|
|

Открывая семинар, первый заместитель Государственного секретаря Министерства здравоохранения Михаил Пасечник подчеркнул, что проблема безопасности ЛС становится все более актуальной во всем мире. Это обусловлено тем, что в медицинской практике увеличивается доля новых препаратов с высокой биологической активностью. Применение таких ЛС может сопровождаться ПР разной степени тяжести. Так, по разным оценкам (Dormann H., 2000), частота возникновения ПР ЛС у госпитализированных больных составляет 1,5–35%, что приводит к увеличению сроков госпитализации от 1 до 5,5 дня. По данным ВОЗ, среди причин смерти ПР ЛС занимают 5-е место в мире после сердечно-сосудистых заболеваний, заболеваний дыхательной системы, онкологических заболеваний и травм. Экономические затраты, связанные с дополнительными мероприятиями, направленными на устранение последствий таких патологических состояний, например, в США составляют около 100 млн долл. Аналогичная ситуация в большинстве стран Европейского Союза. Именно поэтому практически во всех странах мира создаются и функционируют Национальные центры по контролю безопасности ЛС.
Как подчеркнул М. Пасечник, основными путями решения проблемы предупреждения осложнений при медицинском применении ЛС, является как производство и внедрение на фармацевтический рынок новых, более качественных и безопасных лекарственных препаратов, соответствующих требованиям GLP, GCP, GMP, GDP и GPP, так и создание действенной системы контроля за безопасностью ЛС — фармакологического надзора.
Осуществление фармаконадзора за ПР ЛС является одним из приоритетных направлений национальной политики в сфере обращения ЛС. В решении этого вопроса уже имеются определенные сдвиги как правового, так и организационного характера. Однако реальный потенциал, который имеется в Украине, полностью не используется. Во всем мире основное внимание при контроле безопасности ЛС направлено на производителя. В Украине же эта обязанность в основном возлагается на врачей (Закон Украины «О лекарственных средствах», 1996; Приказы МЗ Украины № 347 от 19.12.2000, № 51 от 08.02.2001, № 52 от 08.02.2001, № 292 от 16.07.2001, № 497 от 12.12.2001). В настоящее время разрабатывается национальная лекарственная политика Украины, положения которой соответствуют рекомендациям ВОЗ и учитывают интересы производителей фармацевтической продукции, регулирующих органов, а также потребителей ЛС. Как отметил М. Пасечник, в проекте Закона «О внесении изменений и дополнений в Закон Украины «О лекарственных средствах» особое внимание уделено вопросам фармакологического надзора, учтен международный опыт в сфере контроля за ПР ЛС. В соответствии с одним из положений проекта для обеспечения поступления адекватной и оперативной информации о предполагаемых и возникших ПР ЛС обязанностью производителей ЛС и товаров медицинского назначения станет создание соответствующих структурных подразделений, занимающихся фармаконадзором за ПР ЛС.
Заведующий сектором координации и контроля клинических испытаний лекарственных средств ГФЦ МЗ Украины, доктор медицинских наук, профессор Владимир Мальцев в своем докладе «Организация фармакологического надзора в Украине» отметил, что фармаконадзор (pharmacovigilance — фармакобдительность), согласно Директиве 2001/83/ЕС Европейского парламента и Совета ЕС от 6.11.2001 г. «О своде законов Сообщества в отношении лекарственных препаратов для человека» (раздел IХ «Фармаконадзор», статьи 101–108) (Фармацевтический сектор: основы современного законодательства в Европейском Союзе. — К.: МОРИОН, 2002. — 96 с.), — это государственная система сбора данных, которая на основании полученной информации о ПР на лекарственные препараты в условиях их обычного применения обеспечивает принятие соответствующих регуляторных решений в отношении лицензированных ЛС. Эта система используется для сбора данных, необходимых для осуществления надзора за ЛС, при особом контроле в отношении ПР у человека, а также для научной оценки этой информации. Полученные сведения должны быть сопоставлены с данными по изучению использования лекарственных препаратов. Система фармаконадзора также обеспечивает изучение всех имеющихся данных о неправильном применении ЛС и случаях злоупотребления ими, что может повлиять на оценку соотношения польза/риск при назначении и приеме ЛС.
Проблема безопасного применения ЛС и контроля за процессом создания современной системы фармаконадзора является одной из наиболее важных и актуальных в здравоохранении. Переломным в этом отношении для международного сообщества стал 1961 г. в связи с «талидомидовой трагедией»1. Выявление «грей-синдрома» у детей при применении хлорамфеникола, запрет на использование диэтилстильбэстрола (1971) в связи с развитием злокачественных опухолей половых органов у девочек, матери которых принимали этот препарат, — все это заставило мировую медицинскую общественность и правительственные учреждения сосредоточить усилия на проведении мероприятий, которые обеспечили бы безопасность пациентов как в ходе клинических испытаний (КИ) новых ЛС, так и при их медицинском применении. Следует подчеркнуть, что наряду с приведенными выше «большими катастрофами» было выявлено немало других «мелких» осложнений, которые не вызвали такой широкой резонанс в обществе, однако отрицательно повлияли на качество жизни пациентов. Все эти негативные примеры стали препятствием для дальнейшего продвижения на рынок целого ряда опасных ЛС: клозапина, дипирона, изоксикама, практолола, талидомида, бромкриптина, диэтилстильбэстрола, фенилпропраноламина и др. (Дюкс М., 1995).
Значительное внимание этой проблеме уделяет ВОЗ, которая координирует и поддерживает деятельность национальных центров по контролю безопасности ЛС более чем в 70 странах мира. Центры принимают участие в международной программе по мониторингу ПР ЛС (вступила в действие в 1964 г.) (WHO, 1972). В 2002 г. Украина стала полноправным членом ВОЗ и регулярно предоставляет данные о возникновении ПР ЛС в Центр ВОЗ по мониторингу ПР ЛС (г. Упсала, Швеция). В настоящее время в банке данных Центра ВОЗ более 2 млн сообщений о нежелательных ПР лекарств (НПРЛ). Ежегодно в банк данных ВОЗ поступает около 200 000 сообщений о НПРЛ.
Каковы условия участия в Международной программе ВОЗ по мониторингу лекарств? Кроме наличия национального центра по контролю за безопасностью ЛС, утвержденного МЗ, необходимо наличие программы по сбору спонтанных сообщений и компетентного персонала, которые соответствовали бы требованиям ВОЗ. Основными задачами Центра ВОЗ по мониторингу лекарств являются:
•?сбор, анализ, систематизация сообщений о ПР на международном уровне;
•?формирование международной базы данных о ПР;
•?распространение информации;
•?научно-исследовательская работа (разработка методов и стандартов для оценки риска и пользы и др.);
•?международная гармонизация систем мониторирования ПР ЛС.
Профессор В. Мальцев подробно осветил основные моменты становления системы фармаконадзора в Украине, задачи и направления деятельности Отдела фармакологического надзора ГФЦ МЗ Украины, а также этапы формирования нормативной базы фармаконадзора в Украине, в значительной мере основанной на Руководстве Международной конференции по гармонизации технических требований к регистрации лекарственных средств для человека (ICH GCP, 1996 г.) и Директиве Совета Европейского Экономического Сообщества по вопросам фармакологического надзора 75/319 ЕЭС. Были рассмотрены требования к сбору и обработке данных, необходимых для осуществления особого контроля за ПР ЛС, которые возникают при КИ и медицинском применении, а также к проведению научной оценки этой информации. Подробно эти и другие вопросы изложены в ряде публикаций «Еженедельника АПТЕКА» (№ 5 (376) от 10.02.2003 г., № 45 (366) от 18.11 2002 г.) и в книге «Фармацевтический сектор: фармаконадзор за лекарственными препаратами для человека», выпущенной ГФЦ совместно с издательством «Морион».
Требования к фармаконадзору в соответствии с руководствами ЕС на этапе медицинского применения ЛС включают: системы спонтанных (случайных) сообщений; системы обращения с данными, полученными от производителей, и системы обращения с данными из других источников. Собранные данные о ПР ЛС поступают в Национальный центр по фармаконадзору с последующей процедурой передачи информации о результатах фармаконадзора внутри ЕС/ВОЗ.
Случайное сообщение (Spontaneous Report) — сообщение, в котором описана предполагаемая ПР у пациента, принимающего один или несколько ЛС, и которое не является результатом проведенного КИ. Случайные сообщения получают:
•?компания — производитель ЛС;
•?национальный регуляторный орган;
•?другие организации.
Система спонтанных (случайных) сообщений характеризуется простотой, возможностью подтверждения представленной информации и постоянным контактом с отправителями информации (рис. 1).
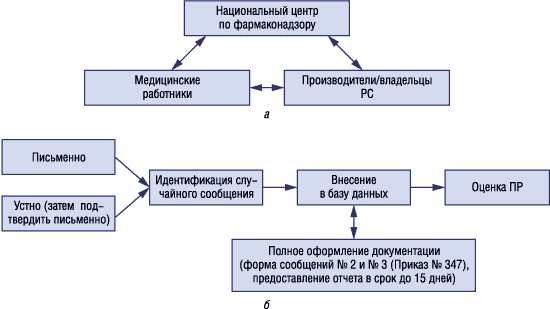 |
Рис. 1. Система спонтанных (случайных) сообщений в странах ЕС и в Украине (адаптировано: Приказ МЗ Украины № 347 от 19.12.2000 г.)
Составными положениями в системе фармаконадзора для производителя/владельца РС являются:
•?подготовка отчетов о случаях ПР;
•?регулярно обновляемые отчеты по безопасности ЛС (PSUR);
•?пострегистрационные исследования, финансируемые производителем/владельцем РС.
Цель предоставления регуляторным органам регулярно обновляемых отчетов по безопасности ЛС или PSUR (Periodic Safety Update Reports) заключается в установлении соответствия информации, поступившей и зарегистрированной в течение отчетного периода, ранее имевшимся данным о безопасности ЛС, что позволяет сделать вывод о целесообразности внесения изменений в информацию о препарате. При подготовке отчетов необходимо:
•?гарантировать точность, своевременность сбора и передачи информации;
•?обеспечить проверку всех данных;
•?гарантировать, что все отчеты при необходимости будут дорабатываться для облегчения проведения оценки причинно-следственной связи;
•?гарантировать предоставление дополнительной информации об объеме продаж или количестве назначений соответствующего ЛС.
Создавая внутреннюю систему фармаконадзора, производитель/владелец РС обязан:
•?включить в штат сотрудника или сотрудников, отвечающих за фармаконадзор (обязательное условие — опыт работы в области фармаконадзора (подготовка), медицинское образование или возможность обратиться к такому лицу);
•?гарантировать, что информация о ПР, которая поступает к производителю/владельцу РС и агентам по продаже ЛС, будет обрабатываться и станет доступной регуляторному органу;
•?регистрировать все серьезные ПР, о которых сообщают медицинские работники, и информировать регуляторный орган немедленно и не позднее 15 дней с момента получения информации;
•?предоставлять дополнительную информацию для оценки соотношения польза/риск в связи с использованием ЛС;
•?предоставлять информацию об объеме продаж или количестве выписанных рецептов на соответствующее (подозреваемое) ЛС;
•?документировать информацию обо всех возможных ПР как в Украине, так и в других странах (банк данных информации о ПР как конкретного производителя, так и банк данных ВОЗ).
В. Мальцев отметил, что система фармаконадзора тесно связана с КИ лекарственных препаратов, так как начинает действовать на этапе медицинского применения ЛС (III–IV фаза КИ). Обеспечение качества КИ ЛС — система мер, способствующая проведению КИ и гарантирующая соответствие полученных данных требованиям протокола, принципам GCP и национальному законодательству, зиждется на двух «китах» — спонсоре (мониторинг и аудит) и государстве (проведение инспекционных проверок). Мониторинг — процедура контроля за ходом КИ и обеспечения его проведения, сбора данных и представления результатов исследования согласно протоколу, стандартным операционным процедурам (СОП), GCP и действующим нормативным требованиям. Монитор — лицо, назначаемое спонсором (производителем ЛС) и контролирующее проведение КИ в соответствии с протоколом. Инспекция — процедура официальной проверки представителем уполномоченного регуляторного органа документов, помещений, записей, а также других материалов, которые рассматриваются представителем регуляторного органа как относящиеся к КИ и которые могут находиться в медицинском учреждении, офисах спонсора, а также другого учреждения.
Клинические постлицензионные исследования по безопасности ЛС (Post-authorisation Safety Study) — это фармакоэпидемиологические или КИ, выполняемые в рамках зарегистрированных показаний к применению ЛС, которые проводятся в целях идентификации или определения количества рисков в отношении безопасности лицензированного ЛС и полностью или частично финансируются фармкомпаниями. Целью проведения постлицензионных КИ является:
•?идентификация проблем, связанных с безопасностью применения ЛС, ранее не являвшихся очевидными;
•?исследования возможного риска (подтверждения наличия причинно-следственной связи);
•?подтверждение предполагаемого профиля безопасности ЛС в условиях реализации;
•?определение количества установленных ПР и идентификация факторов риска.
Проведение таких КИ в первую очередь целесообразно для ЛС с новой химической структурой или новым механизмом действия; препаратов, имеющих высокоспецифическую область применения, что обусловливает необходимость наблюдения специалиста, если существуют неопределенность относительно токсического эффекта у животных, клинической значимости ЛС или профиля безопасности. Постлицензионные КИ проводятся при необходимости более точного определения количества ПР, обнаруженных во время ранее проведенных КИ; с целью изучения факторов риска возникновения ПР ЛС.
Комментируя результаты инспекционных проверок КИ, В. Мальцев отметил, что сектором координации и контроля КИ лекарственных средств ГФЦ МЗ Украины в 2002 г. 3 из 43 КИ ЛС было приостановлено, 1 — прекращено.
В целом, основанием для запрета (остановки) и изъятия ЛС из обращения на территории Украины (Приказ МЗ Украины № 497 от 12.12.01) с последующей остановкой действия или аннулированием регистрационного свидетельства, в случае возникновения ПР являются:
•?сообщения о ПР или смерть людей при применении ЛС;
•?собщения о ПР, не указанных в регистрационных документах;
•?использование ЛС, которое привело или может привести к тяжелым последствиям для здоровья и жизни людей в связи с возникновением серьезной ПР.
Основанием для запрета (остановки) или изъятия из обращения на территории Украины некачественного ЛС является:
•?установление несоответствия качественного и количественного состава ЛС, заявленному в регистрационном свидетельстве;
•?для инфузионных растворов, инъекционных лекарственных форм, которые непосредственно контактируют с кровью, — негативные результаты лабораторных исследований одной серии, для других лекарственных форм — трех серий;
•?установление факта фальсификации ЛС.
В докладе высказан ряд замечаний к работе региональных отделений сектора координации и контроля клинических испытаний лекарственных средств ГФЦ МЗ Украины:
1. Предоставляемая информация о ПР (карты-сообщения № 137/о) является неполной, небрежно оформленной, не поддающейся анализу.
2. Руководители региональных отделений должны контролировать все случаи серьезных ПР (сбор уточненной информации для выявления причинно-следственной связи ПР и подозреваемого ЛС);
3. Не отработан механизм взаимодействия сотрудников региональных отделений с органами областных управлений здравоохранения.
|

Сергей Сур, заместитель главного государственного инспектора Украины по контролю качества лекарственных средств, в докладе «Вопросы фармаконадзора с точки зрения качества лекарственных средств» отметил, что согласно определению ВОЗ, фармаконадзор является наукой и деятельностью, направленной на выявление, оценку и предупреждение ПР или других возможных проблем, связанных с ЛС (The Importance of Pharmacovigilance. Safety Monitoring of medicinal products. WHO, 2002). Одна из таких проблем — контроль качества ЛС (выявление фальсифицированных и субстандартных лекарственных препаратов) — непосредственная задача ГИКК.
Каких же взглядов в отношении качества ЛС, по мнению докладчика, традиционно придерживается большинство врачей и каких — сотрудник Государственной инспекции по контролю качества ЛС?
Врач:
•?состав и свойства ЛС неизменны вчера, сегодня и завтра;
•?при применении ЛС проявляет предвиденный терапевтический эффект;
•?это относится к одним и тем же ЛС, выпускаемым различными производителями.
Сотрудник ГИКК:
•?разные серии одного препарата могут отличаться как в пределах требований АНД, так и относиться к субстандартным ЛС;
•?ЛС могут быть фальсифицированными (содержать недостаточное количество активных ингредиентов, не содержать активных ингредиентов, содержать незадекларированные ингредиенты и т.д.);
•?одни и те же ЛС, выпущенные различными производителями, могут иметь разные свойства.
С. Сур привел сравнительные данные ВОЗ и полученные в Украине по количеству уведомлений о случаях фальсификации ЛС (табл. 1).
Таблица 1
Данные ВОЗ и полученные в Украине по количеству уведомлений о фальсифицированных ЛС
(Варченко В.Г., Пилипенко І.В., Остраниця Т.В. // Вісн. фармакології і фармації. — 2003. — № 1)
|
Уведомления о случаях фальсификации ЛС |
База данных ВОЗ |
Украина |
|
Всего случаев |
771 |
110 |
|
Отсутствуют активные компоненты |
60% |
15,8% |
|
Незадекларированные компоненты |
16% |
16,8% |
|
Правильные компоненты в соответствующих количествах |
7% |
44,2% |
|
Отклонения в количественном содержании активных компонентов |
17% |
23,2% |
Сегодня процент субстандартных и фальсифицированных ЛС на рынке Украины может значительно превышать процент возникновения ПР при применении качественных ЛС. Отсутствие терапевтического эффекта может быть обусловлено:
•?отсутствием или несоответствием содержания активных веществ в ЛС, заявленному в АНД, неоднородностью препарата;
•?наличием токсических примесей, других незадекларированных активных веществ;
•?несоответствием биодоступности препарата (в том числе по фармакотехнологическим показателям).
Система фармакологического надзора внедрена в лечебно-профилактических учреждениях (ЛПУ) НАН и АМН Украины, клинических НИИ и учреждениях МЗ, ЛПУ, непосредственно подчиненных управлениям здравоохранения местных госадминистраций. Не охвачены системой ведомственные и частные ЛПУ, частные врачи и стоматологи, фармацевтические учреждения. Нерешенной остается проблема самоназначений и самолечения.
Информацию о возникших ПР ЛС во всем мире предоставляют:
•?клиники и научно-исследовательские учреждения;
•?врачи всех специальностей (однако по данным The Importance of Pharmacovigilance. Safety Monitoring of medicinal products. WHO, 2002, лишь 5% врачей принимают активное участие в системах фармаконадзора);
•?фармацевты (например, в Японии фармацевты ведут учет аллергических и других ПР ЛС у отдельных пациентов в целях безопасного применения ими этих ЛС, получая информацию от пациентов и из историй болезни (Ямамото Н.//Фармацевт. журн., 2002. — № 2));
•?пациенты (потребители) и общественные организации.
В последнее десятилетие процессы глобализации, развитие свободной торговли и коммуникаций, использование интернета значительно изменили доступ к ЛС и информации о них. Это привело к появлению новых обстоятельств, которые влияют на безопасность ЛС (The Importance of Pharmacovigilance. Safety Monitoring of medicinal products. WHO, 2002):
•?нелегальная продажа ЛС и наркотических средств через интернет;
•?расширение практики самолечения;
•?нерациональная и потенциально небезопасная практика предоставления гуманитарной помощи;
•?рост производства и продаж фальсифицированных и субстандартных ЛС;
•?более широкое применение средств народной медицины за пределами стран, в которых существуют древние культурные традиции их применения (не говоря уже о генетической предрасположенности и иммунной готовности организма);
•?увеличение применения ЛС растительного происхождения и ЛС народной медицины одновременно с традиционными препаратами, в результате чего повышается риск возникновения ПР при их взаимодействии и др.
В связи с этим возникает потребность в расширении сферы фармаконадзора и включения в нее, помимо традиционных ЛС, средств народной медицины, препаратов на основе растительного сырья, препаратов крови, иммунобиологических препаратов, медицинской техники и вакцин.
Участие ГИКК в системе фармаконадзора заключается в сборе информации, поступающей от пациентов и сотрудников аптек, о ПР ЛС и ее передача в ГФЦ, проверка ЛС, которые вызвали ПР, в отношении возможной их фальсификации или на соответствие требованиям АНД, принятие административных мер в соответствии с Приказом МЗ Украины № 497 от 12.12.2001 г. по изъятию ЛС из оборота.
Заместитель заведующего сектором координации и контроля клинических испытаний лекарственных средств ГФЦ МЗ Украины, доктор медицинских наук, профессор Алексей Викторов в докладе «Контроль за побочными реакциями/действиями лекарственных средств при их медицинском применении в Украине» привел общие положения, касающиеся развития ПР ЛС:
•?любое ЛС может вызвать развитие ПР;
•?ПР ЛС различны, могут быть специфичными или повторять клинические проявления болезни;
•?больные (в педиатрии — родители или опекуны, в психиатрии — опекуны) должны быть на 100% информированы о возможных ПР ЛС, особенно при продолжительной фармакотерапии ЛС, которые способны повреждать ткани, клетки, органы;
•?врач обязан регистрировать каждый случай ПР ЛС и своевременно информировать об этом органы государственной регуляторной системы.
К факторам, способствующим возникновению ПР ЛС, относятся:
1. ПР, не связанные с действием ЛС:
•?особенности организма больного (возраст, пол, генетические особенности, склонность к аллергическим реакциям, специфика течения заболевания, вредные привычки);
•?внешние, по отношению к больному, факторы (экологическая среда, условия труда и др.).
2. ПР, связанные с действием ЛС:
•?клинико-фармакологическая характеристика ЛС, адекватность выбора препарата;
•?способ применения препарата;
•?взаимодействие ЛС при полипрагмазии (одновременном назначении нескольких препаратов) (рис. 2).
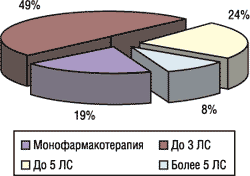 |
Рис. 2. Частота случаев возникновения ПР в зависимости от количества назначаемых ЛС (по данным спонтанных сообщений, 1996–2003 гг.)
Причинами ограниченного поступления информации о ПР ЛС является:
•?неумение распознать симптомы ПР ЛС;
•?отсутствие или ограниченность знаний об особенностях ПР ЛС;
•?упущения в работе регуляторных органов (отсутствие на местах карт спонтанных сообщений и т.д.).
Наличие сообщений о ПР ЛС не является основанием для занесения этой информации в Национальный банк данных. К основным критериям обоснования причинно-следственной связи (табл. 2) между подозреваемым проявлением ПР и применяемым ЛС относят:
Таблица 2
Доказательства наличия причинно-следственной связи (Флетчер Р. и соавт., 1998)
|
Критерий |
Комментарий |
|
Последовательность во времени |
Причина предшествует эффекту |
|
Сила связи |
Высокий относительный риск |
|
Зависимость эффекта от дозы |
При усилении воздействия заболеваемость повышается |
|
Обратимость |
При ослаблении воздействия заболеваемость снижается |
|
Устойчивость |
Эффект наблюдается разными исследователями независимо от места, условий и времени |
|
Биологическое правдоподобие |
Эффект согласуется с современными научными представлениями |
|
Специфичность |
Одна причина приводит к одному эффекту |
|
Аналогия |
Причинно-следственная связь уже установлена для подобного воздействия или заболевания |
•?взаимосвязь между фармакологическими свойствами ЛС и проявлением его ПР;
•?обоснование или исключение других возможных причин.
В целом, за период с 1996 г. по І квартал 2003 г. в ГФЦ поступило следующее количество спонтанных сообщений о ПР на ЛС (рис. 3).
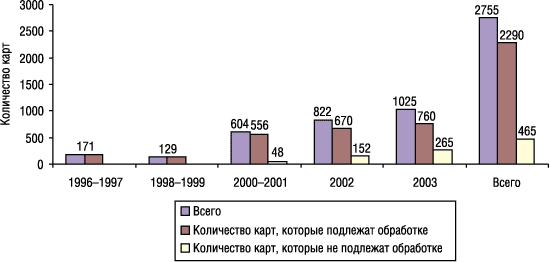 |
Рис. 3. Динамика поступления спонтанных сообщений о случаях ПР при использовании ЛС (по данным отдела фармацевтического надзора ГФЦ, 1996–2003 гг.)
Причем в общей структуре данных о возникших ПР ЛС частота развития ПР в связи с применением ЛС отечественного и зарубежного производства примерно соответствует количественному соотношению отечественной и зарубежной продукции, представленной на фармацевтическом рынке Украины (рис. 4).
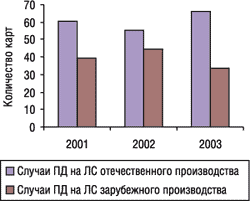 |
Рис. 4. Распределение случаев ПД ЛС в зависимости от происхождения ЛС (по данным спонтанных сообщений, 2001–2003 гг.)
А. Викторов напомнил о необходимости выполнения производителями Приказа МЗ Украины № 347 от 12.19.2000 г. «Об утверждении Инструкции об осуществлении надзора за побочными реакциями/действиями лекарственных средств», согласно которому:
п. 3.1. Информация о побочных реакциях/действиях лекарственных средств поступает в Центр от:
— производителей/владельцев регистрационного свидетельства или их уполномоченных представителей
Комментарии: По состоянию на 20.05.2003 г. от отечественных производителей не поступило ни одного сообщения о ПР ЛС (от представительств зарубежных компаний поступило 37 сообщений о случаях ПР ЛС в Украине и около 1500 — о случаях ПР ЛС за ее пределами).
п. 5.3. Производители/владельцы РС (или их уполномоченные представители) ЛС, разрешенного к медицинскому применению, обязаны предоставлять Центру информацию о побочном действии ЛС в установленной форме в соответствии с приложением 4 к настоящей Инструкции (форма 3).
п. 5.4. Производители/владельцы РС (или их уполномоченные представители) обязаны предоставлять Центру обобщенный отчет относительно побочного действия ЛС:
•?один раз в 6 месяцев — в течение 2 лет после получения РС;
•?ежегодно — в течение последующих 3 лет;
•?в дальнейшем — 1 раз в 5 лет (при условии перерегистрации ЛС).
Комментарии: По состоянию на 20.05.2003 г. Центр получил более 380 писем от отечественных производителей о предоставлении им информации о наличии/отсутствии случаев ПР ЛС своего производства. Центром только в течение 2002–2003 гг. была подготовлена информация о 687 ЛС отечественного и 74 ЛС зарубежного производства в связи с их перерегистрацией в Украине.
Подводя итоги работы ГФЦ по взаимодействию с производителями, профессор А. Викторов подчеркнул, что:
1. Отечественные производители ЛС не выполняют как данный приказ, так и другие соответствующие приказы МЗ Украины (№ 292 от 16.07.2001 г. «Об усовершенствовании организации предоставления информации о побочной реакции лекарственных средств») и пока еще не принимают участия в создании Государственной системы фармакологического надзора.
2. Собственная система контроля за безопасностью продукции при ее медицинском применении у них отсутствует.
3. Практически отсутствует взаимодействие с Центром в сфере безопасности лекарств при их медицинском применении.
4. Производители пассивно реагируют на заключения экспертов Центра, которые имеют прямое отношение к качеству жизни пациентов при медицинском применении их продукции, а следовательно, и более широкому аспекту — правам человека.
Заведующая отделом фармакологического надзора ГВЦ МЗ Украины, кандидат медицинских наук Марина Шараева в докладе «Источники информации и порядок сообщений о побочном действии лекарственных средств при медицинском применении» отметила, что в обязанности производителей ЛС входит:
•?проведение фармаконадзора;
•?введение должности уполномоченного лица для проведения фармаконадзора на каждом предприятии;
•?составление, регулярное обновление и представление в регулирующие органы отчетов (регулярно обновляемые отчеты по безопасности (PSUR)). Отчеты служат основой для перерегистрации и входят в регистрационное досье;
•?публикация информации о ПР ЛС в специальном бюллетене;
•?возможное аннулирование РС на основании данных фармаконадзора;
•?предоставление необходимой помощи регуляторным органам в проведении процедуры сбора и оценки информации о ПР ЛС («прозрачность» данных, доступность информации);
•?составление экстренных сообщений;
•?сотрудничество с ВОЗ.
Обязанности уполномоченного лица по фармаконадзору на предприятии заключаются в:
•?создании и поддержании в действии системы сбора и оценки данных по подозреваемым ПР ЛС;
•?выполнении своих обязанностей в отношении оповещения о предполагаемых ПР;
•?выполнении своих правовых обязательств в отношении составления и предоставления регулярно обновляемых отчетов по безопасности (PSUR);
•?предоставлении дополнительной информации на запросы регуляторных органов, необходимой для оценки пользы и риска при применении ЛС;
•?определении и оценке сигналов о подозреваемом ПР ЛС (сигнал — предостережение из любого источника, о том что ЛС может ассоциироваться с ранее невыясненным вредным воздействием, или известное вредное воздействие может количественно или качественно отличаться от уже известных ожиданий).
Заведующий кафедрой клинической фармации Национального фармацевтического университета, доктор мелицинских наук, профессор Игорь Зупанец, заведующий кафедрой клинической фармакологии Украинской государственной стоматологической академии, доктор медицинских наук, профессор Виктор Бобырев и заведующий отделом клинической фармакологии с лабораторией функциональной диагностики Института кардиологии им. Н.Д. Стражеско АМН Украины, доктор медицинских наук, профессор Алексей Викторов в докладе «Задачи по преподаванию вопросов безопасности лекарств студентам, врачам и провизорам на современном этапе» ознакомили с новыми учебными планами, в которые включены вопросы, касающиеся изучения системы фармаконадзора и безопасности ЛС в практике врачей и фармацевтов.
На семинаре с докладом «Фармацевтический надзор в динамике: развитие на современном этапе» выступили Рональд Мейбум (Центр ВОЗ по мониторингу ЛС, г. Упсала, Швеция), а также заведующий отделом Института эндокринологии и обмена веществ им. В.П. Комиссаренко АМН Украины, доктор медицинских наук, профессор Вадим Корпачев («Проблема безопасности инсулиновых препаратов»). Ввиду актуальности тем выдержки из этих докладов будут опубликованы отдельно в ближайших номерах «Еженедельника АПТЕКА».
Максим Плошенко
Фото Евгения Чорного
1 В 1961 г. в Западной Германии участились случаи возникновения фокомегалии (гипопластические или апластические деформации конечностей) у новорожденных. Причиной этого стал препарат талидомид — снотворное ЛС небарбитуровой природы, тератогенное действие которого проявляется в I триместре беременности. Выяснилось, что в Западной Германии с такими дефектами родились около 10 000 детей, из которых выжили 5000 (высокая смертность обусловлена другими деформациями), Великобритании — 600 детей, из которых выжили 400. В США препарат не был допущен на рынок в связи с наличием данных об индуцированном гипотериозе и периферической невропатии. Однако несколько случаев фокомегалии у детей, матери которых принимали участие в КИ, были зарегистрированы в тот же период (тесты на тератогенность еще не были общепринятыми). Позднее экспериментально было подтверждено, что талидомид является тератогенным для животных, если его применять в ранние сроки беременности. Случаи с талидомидом послужили толчком к внедрению во всем мире системы контроля за ЛС в целях более тщательного доклинического исследования и клинической оценки перед выходом ЛС на фармацевтический рынок, а также усиления контроля за ПР ЛС и развития методов выявления и учета ПР ЛС.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим