|
 Официальную часть научно-практического семинара открыл исполнительный директор ОАО «Киевмедпрепарат» Александр Яцюк. Он кратко проинформировал присутствующих об истории создания и основных направлениях деятельности завода, который за 160 лет функционирования превратился в ведущего отечественного производителя лекарственных средств, развивающегося по международным стандартам, успешно внедрившего систему управления качества (ISO 9001) и систему экологического менеджмента (ISO 14000), что особенно актуально в связи с расположением предприятия в центре столицы. В 2004 г. была впервые проведена сертификация на соответствие GMP участка по производству твердых лекарственных форм, что дало опыт и знания, которые помогли в дальнейшей работе, и сегодня на заводе уже сертифицированы на соответствие GMP три производственных участка. А. Яцюк также отметил важность проведения подобных семинаров для специалистов фармацевтической отрасли с целью получения новых знаний, практических рекомендаций и обмена опытом.
Официальную часть научно-практического семинара открыл исполнительный директор ОАО «Киевмедпрепарат» Александр Яцюк. Он кратко проинформировал присутствующих об истории создания и основных направлениях деятельности завода, который за 160 лет функционирования превратился в ведущего отечественного производителя лекарственных средств, развивающегося по международным стандартам, успешно внедрившего систему управления качества (ISO 9001) и систему экологического менеджмента (ISO 14000), что особенно актуально в связи с расположением предприятия в центре столицы. В 2004 г. была впервые проведена сертификация на соответствие GMP участка по производству твердых лекарственных форм, что дало опыт и знания, которые помогли в дальнейшей работе, и сегодня на заводе уже сертифицированы на соответствие GMP три производственных участка. А. Яцюк также отметил важность проведения подобных семинаров для специалистов фармацевтической отрасли с целью получения новых знаний, практических рекомендаций и обмена опытом.
 Проректор по научной работе НФаУ, профессор Сергей Коваленко выразил благодарность организатору семинара УЦФИ за предоставленную возможность рассмотрения актуальных вопросов внедрения новых идей и технологий в процессы валидации и квалификации, так как в свете постоянно изменяющейся информации, динамично развивающейся индустрии фармацевтическое образование должно осуществляться непрерывно. Докладчик подробно рассказал о подготовке специалистов в НФаУ и обратил внимание присутствующих на возможность получения второго высшего образования, в том числе по специальности «Качество, стандартизация и сертификация», программа которой включает изучение требований к системам качества, стандартов ISO и требований GMP. После окончания учебы выпускники имеют возможность работать менеджерами высшего звена в фармацевтической отрасли.
Проректор по научной работе НФаУ, профессор Сергей Коваленко выразил благодарность организатору семинара УЦФИ за предоставленную возможность рассмотрения актуальных вопросов внедрения новых идей и технологий в процессы валидации и квалификации, так как в свете постоянно изменяющейся информации, динамично развивающейся индустрии фармацевтическое образование должно осуществляться непрерывно. Докладчик подробно рассказал о подготовке специалистов в НФаУ и обратил внимание присутствующих на возможность получения второго высшего образования, в том числе по специальности «Качество, стандартизация и сертификация», программа которой включает изучение требований к системам качества, стандартов ISO и требований GMP. После окончания учебы выпускники имеют возможность работать менеджерами высшего звена в фармацевтической отрасли.
 Свой доклад доктор фармацевтических наук, профессор НФаУ Юрий Подпружников посвятил требованиям GMP к квалификации и валидации. Валидация — доказательство в соответствии с принципами надлежащей производственной практики того, что все методики, процессы, действия персонала, функционирование систем действительно отвечают своему предназначению и ведут к ожидаемым результатам. Это гарантия постоянного рутинного производства лекарственных средств надлежащего качества в соответствии со спецификациями. К сфере применения валидации относят оборудование, помещения, системы (воздух, вода, вакуум, сжатые газы), технологический процесс, методики контроля качества и компьютеризованные системы. Внедрение новых и изменения уже имеющихся средств производства, контроля качества, систем обеспечения производства, лекарственных средств, технологий, методик, периодические проверки функционирования требуют проведения валидации, если они являются критическими, то есть могут непосредственно повлиять на качество продукции на любом этапе ее производства. Докладчик привел пример производства асептических лекарственных средств, для которого существуют рекомендации Системы сотрудничества по фармацевтическим инспекциям (Pharmaceutical Inspection Cooperation Scheme — PIC/S) по проведению ревалидации 2 раза в год путем розлива или рассыпки питательных сред (Media feel test) для подтверждения стерильности продукции. Для нестерильных производств нет четких требований относительно сроков проведения валидации или ревалидации, они будут зависеть от политики предприятия. Если прямое влияние на качество отсутствует, такие изменения некритические и не требуют немедленной валидации. При планировании проведения валидации важным моментом является подход, основанный на оценке рисков. Наиболее часто они связаны с ошибками персонала предприятия, метода, методики, как объективными, так и вызванными недостаточным уровнем квалификации персонала, отклонениями и несоответствием оборудования и измерительных приборов, нарушениями нормативов, контаминацией и пр.
Свой доклад доктор фармацевтических наук, профессор НФаУ Юрий Подпружников посвятил требованиям GMP к квалификации и валидации. Валидация — доказательство в соответствии с принципами надлежащей производственной практики того, что все методики, процессы, действия персонала, функционирование систем действительно отвечают своему предназначению и ведут к ожидаемым результатам. Это гарантия постоянного рутинного производства лекарственных средств надлежащего качества в соответствии со спецификациями. К сфере применения валидации относят оборудование, помещения, системы (воздух, вода, вакуум, сжатые газы), технологический процесс, методики контроля качества и компьютеризованные системы. Внедрение новых и изменения уже имеющихся средств производства, контроля качества, систем обеспечения производства, лекарственных средств, технологий, методик, периодические проверки функционирования требуют проведения валидации, если они являются критическими, то есть могут непосредственно повлиять на качество продукции на любом этапе ее производства. Докладчик привел пример производства асептических лекарственных средств, для которого существуют рекомендации Системы сотрудничества по фармацевтическим инспекциям (Pharmaceutical Inspection Cooperation Scheme — PIC/S) по проведению ревалидации 2 раза в год путем розлива или рассыпки питательных сред (Media feel test) для подтверждения стерильности продукции. Для нестерильных производств нет четких требований относительно сроков проведения валидации или ревалидации, они будут зависеть от политики предприятия. Если прямое влияние на качество отсутствует, такие изменения некритические и не требуют немедленной валидации. При планировании проведения валидации важным моментом является подход, основанный на оценке рисков. Наиболее часто они связаны с ошибками персонала предприятия, метода, методики, как объективными, так и вызванными недостаточным уровнем квалификации персонала, отклонениями и несоответствием оборудования и измерительных приборов, нарушениями нормативов, контаминацией и пр.
Рекомендации относительно оценки рисков и собственно риск-менеджмента прописаны в документе Q9 Международной конференции по гармонизации технических требований по регистрации лекарственных средств для человека (ICH), они подробно рассматривались на одном из прошедших семинаров и были освещены на страницах нашего издания (см. «Еженедельник АПТЕКА» № 1 (572) от 08.01.2007 г.). На сегодня уже существует методология оценки рисков, которая включает такие методы, как FMEA (Failure Mode Effect Analysis) — анализ результатов возможных отказов, НАССP (Hazard Analysis and Critical Control Points) — система анализа факторов риска и критических точек контроля, PHA (Preliminary Hazard Analysis) — предварительный анализ опасности и др. На сайте ec.europa.eu можно познакомиться с приложением 20 проекта руководства по GMP ЕC, также содержащем рекомендации производителю по проведению оценки риска качества (Quality Risk Management — QRM). В настоящее время группа специалистов во главе с Николаем Ляпуновым готовит в виде нормативного документа рекомендательного характера материалы украинского актуализированного руководства по GMP, полностью соответствующего европейскому документу.
Ю. Подпружников подробно раскрыл тему планирования процесса валидации. Основной план валидации предприятия — VMP (Validation Master Plan) представляет собой общий взгляд на проблему валидации, пути ее решения и может быть составлен как для отдельных технологических макронаправлений, так и для предприятия в целом. План содержит несколько частей. Во вступительной части раскрывается политика валидации, цель плана и общие сведения о предмете валидации (предприятие, участок, система и т.д.). В следующей части определяется организационная структура и область деятельности персонала, связанного с данным процессом, с перечнем конкретных должностей, распределением обязанностей. Краткое описание технических средств, систем, оборудования, процессов, которые подлежат валидации с учетом ее вида, заключается в следующей части VMP. Описание критериев приемлемости в соответствии с детальными мастер-планами (помещения, основные и вспомогательные системы, технологическое оборудование, контроль качества и пр.), формы документации (планы, протоколы, отчеты); планирование, порядок и график проведения валидации; контроль изменений, отклонений и несоответствий; документация предприятия, перекрестные ссылки и сокращения также входят в VMP. Более подробно докладчик рассмотрел критерии приемлемости технических средств, систем, оборудования, процессов, которые подлежат валидации. Эти критерии также относятся к менеджменту рисков и устанавливаются с учетом моделирования ситуации так называемого наихудшего случая во время технологического процесса (например остановка упаковочного аппарата). Существуют верхний и нижний допустимые пределы рабочих параметров оборудования или ведения технологического процесса (от — до или не более — не менее). Если во время проведения валидации процесса в этих граничных пределах достигается необходимый результат, таким образом доказывается его устойчивость, и в середине допустимого предела процесс будет протекать заведомо правильно.

Также в докладе были освещены требования к системе документации, необходимой в процессе проведения валидации, и последовательность ее оформления. Ю. Подпружников акцентировал внимание присутствующих на необходимости письменного разрешения при переходе на каждую последующую стадию валидации или квалификации.
Участники семинара ознакомились с основными этапами проведения валидации. Квалификация проекта (design-qualification — DQ) — первая стадия валидации, проводится сотрудниками предприятия либо приглашенными специалистами относительно нового производства, единицы технологического оборудования, систем либо реконструкции уже имеющегося, позволяет оценить соответствие проекта требованиям GMP. В результате анализа оформляется отчет о проведении квалификации проекта, после чего возможен переход к следующей стадии — квалификации монтажа (installation qualification — IQ), также применимой к единицам оборудования, системам, квалификации функционирования с целью подтверждения правильности их установки, комплектации, наличия всех необходимых контрольно-измерительных приборов с сертификатами калибровки или поверки и т.д. Следующий этап — квалификация функционирования (operational qualification — OQ) включает следующие элементы: испытания на основе знаний, охват верхнего и нижнего пределов рабочих параметров (наихудший случай), проверка функциональных возможностей (параметров) оборудования, работы элементов управления оборудованием и аварийных систем, проверка правильности работы оборудования и его систем, показателей контрольно-измерительных приборов, проверка производственной мощности, наличия и правильности составления инструкций по обслуживанию и эксплуатации оборудования. Согласно проведенной квалификации делаются выводы с оформлением протоколов и отчетов. Квалификацию эксплуатации (performance qualification — PQ) используют при производстве нестерильных препаратов согласно рекомендациям PIC/S PI 006-3 ( ), возможно ее совмещение с валидацией технологического процесса, которая проводится с целью проверки возможности обеспечения производства продукции или работы системы необходимого качества (проверка качества работы оборудования — качество продукции, которая производится на данном оборудовании). Докладчик остановился на рассмотрении каждого варианта проведения валидации процесса. Перспективная валидация (до реализации выпущенной продукции) проводится с целью подтверждения правильности регламентированных параметров технологических процессов, методик (инструкций) по осуществлению операций в соответствии с предназначением оборудования; возможности оборудования обеспечить соблюдение всех параметров ведения техпроцессов и качества продукта; возможности (способности) персонала обеспечить выполнение (соблюдение) операций; воспроизводимости (точности воспроизведения) параметров техпроцесса и обеспечение при этом необходимых показателей качества. Сопутствующая валидация (во время серийного выпуска продукции) заключается в контроле по всем запланированным в протоколе валидации параметрам не менее 3 последовательно выпущенных и не поступивших в реализацию серий препарата с оформлением протоколов и отчетов для каждой серии, далее делают выводы об отсутствии отклонений и соответствии критериям, после чего процесс считается провалидированным. Ретроспективная валидация проводится в процессе производства и реализации препарата с исследованием протоколов производства не менее 10–30 серий препарата по критериям приемлемости и возможным отклонениям. Этот вид валидации не рекомендуется проводить для производства стерильных препаратов.
Что касается валидации аналитических методик, Ю. Подпружников кратко обозначил изменения, требующие доказательства их приемлемости и, соответственно, ревалидации: изменение схем синтеза активных фармацевтических субстанций, состава готовой лекарственной формы и вспомогательного вещества, изменение методик.
Значительная часть доклада была посвящена сложному вопросу валидации компьютеризованных систем (далее — КС), связанных с аналитическим либо технологическим оборудованием. Общие требования к проведению данной валидации заключены в дополнении 11 к GMP ЕС (EU Annex 11 to the EU guidelines of Good Manufacturing Practice for Medicinal Products; ec.europa.eu). Более подробный документ с практическими рекомендациями — «Good Practices For Computerised Systems In Regulated «GXP» Environments PIC/S Guidance Pi 011-3», — согласно которому жизненный цикл КС включает следующие этапы:
- планирование (техзадания);
- установление спецификаций (чему она должна соответствовать);
- программирование;
- тестирование;
- настройки;
- документирование;
- эксплуатация;
- контроль;
- модификация.
Валидация КС является частью их жизненного цикла. Согласно требованиям GMP необходимо составить и постоянно актуализировать подробное описание КС (включая при необходимости схемы). Это техническая документация, доступная пониманию пользователя, в которой описаны принципы, цели, меры безопасности и сфера применения системы, основные особенности той области, где используется компьютер, а также взаимодействие этой системы с другими системами и процедурами.
Согласно документу «Good Practices For Computerised Systems In Regulated «GXP» Environments PIC/S Guidance Pi 011-3» КС состоит из компьютерной системы и контролируемой функции либо процесса, которые находятся в операционной окружающей среде, включающей другие сети, системы, среды, персонал, оборудование и процедуры.
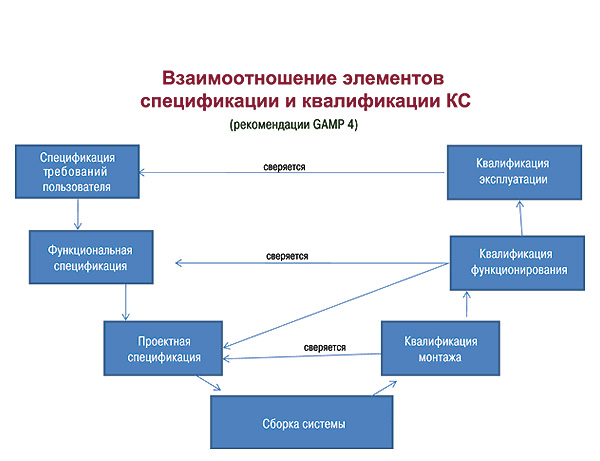
Все КС должны пройти документированную процедуру перспективной валидации или квалификации с оформлением протоколов и отчетов с целью доказательства их применимости. Первый шагом проведения валидации является спецификация требований пользователя (User Requirement Specifications — URS), которая содержит описание пожеланий, требований к КС со стороны пользователя и их аргументация. Следующий шаг — функциональная спецификация (Functional Specifications), которая составляется совместно пользователем и специалистом по компьютерным технологиям и является описанием того, как будет обеспечено соответствие КС требованиям URS. Проектная спецификация (Design Specification) описывает технические характеристики и программные составляющие КС и разрабатывается специалистом по компьютерным технологиям.
Взаимоотношение элементов спецификации и квалификации КС отображено в рекомендациях «Good Automated Manufacturing Practice» (GAMP 4) международной инжиниринговой организации ISPE.
 Доклад директора ДП «Научно-экспертный фармакопейный центр», профессора Александра Гризодуба был посвящен валидации аналитических методик. В Государственную Фармакопею Украины (ГФУ)–2008 впервые внесены рекомендации по проведению валидации методик контроля качества лекарственных средств, а именно, в приложении 2 к ГФУ–2008 в общую статью «Валідація аналітичних методик і випробувань» введен раздел «D. Рекомендації щодо критеріїв при проведенні валідації для методик кількісного аналізу». Данные рекомендации разработаны Фармакопейным центром и уже около 5 лет успешно применяются в Украине. За это время апробировано более 50 методик, на основе которых создано руководство по валидации методик анализа лекарственных средств.
Доклад директора ДП «Научно-экспертный фармакопейный центр», профессора Александра Гризодуба был посвящен валидации аналитических методик. В Государственную Фармакопею Украины (ГФУ)–2008 впервые внесены рекомендации по проведению валидации методик контроля качества лекарственных средств, а именно, в приложении 2 к ГФУ–2008 в общую статью «Валідація аналітичних методик і випробувань» введен раздел «D. Рекомендації щодо критеріїв при проведенні валідації для методик кількісного аналізу». Данные рекомендации разработаны Фармакопейным центром и уже около 5 лет успешно применяются в Украине. За это время апробировано более 50 методик, на основе которых создано руководство по валидации методик анализа лекарственных средств.
А. Гризодуб напомнил, что в соответствии с требованиями ГФУ, гармонизованной с Европейской Фармакопеей, все методики контроля качества лекарственных средств должны быть валидированы, однако в общей статье ГФУ–2001 «Валідація аналітичних методик і випробувань» излагаются лишь общие принципы, а необходимые критерии приемлемости и процедура проведения валидации должны разрабатываться самими предприятиями для конкретных методик с учетом их специфики. В связи с этим фармацевтическими производителями могут быть предложены разные критерии и подходы, которые формально не противоречат требованиям ГФУ, однако будут давать разные выводы по валидации. В соответствии с одним подходом (производителя) методика может считаться валидированной, а в соответствии с другим (контролера) — нет.
Основная цель введения рекомендаций по проведению валидации аналитических методик в ГФУ — это стандартизация данной процедуры и используемых критериев приемлемости, что позволит избежать противоречивых выводов о валидации в разных лабораториях. Докладчик отметил, что предлагаемый подход носит рекомендательный характер, предприятия могут использовать любые другие подходы и критерии, однако им в этом случае придется их научно обосновывать.
Теоретический базис рекомендаций по валидации аналитических методик разработан Фармакопейным центром и опирается на 5 главных принципов:
–первый заключается в использовании линейной модели сложения неопределенностей (согласно ГФУ–2004 «Статистичний аналіз результатів хімічного експерименту»), на которую опирается вся дальнейшая процедура валидации.
- Второй принцип включает стандартизацию процедуры проведения валидации аналитических методик с целью одновременного определения показателей линейности, точности, правильности, что позволяет существенно сократить объем эксперимента. Для лекарственных средств типичным является использование непрямых методов в варианте метода стандарта (спектрофотометрия, хроматография), для которого и были разработаны данные рекомендации.
- Согласно третьему принципу ГФУ–2008 рекомендует представлять концентрацию и аналитические сигналы не в реальных величинах, а в процентах к номинальному (или нормируемому) значению — в нормализованных координатах. Это позволяет сформулировать единые критерии, связанные только с допусками содержания и не зависящие от специфики конкретных веществ.
- Четвертый принцип рекомендаций основан на последовательном систематическом применении принципа незначимости для обоснования критериев приемлемости, который формулируется таким образом: неопределенность методики анализа (или ее составляющая) не должна значимо влиять на принятие решений о качестве лекарственного средства.
- Пятый принцип предусматривает прогноз полной неопределенности анализа. Для подтверждения воспроизводимости методики недостаточно результатов проведения ее валидации в одной лаборатории, поскольку уровень оборудования лаборатории может быть гораздо выше допустимых по Фармакопее требований, потому необходим прогноз полной неопределенности методики в соответствии с этими требованиями.
А. Гризодуб привел практический пример проведения валидации методики спектрофотометрического анализа таблеток амброксола и в завершение доклада подчеркнул, что дополнительные сведения о валидации аналитических методик можно почерпнуть из соответствующих научных публикаций.
 С докладом, посвященным контролю качества очистки оборудования, выступила руководитель департамента развития и опытного производства ООО «Фарма Старт» Наталья Асмолова. Она уточнила, что валидация очистки подразумевает документированное доказательство эффективности удаления остатков с производственного оборудования до определенного допустимого уровня согласно одобренной процедуре очистки. Основная причина ее проведения — предотвращение контаминации фармацевтических продуктов, производимых на одном и том же оборудовании, а цель — доказательство их качества и безопасности. Основные регуляторные требования к проведению валидации очистки изложены в нормативных и методических документах PIC/S, Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administartion).
С докладом, посвященным контролю качества очистки оборудования, выступила руководитель департамента развития и опытного производства ООО «Фарма Старт» Наталья Асмолова. Она уточнила, что валидация очистки подразумевает документированное доказательство эффективности удаления остатков с производственного оборудования до определенного допустимого уровня согласно одобренной процедуре очистки. Основная причина ее проведения — предотвращение контаминации фармацевтических продуктов, производимых на одном и том же оборудовании, а цель — доказательство их качества и безопасности. Основные регуляторные требования к проведению валидации очистки изложены в нормативных и методических документах PIC/S, Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administartion).
Загрязнение (контаминация) выражается количеством остатков в среде, оборудовании или материале. Контаминацию обычно выражают как концентрацию (число, объем) остатков на определенной площади (плоскостная) или в определенном объеме (объемная контаминация). С такой точки зрения чистота понимается как отсутствие остатков в среде, на оборудовании или материале. На практике, в свою очередь, трудно доказать отсутствие загрязнения (за редким исключением), поэтому определение чистоты звучит как низкая степень контаминации, которая ниже определенной предельной концентрации остатков (или критерия приемлемости). Такое понятие чистоты имеет преимущество в том, что ее можно практически достичь и оценить.
В зависимости от типа остатков загрязнение бывает физическое (механическое), химическое (растворимое) и биологическое. В зависимости от способа рассеяния остатков различают равномерное загрязнение, встречающееся редко и характеризующееся простотой пробоотбора и количественной оценки, и неравномерное, для которого необходим тщательный выбор порядка пробоотбора и характерна сложность проведения количественной оценки вплоть до ее полной невозможности. При проведении валидации очистки определяется характер и способ загрязнения, способ рассеяния и доказывается эффективность выполненной очистки.
Большинство процедур очистки направлено на удаление с поверхности оборудования, приспособлений и оснастки остатков следующих типов: исходных действующих веществ и промежуточных продуктов, требующих удаления в первую очередь, с учетом воздействия на пациентов, вспомогательных веществ, частиц, технических, моющих, дезинфицирующих средств, микроорганизмов и пирогенов. Для всех типов остатков необходимо устанавливать критерии приемлемости. При установке допустимых пределов для химических остатков можно использовать различные подходы, основанные на комбинации терапевтической дозы и фактора безопасности, токсичности, пределе аналитического обнаружения остатка, приемлемой концентрации остатка в продукте. Н. Асмолова подробно остановилась на каждом из этих подходов с приведением практических примеров расчетов максимально допустимого переноса остатка в последующий препарат. Она также отметила, что основной проблемой валидации процесса очистки является отбор проб остатков с поверхности оборудования, для правильного выполнения которого необходимо подготовить схему пробоотбора с учетом труднодоступных мест конструкции и с дальнейшим описанием точек отбора в протоколе валидации очистки.
В докладе подробно рассматривались преимущества и недостатки различных методов отбора проб и их практическое применение на предприятии «Фарма Старт».
 Большой интерес у участников семинара вызвал доклад менеджера по управлению валидацией корпорации «Артериум» Евгения Резцова, в котором рассматривались практические аспекты проведения валидации/ревалидации технологического процесса в корпорации. Специалисты ознакомились с организационной структурой службы обеспечения качества и управления влиянием на окружающую среду, нормативными документами корпорации «Артериум» в области управления валидационными процессами, последовательностью проведения валидации технологического процесса, учетом и анализом данных, полученных в результате валидации.
Большой интерес у участников семинара вызвал доклад менеджера по управлению валидацией корпорации «Артериум» Евгения Резцова, в котором рассматривались практические аспекты проведения валидации/ревалидации технологического процесса в корпорации. Специалисты ознакомились с организационной структурой службы обеспечения качества и управления влиянием на окружающую среду, нормативными документами корпорации «Артериум» в области управления валидационными процессами, последовательностью проведения валидации технологического процесса, учетом и анализом данных, полученных в результате валидации.
 На семинаре с докладами выступили специалисты ведущих чешских фирм «Фавея Инжиниринг» и «Блок». О валидации и возможностях изоляторной технологии рассказал представитель фирмы «Блок» Вирт Зденек. Он выделил следующие факторы риска, связанные с рабочим процессом и требующие внедрения изоляторных технологий: химические (запыленность, газы, пары, жидкости, жидкие аэрозоли), биологические (бактерии, вирусы, плесень), специфические условия окружающей среды (низкая относительная влажность, содержание кислорода и пр.). Для защиты рабочего места от этих факторов рекомендуется использовать такие изоляторные технологии, как вытяжные шкафы, боксы безопасности «Biohazard», открытые и закрытые барьеры, изоляторы с предусмотренным классом герметичности 1–4, которые могут предоставить и установить специалисты фирмы «Блок».
На семинаре с докладами выступили специалисты ведущих чешских фирм «Фавея Инжиниринг» и «Блок». О валидации и возможностях изоляторной технологии рассказал представитель фирмы «Блок» Вирт Зденек. Он выделил следующие факторы риска, связанные с рабочим процессом и требующие внедрения изоляторных технологий: химические (запыленность, газы, пары, жидкости, жидкие аэрозоли), биологические (бактерии, вирусы, плесень), специфические условия окружающей среды (низкая относительная влажность, содержание кислорода и пр.). Для защиты рабочего места от этих факторов рекомендуется использовать такие изоляторные технологии, как вытяжные шкафы, боксы безопасности «Biohazard», открытые и закрытые барьеры, изоляторы с предусмотренным классом герметичности 1–4, которые могут предоставить и установить специалисты фирмы «Блок».
 Главный инженер компании «Фавея Инжиниринг» Франтишек Крженек посвятил свой доклад квалификации проектов систем водоподготовки в фармацевтической промышленности и практическому опыту валидации этих систем. Вода является наиболее употребляемой жидкостью в фармацевтической промышленности как составная часть лекарственных средств и агент очистки оборудования, помещений и систем. В зависимости от способа получения и степени чистоты вода классифицируется по следующим видам: очищенная (Purified Water — PW), высокоочищенная (High Purified Water —HPW), для инъекций (Water for injection — WFI). Докладчик подробнее остановился на наиболее распространенном способе производства воды очищенной — электродеионизации, в основе которого лежит применение ионообменных мембран, ионитов и электродов без необходимости воспроизведения химическими реактивами, в результате чего происходит безопасное и более экономичное производство воды высокого качества. Ф. Крженек подчеркнул, что технологии, используемые для очистки и распределения воды очищенной, зачастую оказывают гораздо меньшее влияние, чем такие факторы, как качество монтажа системы, процедуры отбора проб и анализа воды, эксплуатации и обслуживания, хранения данных и т.д. Системы подготовки и распределения воды на фармацевтическом предприятии относят к критичным, поэтому для них важно прохождение всех стадий квалификации, определение параметров функционирования и их критических значений, а также критериев приемлемости.
Главный инженер компании «Фавея Инжиниринг» Франтишек Крженек посвятил свой доклад квалификации проектов систем водоподготовки в фармацевтической промышленности и практическому опыту валидации этих систем. Вода является наиболее употребляемой жидкостью в фармацевтической промышленности как составная часть лекарственных средств и агент очистки оборудования, помещений и систем. В зависимости от способа получения и степени чистоты вода классифицируется по следующим видам: очищенная (Purified Water — PW), высокоочищенная (High Purified Water —HPW), для инъекций (Water for injection — WFI). Докладчик подробнее остановился на наиболее распространенном способе производства воды очищенной — электродеионизации, в основе которого лежит применение ионообменных мембран, ионитов и электродов без необходимости воспроизведения химическими реактивами, в результате чего происходит безопасное и более экономичное производство воды высокого качества. Ф. Крженек подчеркнул, что технологии, используемые для очистки и распределения воды очищенной, зачастую оказывают гораздо меньшее влияние, чем такие факторы, как качество монтажа системы, процедуры отбора проб и анализа воды, эксплуатации и обслуживания, хранения данных и т.д. Системы подготовки и распределения воды на фармацевтическом предприятии относят к критичным, поэтому для них важно прохождение всех стадий квалификации, определение параметров функционирования и их критических значений, а также критериев приемлемости.
Докладчик отметил, что квалификация системы водоподготовки предприятия осуществляется еще с этапа создания концептуального и рабочего проектов системы с подробным анализом рабочей документации. Главный принцип, которому должны соответствовать системы распределения воды, — минимизация возможности создания условий, приемлемых для развития микроорганизмов. Для этого системы должны быть правильно сконструированы с использованием систем очистки, при отсутствии влияния на продукт контактирующих с ним поверхностей, и эксплуатация систем должна проводиться квалифицированным персоналом под непрерывным микробиологическим контролем и обязательной санацией через определенные промежутки времени. Процедуры эксплуатации, очистки и обслуживания систем водоподготовки должны быть надлежащим образом оформлены с обязательной регистрацией проведенных мероприятий и сохранением полученных данных. Некоторые составляющие систем водоподготовки, производимые фирмой «Фавея Инжиниринг», и элементы уже внедренных в производство проектов, были продемонстрированы участникам в виде слайдов.
Наряду с теоретической частью для специалистов была проведена познавательная экскурсия в один из производственных цехов предприятия «Киевмедпрепарат», сертифицированный на соответствие стандартам GMP. Присутствующие также приняли участие в дискуссии по поводу предложенной ситуационной задачи, после которой было проведено тестирование с целью проверки полученных знаний, с чем все без исключения успешно справились. И напоследок с целью усовершенствования дальнейших мероприятий организаторы провели анонимное анкетирование, где каждый смог высказать свои замечания относительно актуальности тематики, уровня освещения материала докладчиками, а также внести пожелания и рекомендации.
В свете грядущих перемен трудно переоценить важность проведенного семинара, доказанную интересом к нему столь значительного количества представителей отечественных фармкомпаний, а уровень организации и информационной насыщенности мероприятия уже который раз свидетельствует о высоком профессионализме сотрудников УЦФИ. n
Анна Бармина, фото Любови Столяр
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим