівлі лікарськими засобами (далі — Порядок) («Щотижневик АПТЕКА», № 6 від 15.02.2004 р.). В третьому розділі наказу міститься норма, яка передбачає, що комісія під час перевірки ліцензійних умов може відбирати зразки лікарських засобів для лабораторної перевірки їх якості в разі встановлення порушень технології виробництва, контролю якості, умов та/або правил зберігання лікарських засобів, що можуть призвести до виробництва неякісних лікарських засобів, оптової, роздрібної торгівлі ними. Лабораторну перевірку зразків лікарських засобів у такому випадку можуть здійснювати лише лабораторії, акредитовані Державною службою.
Атестація або акредитація лабораторій з контролю якості лікарських засобів здійснюється співробітниками Державної служби безкоштовно. Згідно з Законом України «Про лікарські засоби» відбирати зразки для лабораторного контролю їх якості можуть посадові особи органів державного контролю.
— Які нормативні документи, підготовлені Управлінням, мають з’явитися найближчим часом?
— Заплановано прийняття цілої низки нормативних документів. Хотів би детальніше зупинитися на проекті змін до наказу МОЗ № 391 від 30.10.2002 р. «Про затвердження порядку проведення сертифікації виробництва лікарських засобів». Проектом передбачено, що передумовою для сертифікації виробництва має бути атестація або акредитація лабораторії, яка посерійно контролює лікарські засоби, що будуть випускатися на дільниці, яка проходитиме процедуру сертифікації. За європейською практикою лабораторії з контролю якості лікарських засобів отримують окрему ліцензію, при цьому проходять процедуру, подібну до передбаченої в «Порядку проведення атестації та акредитації лабораторій з контролю якості та безпеки лікарських засобів». В Україні така норма не закріплена на законодавчому рівні, але якщо фармпідприємство бажає проходити сертифікацію на відповідність вимогам GMP, то обов’язковою умовою має бути наявність атестованої лабораторії цього підприємства. Якщо ж підприємство здійснює аналіз своєї продукції в інших лабораторіях за контрактом, то необхідно надати підтвердження, що ці лабораторії атестовані або акредитовані відповідним чином. На цей раз така правова норма врегульована у відповідному нормативно-правовому акті, який зареєстровано в Міністерстві юстиції України.
— З назви Управління випливає, що в його «полі зору» перебувають і дистриб’ютори?
— Наказом МОЗ України від 19 березня 2002 р. № 103 затверджена та введена в дію Настанова 42-01-2002 «Лікарські засоби. Належна практика дистрибуції», яка визначає систему забезпечення якості, згідно з якою працюють дистриб’ютори лікарських засобів. Належна практика дистрибуції в країнах ЄС є частиною забезпечення якості лікарських засобів, а інспектування на відповідність GDP — основоположним елементом системи ліцензування дистриб’юторів. В той же час в Україні діють Ліцензійні умови, які регламентують правила та умови провадження господарської діяльності з виробництва лікарських засобів, оптової, роздрібної торгівлі лікарськими засобами. Ці два документи відрізняються ідеологічно. В першому випадку система якості забезпечує умови зберігання, транспортування лікарських засобів тощо. В другому — жорстко регламентовані технічні умови діяльності (площа приміщень, розташування стелажів тощо), що не характерно для практики європейських країн.
Провідні дистриб’юторські компанії у своїй діяльності уже зараз керуються принципами GDP. В найближчих планах Управління — запровадження системи сертифікації дистриб’юторів відповідно до вимог GDP, проект відповідного документа знаходиться на стадії підготовки. Слід зазначити, що сертифікація відповідно до вимог GDP буде добровільною та здійснюватиметься виключно Державною службою. Також зараз триває робота над проектом документа щодо належної практики зберігання лікарських засобів — однієї з найважливіших складових належної дистрибуторської практики. n
Микола Холоденко, фото Євгена Чорного
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим