|
Провідне місце у здійсненні моніторингу за безпекою ЛЗ належить виробнику. Адже якщо до XVII–XVIII ст. відповідальність за безпеку ЛЗ ніс лікар, у XVIII–XIX ст. — лікар і провізор, то, починаючи з XX ст., практично в усьому світі відповідальність за безпеку ЛЗ в першу чергу несе виробник, і вже потім — лікар і провізор. З метою вдосконалення та розвитку системи нагляду за безпекою ЛЗ в Україні, її гармонізації з міжнародними вимогами МОЗ України у 2006 р. затвердило Порядок здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування (наказ МОЗ України від 27.12.2006 р. № 898). Підґрунтям для створення цього підзаконного акта стали такі документи: Закон України «Про лікарські засоби», Порядок державної реєстрації (перереєстрації) лікарських засобів, а також норми міжнародної практики з цих питань, а саме — відповідна настанова Міжнародної конференції з гармонізації технічних вимог до реєстрації лікарських засобів для людини (ICH E2C(R1)), Директива Європейського парламенту і Союзу від 06.11.2001 р. № 2001/83, Постанова (Регламент) ЄС від 22.07.1993 р. № 2309/93 та 9-й том законодавства ЄС про лiкарськi засоби для людини (EudraLex, Volume 9A — Pharmacovigilance for Medicinal Products for Human Use).
Необхiднiсть здійснення фармакологічного нагляду виробником ЛЗ обумовлена не тільки юридичними й міжнародними нормами, але й етичними принципами компаній. Відзначимо, що у виробників ЛЗ, що представляють свої продукти на фармацевтичному ринку не одне десятиліття, вимоги корпоративної політики щодо безпеки своєї продукції досить високі, що у значній мірі визначає імідж компанії. Так, завдяки функціонуванню системи фармакологічного нагляду компанії добровільно вилучають з ринку препарати, у яких співвідношення ризик/користь зміщується у бік ризику, або вносять зміни/доповнення в інструкції для медичного застосування ЛЗ на підставі власних спостережень, рекомендацій регуляторних органів, даних досліджень і сигнальної інформації.
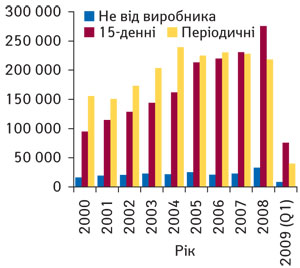
Тому для здійснення моніторингу безпеки ЛЗ виробник або його представник повинен мати належну систему фармакологічного нагляду, що дозволяє отримати оперативну інформацію про всі випадки виникнення ПР. У США у 2008 р. 93,8% звітів та повiдомлень про ПР ЛЗ в Управління з контролю за харчовими продуктами та лікарськими засобами (Food and Drug Administration — FDA) поступило від виробників ЛЗ () (рисунок). Слiд зазначити, що у США перiодичні звiти протягом перших трьох рокiв з мiжнародної дати «народження» препарату подають щоквартально, а потiм — щорiчно (21CFR314.80). До речi, в ЄС перiодичнi звiти подають кожнi пiвроку до початку маркетингу та впродовж двох перших рокiв знаходження препарату на ринку ЄС, раз на рiк — у наступнi 2 роки, а потiм — 1 раз на 3 роки. У нашiй країнi звiт подають один раз на 6 міс — протягом перших 2 років після одержання реєстраційного посвідчення, один раз на рік — протягом наступних 3 років, а далі — один раз на 5 років за умови знаходження ЛЗ на фармацевтичному ринку України.
|
Метою здійснення виробником фармакологічного нагляду є визначення можливих ризиків при застосуванні ЛЗ і прийняття відповідних заходів для захисту здоров’я населення.
Серед основних завдань системи фармаконагляду компанiї — виробника ЛЗ (її представництва) слід зазначити:
- організацію виявлення і обліку ПР;
- верифікацію, систематизацію та аналіз повідомлень, які поступили;
- виявлення підвищення частоти вже відомих ПР;
- інформування регуляторних органів про виявлені ПР у встановленому порядку;
- інформування медичних працівників про ПР та інші проблеми, пов’язані з ускладненнями при застосуванні ЛЗ;
- внесення змін або доповнень в інструкції для медичного застосування;
- організація і проведення післяреєстраційних досліджень з метою отримання додаткової інформації про безпеку ЛЗ.
Систематизація та аналіз інформації про ПР ЛЗ тісно пов’язані з процесами узагальнення і оновлення інформації про безпеку препаратів. На підставі власних даних щодо безпеки застосування ЛЗ, інформації, отриманої від лікарів, регуляторних органів, з даних літератури, при проведенні клінічних досліджень, досліджень щодо безпеки у післяреєстраційний період виробник (або його представник) складає регулярно оновлюваний звіт з безпеки (periodic safety update reports — PSUR; далі — звіт з безпеки).
Концепція звіту з безпеки була розроблена ІІ робочою групою Союзу міжнародних організацій з медичної науки (Council for International Organizations of Medical Sciences — CIOMS) і передбачає його стандартний формат і зміст. Звіт з безпеки — це письмовий звіт, який містить регулярно оновлювану інформацію. Структура та вимоги до заповнення його розділів в Україні затверджені зазначеним вище наказом МОЗ України від 27.12.2006 р. № 898 та повністю гармонiзованi зі свiтовими вимогами.
Основною метою звіту з безпеки є виявити, чи відповідають дані, отримані за звітний період, тим, які зазначені в інформації з безпеки ЛЗ у всіх країнах, де зареєстрований підзвітний препарат.
З метою навчання виробників ЛЗ (або їх представників) в Україні правилам та підходам до створення звітів з безпеки лише протягом 2007–2008 рр. Управлінням післяреєстраційного нагляду ДП «Державний фармакологічний центр» МОЗ України було проведено 5 навчальних семінарів, видано методичні рекомендації «Принципи складання та порядок подання виробником інформації про побічні реакції лікарського засобу» (Київ, 2007 р.). Протягом 2008 р. проведено 217, а за І кв. 2009 р. — 86 консультацій відповідальним за здійснення фармаконагляду у представництвах та на підприємствах з питань фармакологічного нагляду та створення звітів з безпеки.
Результатом проведеної роботи можна вважати надання звітів з безпеки як при перереєстрації, так і проміжних вітчизняними та зарубіжними виробниками ЛЗ (таблиця).
|
Таблиця |
Надходження звітів з безпеки (2007–2009 рр.) |
|
Період |
Кількість звітів |
|
2007 р. |
1334 |
|
2008 р. |
1891 |
|
січень–квiтень 2009 р. |
916 |
Отже, питання створення звітів з безпеки в Україні можна вважати вирішеним.
Однак проблеми існують. При створенні звітів з безпеки виробникам як оригінальних, так і генеричних ЛЗ слід враховувати ПР, виявлені у післяреєстраційний період. Ці дані, тим більше, якщо вони відображені у звіті з безпеки та оцінені як достовірні, повинні бути вчасно, по мірі їх виявлення, відображені у короткій характеристиці препарату (Summary of Product Characteristics — SPC) та інших документах компанії. Такий підхід виробників ЛЗ (або його представників) дозволить уникнути розбіжностей між даними звіту з безпеки, SPC та CCDS (Company core data sheet) — власною підбіркою даних компанії щодо препарату, а також зайвих запитань з боку експертного органу.
Станом на сьогодні в Україні, на жаль, існують розбіжності в інформації з безпеки оригінальних та генеричних ЛЗ. Оскільки генерик по суті є копією оригінального ЛЗ, цього не повинно бути. Отже виробникам генеричних ЛЗ слід усунути таку невідповідність та відобразити це у SPC, звіті з безпеки та інших документах компанії.
Один звіт з безпеки може стосуватися усіх лікарських форм і дозування ЛЗ, а також усіх показань до застосування для однієї діючої речовини у різних лікарських формах. У цьому разі в рамках одного звіту доцільним було б окремо представити дані з безпеки для різних форм випуску, враховуючи, що відрізняється спосіб введення, біодоступність ЛЗ тощо. Проведення аналізу ПР ЛЗ різних форм випуску може стати підставою до внесення змін та доповнень в інструкцію для медичного застосування для конкретної, а не для усіх форм випуску ЛЗ.
Важливо також, що за ідентичності якісного складу препаратів як по діючих, так і по допоміжних речовинах, але за відмінності по кількісному складу, інформація з безпеки повинна бути ідентичною, за виключенням тієї, що стосується дозозалежних ефектів. За наявності відмінностей у складі допоміжних речовин ця інформація також повинна бути відображена, адже допоміжні речовини можуть викликати появу певних ПР чи навпаки.
При підготовці звіту з безпеки для комбінованих препаратів слід враховувати дані з безпеки застосування кожної з діючих речовин, зокрема інформацію про ПР, протипоказання, особливості застосування в окремих груп пацієнтів тощо.
За минулий рік рівень складання локальних звітів деяких вітчизняних та зарубіжних виробників ЛЗ значно зріс, проте актуальними лишаються такі зауваження:
- неправильно вказуються дати звітного періоду, міжнародна дата «народження» ЛЗ, дата складання звіту;
- у розділі 2 («Положення щодо ліцензування в інших країнах світу») часто вказується тільки Україна, а слід зазначати інші країни, де також зареєстрований підзвітний препарат;
- у розділі 5 («Вплив ЛЗ на пацієнта») не зазначається експозиція пацієнтів, частота ПР або інші показники, які б дозволили оцінити вплив ЛЗ на пацієнта, а лише вказуються обсяги продажу підзвітного ЛЗ;
- у розділі 6 («Представлення індивідуальних історій хвороби») відсутня інформація про ПР ЛЗ за даними літератури;
- у розділі 7.3 («Опубліковані дані про дослідження з безпеки») слід надавати опубліковані дані про дослідження з безпеки/огляд літератури, які стосуються не лише підзвітного ЛЗ під певною торговою назвою, але й інших ЛЗ з тією ж самою діючою речовиною;
- у розділі 9 («Загальна оцінка безпеки») слід надавати інформацію про випадки передозування, випадки зловживання, помилкового, нераціонального застосування ЛЗ, випадки застосування препарату під час вагітності та лактації, а не дані з інструкції для медичного застосування.
За кількістю зареєстрованих ПР, особливо отриманих за даними виробника, звіти з безпеки вітчизняних та зарубіжних виробників відрізняються. Це залежить від багатьох чинників, у першу чергу — від організації належної системи функціонування фармакологічного нагляду на виробництві чи у представництві. Звертає на себе увагу той факт, що більшість ПР, представлених у звітах з безпеки на ЛЗ зарубіжного виробництва, надійшла з інших країн світу, а не з України. Отже, збір інформації про ПР ЛЗ представництвами зарубіжних компаній в Україні потребує значних зусиль та вдосконалення.
Наприклад, у звіті з безпеки одного ЛЗ зарубіжного виробництва для лікування пептичної виразки, який зареєстрований майже у 80 країнах світу, надана інформація про понад 1400 ПР (серйозних та несерйозних), з них 384 — шлунково-кишкові розлади, 143 — зміни з боку шкіри та підшкірної клітковини. При цьому у даному звіті не представлено жодних даних щодо випадків ПР, зареєстрованих в Україні. У базі даних Управління післяреєстраційного нагляду ДП «Державний фармакологічний центр» МОЗ України зареєстровано на дану діючу речовину лише 7 ПР, отриманих від лікарів, з них 4 — шлунково-кишкові розлади, 3 — зміни з боку шкіри та підшкірної клітковини. Однак це не є свідченням того, що ПР не виникали у більшої кількості пацієнтів, яким було призначено цей препарат. Останнє ще раз свідчить про необхідність більш активно здійснювати фармаконагляд представництвам та виробникам ЛЗ в Україні.
Не завжди об’єктивними та обґрунтованими є і коментарі до наданої інформації. Адже у звіті з безпеки надається лише точка зору виробника ЛЗ на проблеми з безпеки, який досить часто має тенденцію до мінімізації та розмивання виявлених проблем. Для того щоб об’єктивно оцінити реальну небезпеку підзвітного препарату, виробник повинен встановити причинно-наслідковий зв’язок між ПР та ЛЗ. На жаль, іноді виробник, попри усі існуючі критерії щодо встановлення причинно-наслідкового зв’язку між ПР та ЛЗ, здійснює це упереджено, необ’єктивно, з явним «фоновим уболіванням» за свій препарат. Нагадаємо, що критерії оцінки причинно-наслідкового зв’язку добре відомі та доступні усім, хто займається фармаконаглядом, а експертиза наданої у звіті інформації проводиться об’єктивно і дуже ретельно. Отже, упереджений підхід виробника до оцінки причинно-наслідкового зв’язку між ПР та ЛЗ лише ускладнює оцінку безпеки підзвітного препарату.
Ще про один нюанс щодо змісту деяких розділів звіту з безпеки ЛЗ зарубіжного виробництва. Майже всі звіти з безпеки на ЛЗ зарубіжного виробництва створюються у генеральному офісі компанії, куди надходить інформація про випадки ПР, виявлені в усіх країнах світу, де маркетується підзвітний препарат.
Існують певні міжнародні правила щодо створення звітів з безпеки, які передбачають, що збір даних з безпеки припиняється не менше ніж за 60 днів до дати створення звіту відповідно до міжнародної дати «народження» ЛЗ. Цей час (60 днів) потрібен, щоб провести різнобічний аналіз отриманої інформації, зібраної за період спостереження. Отже представництва усіх країн світу повинні надати до головного офісу усю інформацію про випадки ПР, яка їм відома, до моменту закриття бази даних. Останнє передбачає завчасні дії відповідального за фармаконагляд у представництві щодо своєчасного надання інформації про ПР ЛЗ з певної країни до головного офісу. Діючи у такий спосіб, представництва різних країн вчасно забезпечать генеральний офіс об’єктивною інформацією щодо безпеки підзвітного ЛЗ і ця інформація буде відображена у звіті з безпеки, а також в інших документах компанії (SPC, CCDS тощо). Останнє дозволить уникнути зайвого та невчасного листування регіонального представництва з генеральним офісом у разі виникнення питань з боку експертного органу, якщо останньому відома інформація, яка не відображена у звіті з безпеки.
Звертають увагу на себе ще і такі факти: інколи існує невідповідність між даними, представленими у розділі 4 («Зміни у наданій інформації з безпеки») звіту з безпеки, та тими, що містяться в інструкції для медичного застосування. Так, у звіті з безпеки представник/виробник ЛЗ повідомляє, що ним прийнято рішення про внесення змін та доповнень в інформацію з безпеки ЛЗ, а в інструкції для медичного застосування це не відображено. Останнє призводить до виникнення питань та зауважень з боку експерта.
Або така ситуація: з матеріалів звіту стає відомо, що в інших країнах (виключаючи країну-виробника) за вимогою їх експертних органів були внесені значущі зміни та доповнення до інформації з безпеки підзвітного ЛЗ. Коли виробникові повідомляється про те, що такі самі зміни та доповнення до інформації з безпеки підзвітного ЛЗ слід внести і в Україні, то виникає опір з боку представництва, що вiдмовляється змінювати інформацію з безпеки локально в Україні, мотивуючи це тим, що вона повністю співпадає з SPС країни-виробника. Виникає зачароване коло: з одного боку — в певних країнах світу головний офіс погодився з пропонованими експертним органом змінами та доповненнями та вніс їх, а в Україні для його представництва це — проблема з проблем.
Постає питання: наскільки такі підходи компанії є коректними відносно українських пацієнтів — потенційних споживачів ЛЗ. Зазначимо, що медичне застосування ЛЗ в Україні базується на національному законодавстві, гармонізованому з європейськими стандартами, в основу яких покладено принцип верховенства права та пріоритет забезпечення та захисту життя і здоров’я людини. Оскільки фармакотерапія є базовою складовою медичної допомоги, то при підготовці інструкції для медичного застосування ЛЗ треба керуватися приписами таких правових актів, як Конституція України (ст. 3), Цивільний кодекс України (ст. 283; ст. 284, п. 24), Закон України «Про захист прав споживачів» (ст. 1, п. 1), Закон України «Основи законодавства України про охорону здоров’я» (ст. 6; ст. 39; ст. 43).
Хотілося б, щоб інформація з безпеки оновлювалася централізовано у компаніях по мірі її виявлення та оцінки з наступним відображенням в усіх значущих документах компанії. Винятком можуть бути ситуації, коли йдеться про генетичні чи фенотипічні особливості медичного застосування підзвітного ЛЗ, що потребує внесення змін в інформацію про ЛЗ лише у певних країнах світу. Адже при проведенні терапії лікар та пацієнт мають право володіти у повному обсязі вірогідною інформацією щодо ефективності та безпеки ЛЗ, яка відома виробникові.
ДП «Державний фармакологічний центр» МОЗ України,
Національний фармацевтичний університет, Харків,
О.П. Вікторов, О.В. Матвєєва,
І.О. Логвіна, О.В. Вірста, Д.С. Полякова
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим