 Як відомо, Європа вже майже рік живе за правилами нового Регламенту щодо медичних виробів 2017/745 (MDR).
Як відомо, Європа вже майже рік живе за правилами нового Регламенту щодо медичних виробів 2017/745 (MDR).
Наразі вироби, які були законно розміщені на ринку згідно з попередніми Директивами Ради ЄС від 14.06.1993 р. № 93/42/ЄЕС щодо медичних виробів (MDD) та від 20.06.1990 р. № 90/385/ЄЕС щодо активних медичних виробів, які імплантують (AIMDD) до дати набуття чинності MDR, можуть залишатися на ринку до закінчення строку дії їх ЄC-сертифікатів, але не довше, ніж до травня 2024 р.
Проте це є лише перехідним періодом і всі виробники, що мають СЕ-маркування, вже сьогодні адаптують свою систему якості та технічну документацію на вироби відповідно до вимог нового регулювання.
В Україні вже декілька років поспіль МОЗ України, відповідно до щорічних планів діяльності з підготовки проєктів регуляторних актів, розробляє проєкт постанови Кабінету Міністрів України про затвердження нового технічного регламенту щодо медичних виробів, гармонізованого з MDR. Це викликає певне занепокоєння в учасників ринку медичних виробів в Україні.
Дійсно, MDR висуває вищі вимоги до медичних виробів, ніж попередні директиви (це стосується і рекласифікації виробів, і більш суворих вимог до клінічних даних, документації та до постмаркетингового нагляду).
Окрім іншого, MDR деталізує та розмежовує обов’язки учасників життєвого циклу виробу — виробників, уповноважених представників, імпортерів, дистриб’юторів (розповсюджувачів). З нашої точки зору, таке врегулювання повноважень та розмежування ролей є актуальним.
Протягом тривалого часу висловлювалися занепокоєння щодо відсутності чіткого роз’яснення ролі уповноваженого представника. Виробники часто ототожнювали роль уповноваженого представника та дистриб’юторів (імпортерів) та делегували останнім права та обов’язки уповноваженого представника. Водночас ці ролі є цілком різними.
Згідно з визначенням чинного Технічного регламенту щодо медичних виробів, затвердженого постановою КМУ від 02.10.2013 р. № 753: уповноважений представник виробника — будь-яка юридична особа або фізична особа — підприємець, що є резидентом України або зареєстрована відповідно до законодавства України, представництво іноземного суб’єкта господарювання, що має належним чином підтверджені повноваження від виробника вчиняти юридичні дії від його імені стосовно обов’язків виробника, встановлених відповідним технічним регламентом.
Технічний регламент щодо медичних виробів № 753, гармонізований із Директивою 93/42 (MDD), не міcтить визначення та не згадує за текстом таких учасників ринку, як імпортери та дистриб’ютори (розповсюджувачі).
МDR, навпаки, визначає роль кожного учасника. Якщо тезисно, то MDR розмежовує ролі таким чином:
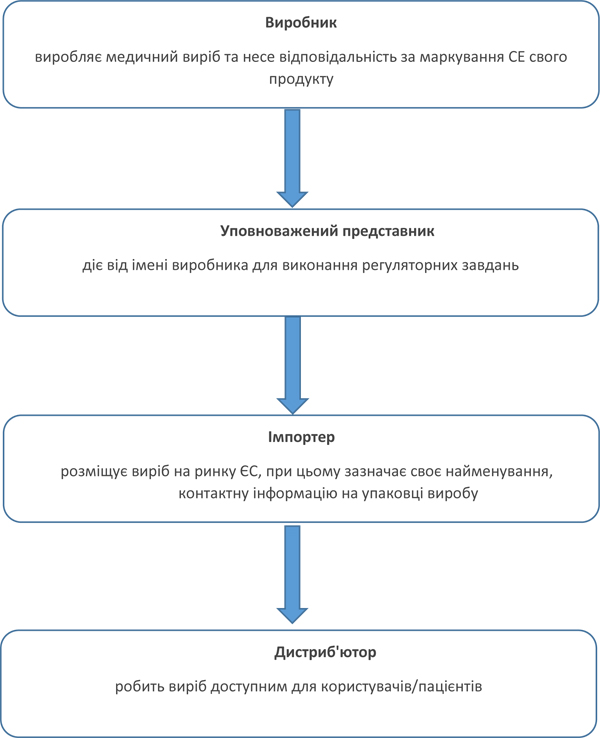
Щодо ролі уповноваженого представника, то MDR визначає, що доручення виробника на призначення уповноваженого представника має містити такі повноваження:
- верифікація факту складання декларації про відповідність вимогам ЄС і технічної документації та, якщо це застосовно, факту проведення виробником належної процедури оцінювання відповідності;
- забезпечення доступу компетентних органів до копій технічної документації, декларації про відповідність вимогам ЄС;
- виконання вимог щодо UDI (Unique Device Identification) реєстрації, а також перевірка факту виконання виробником обов’язків щодо реєстрації;
- надання за запитом компетентного органу всієї інформації і документації, необхідних для підтвердження відповідності виробу;
- направлення виробнику запитів компетентних органів про надання зразків або доступу до виробу, а також верифікація факту надання компетентному органу зразка чи доступу до виробу;
- співпраця з компетентними органами щодо будь-яких попереджувальних та коригувальних заходів, спрямованих на усунення або, якщо це неможливо, зниження ризиків від медичного виробу.
Якщо порівнювати з попередніми Директивами щодо медичних виробів (MDD/AIMDD), то, окрім іншого, MDR висуває більш жорсткі вимоги щодо постмаркетингового нагляду, зобов’язуючи виробників, зокрема, здійснювати збір, аналіз та звітування щодо даних з безпеки виробів на регулярній основі (для виробів IIa, IIb, III класу).
У цьому контексті зазначимо, що згідно з MDR уповноважений представник отримує та обробляє скарги та повідомлення пацієнтів, медичних працівників, користувачів щодо медичного виробу від імпортерів, дистриб’юторів (розповсюджувачів).
Очевидно, що для виконання зазначених завдань уповноважений представник має володіти певною компетенцією, знаннями, досвідом.
Напевно, з цією метою MDR визначає, що уповноважені представники повинні постійно і безперервно мати у своєму розпорядженні принаймні одну особу, відповідальну за дотримання регуляторних вимог, що має необхідні експертні знання щодо регуляторних вимог до медичних виробів. Наявність необхідних експертних знань може бути підтверджена однією з таких кваліфікацій:
a) дипломом, сертифікатом або іншим підтвердженням офіційної кваліфікації, виданими після здобуття університетського ступеня або проходження курсу навчання у сфері права, медицини, фармації, техніки або в іншій відповідній науковій сфері, а також щонайменше одним роком професійного досвіду роботи у відділі нормативно-правового регулювання або в системах управління якістю, пов’язаною з медичними виробами;
b) чотирма роками досвіду роботи у відділі нормативно-правового регулювання або у системах управління якістю, пов’язаною з медичними виробами.
Ця вимога (мати особу, відповідальну за дотримання регуляторних вимог) застосовується MDR до виробника та уповноваженого представника, що підкреслює регуляторну роль останнього, а не просто посередницьку чи торгову.
Відтак MDR визначає та деталізує роль уповноваженого представника, розмежовує його функції та повноваження від повноважень імпортерів та дистриб’юторів.
Якщо згадані положення буде закріплено в новому Технічному регламенті щодо медичних виробів, це надасть учасникам українського ринку медичних виробів більшого розуміння їх функцій, повноважень та зони відповідальності.
Для того щоб якісно та компетентно виконувати свої зобов’язання, європейські уповноважені представники впроваджують систему управління якістю, подібну до системи якості виробників, впроваджують спеціальні процедури, які регламентують питання зв’язку із замовником, ведення документообігу, записів, розгляду скарг, проведення пост-маркетингового нагляду, вилучення продукції з обігу та ін.
В Україні наразі лише одиниці уповноважених представників мають уявлення про таку систему якості або вже впровадили її.
Відтак, є надія, що прийняття нового технічного регламенту щодо медичних виробів деякою мірою підвищить компетенцію уповноважених представників та якість їх послуг.
Дар’я Абулова,
спеціаліст з регуляторних питань компанії JERELO
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим