|
Закон о ценовой конкуренции на ЛС и восстановлении срока действия патентов, который иначе называют поправкой Ваксмена — Хетча к федеральному закону о пищевых продуктах и ЛС, потребовал от Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) публиковать ежемесячно обновляемый список всех получивших разрешение на маркетинг безрецептурных и рецептурных ЛС. Этой цели служит издание «Получившие разрешение на маркетинг ЛС с оценками терапевтической эквивалентности» («Оранжевая книга») (). В списке препаратов референтные, с которыми заявители должны сравнивать свои продукты при помощи исследований биоэквивалентности, помечены словом «yes» в соответствующей колонке. Для препаратов в различных лекарственных формах предусмотрены разные РП. Устанавливая единственный, которому должны быть биоэквивалентны все генерические версии, FDA стремится избежать вариабельности свойств генерических препаратов вследствие наличия нескольких референтных.
Если одобрены заявки (полные) на получение разрешения на маркетинг (New drug application — NDA) препаратов с одним и тем же действующим веществом в одинаковых лекарственных формах, каждый из них получает рыночную эксклюзивность, независимо от того, будет ли он считаться референтным. Компания, которая намерена создать генерическую версию препарата, зарегистрированного на основании NDA, и при этом не считающегося референтным, может обратиться в FDA с ходатайством (citizen petition), посредством специальной процедуры, описанной в законодательстве (CFR 10.25(a) CFR 10.30). В случае удовлетворения ходатайства референтным будет признан еще один препарат, и заявитель сможет использовать его в таком качестве. Кодирование терапевтической эквивалентности (AB, AB1, AB2 и т.п.) применяют в отношении многоисточниковых препаратов, тогда как моноисточниковые (то есть единственный продукт с таким действующим веществом в определенной дозировке и лекарственной форме) не обозначают кодом.
В предисловии к «Оранжевой книге» объясняется также, что два биоэквивалентных друг другу препарата могут быть обозначены как референтные, если биоэквивалентность двух зарегистрированных на основании NDA (!) препаратов может быть установлена путем исследования in vitro.
Во избежание недоразумений FDA рекомендует обращаться в Службу генерических препаратов (Office of generic drugs — OGD) для получения подтверждения правильности выбора РП.
Каков же статус «Оранжевой книги»? Об этом также говорится в предисловии к ней.
Информируя о своей оценке терапевтической эквивалентности препаратов при помощи списка, FDA предлагает свои рекомендации общественности, специалистам и уполномоченным органам по выбору ЛС. Такую оценку не нужно рассматривать как запрет использовать тот или иной препарат либо как свидетельство того, что один из них предпочтительнее другого. Это только научное заключение, тогда как практика генерической замены, имеющая целью экономию средств, основывается также на социальных и экономических аспектах.
Безусловно, в «Оранжевой книге» интересна система буквенных кодов, описывающих терапевтическую эквивалентность. Она позволяет быстро определить, установлена ли биоэквивалентность определенного препарата референтному (первая буква) и получить дополнительную информацию об оценке FDA (вторая буква). Две основные категории, к которым могут быть отнесены многоисточниковые препараты, обозначают первыми буквами: «А» или «В».
К категории «А» относят препараты, терапевтически эквивалентные другим фармацевтически эквивалентным продуктам. Их действительные или потенциальные проблемы с биоэквивалентностью были разрешены путем проведения исследований in vivo и/или in vitro. В отношении этих препаратов нужно действовать следующим образом:
1) для препаратов с такими действующими веществами и в таких лекарственных формах, для которых нет известных или предполагаемых проблем с биоэквивалентностью in vivo, необходима информация для доказательства биоэквивалентности между фармацевтически эквивалентными продуктами (она считается доказанной в случае некоторых лекарственных форм, например растворов, или проверяется in vitro). Терапевтически эквивалентными эти продукты считаются, поскольку они производятся в условиях GMP и соответствуют другим требованиям согласно одобренной заявке. Такие препараты обозначают буквами AA, AN, AO, AP или АТ в зависимости от лекарственной формы;
2) для препаратов, признанных эффективными в ходе выполнения обзора «Имплементация изучения эффективности ЛС» («Drug Efficacy Study Implementation — DESI», изучены препараты, одобренные FDA с 1938 по 1962 г.), в отношении которых FDA идентифицировало существующие или потенциальные проблемы с биоэквивалентностью, и препаратов, одобренных после 1962 г. в лекарственных формах, сулящих потенциальные проблемы биоэквивалентности, суждение о терапевтической эквивалентности может быть вынесено только если заявка содержит адекватные научные доказательства, полученные при исследовании биоэквивалентности in vivo и/или in vitro; в таких случаях применяют обозначение АВ1.
|
При помощи кода «В» обозначают препараты, которые FDA в настоящее время считает неэквивалентными терапевтически другим фармацевтически эквивалентным продуктам, то есть действительные или потенциальные проблемы эквивалентности не могут быть разрешены путем адекватного установления биоэквивалентности; часто проблема заключается в определенной лекарственной форме, а не действующем веществе; в таких случаях применяют обозначения В*, BC, BD, BE, BN, BP, BR, BS, BT, BX. Код «В*» используют, если продукт требует дальнейшего изучения FDA, например, если ранее был присвоен код с буквой «А» или «В», а впоследствии получена информация, которая ставит под вопрос выполненную ранее оценку биоэквивалентности. То есть иногда возможна смена кодировки препарата.
В Канаде с 1978 г. законодательным порядком утверждена возможность подавать сокращенную заявку на получение разрешения на маркетинг «препарата-копии» (Abbreviated New Drug Submission — ANDS) (C.R.C. 1978), полагаясь на данные об эффективности инновационного препарата. При этом в качестве референтного, на основании разрешения министра здравоохранения, может быть использован инновационный продукт, приобретенный вне страны.
В отличие от США в ЕС нет списка РП, и заявитель выбирает их совместно с регуляторными органами. Согласно Директиве 2001/83/ЕС референтным может считаться продукт, получивший разрешение на маркетинг согласно положениям той же Директивы. При этом он обязательно должен быть зарегистрирован на основании полного досье (здесь и далее — Notice to applicants; November 2005). Досье препарата с действующим веществом, медицинское применение которого хорошо изучено; содержащего известные активные субстанции, которые раньше в таком сочетании не использовались с терапевтической целью, или зарегистрированного на основании информированного согласия, может быть использовано в качестве референтного. Напротив, досье генерического препарата не содержит всей необходимой информации об ЛС. Поэтому при регистрации генерика нельзя полагаться на досье генерического препарата.
В качестве референтного выбирают исключительно препарат, получавший разрешение на маркетинг. При этом период действия последнего может быть законченным.
Согласно статье 10(1) Директивы 2001/83/ЕС для того, чтобы считать препарат референтным в определенной стране-члене, достаточно, чтобы препарат получал разрешение на маркетинг в ЕС. Заявителю только следует указать, в какой стране зарегистрирован или был зарегистрирован референтный препарат. Условием является также то, что в стране происхождения препарата истек срок его рыночной эксклюзивности. Регуляторные органы разных стран-членов по соответствующим запросам должны предоставлять друг другу в течение месяца подтверждение того, что факт регистрации имел место, а также всю необходимую документацию.
Для нас очень важно, что в Соглашении о вступлении (The treaty of accession) (2003 г.) европейские новички (Чешская Республика, Эстония, Кипр, Латвия, Литва, Венгрия, Мальта, Польша, Словения и Словакия) договорились о том, что полученные ранее на основании национальных законодательств разрешения на маркетинг будут считаться действительными. Но до тех пор, пока они не будут одобрены согласно новым правилам, соответствующие препараты нельзя использовать в качестве референтных ().
|
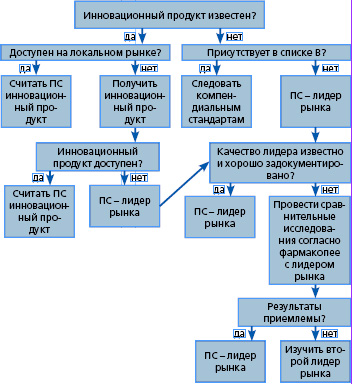
Критерии выбора препаратов сравнения (ПС) и их список с торговыми наименованиями предлагает ВОЗ. В настоящее время список пересматривается, и в последней серии технических отчетов ВОЗ (WHO Technical report series — TRS, No. 937, 2006) соответствующее приложение (Annex) 11 отсутствует. Новая версия, созданная на основе дополненной в соответствии с пожеланиями Международной федерации фармацевтических производителей и ассоциаций (International Federation of Pharmaceutical Manufacturers and Associations — IFPMA) более ранней (TRS, No. 902, 2002), одобрена экспертным комитетом (WHO Expert Committee on Specifications for Pharmaceutical Preparations) и, постоянно пополняясь, будет доступна на веб-сайте.
ВОЗ создает список ПС в качестве помощи регуляторным органам и фармацевтическим компаниям в выборе подходящих РП для прохождения процедуры выдачи разрешения на маркетинг многоисточниковыми (генерическими) препаратами. Эта информация может также использоваться при формировании закупок ЛС.
Для случаев, когда ПС (фармацевтический препарат, с которым новый препарат будет взаимозаменяем при клиническом использовании) четко не определен, предложены критерии, согласно которым можно принимать соответствующие решения (рис.).
Инновационный продукт обычно наиболее разумно использовать в качестве РП, поскольку его качество, безопасность и эффективность оценены во многих пре- и клинических исследованиях и установлена связь этих свойств с особенностями фармацевтической разработки. Но, несмотря на это, общее соглашение по критериям создания международного списка РП еще не выработано, и самого списка не существует. В качестве ПС, кроме инновационного, выбирают и «ведущий» на рынке продукт. Поэтому препараты, используемые в качестве РП в разных странах, могут отличаться.
Список ПС, относящихся к Примерному перечню основных лекарств ВОЗ, состоит из двух частей:
- список А, с информацией о препарате (торговое наименование и страна, на рынок которой он был выведен впервые), предоставленной производителем инновационного продукта;
- список В содержит продукты, информация о которых не предоставлена производителем инновационного или лидирующего на рынке продукта.
Используя список, компания прежде всего должна определить, присутствует ли на рынке ее страны инновационный продукт из списка А и в случае наличия использовать его в качестве РП. Если такой препарат отсутствует, компании следует получить его с рынка той страны, где имеются наиболее полные данные о нем (страна, где препарат впервые попал на рынок). Если продукт отнесен к списку В, то для фармразработки компании придется воспользоваться данными национальной, региональной или Международной фармакопеи относительно действующего вещества и/или продукта, а также официальным референтным текстом. При этом не выполняют оценку эквивалентности с другим препаратом.
|
В список В включены также продукты, компания-оригинатор которых может быть идентифицирована, или доступен лидирующий на рынке продукт, но информации для включения его в список А недостаточно, то есть разработчик прекратил существование или недоступен. В таких случаях применяется такой же подход, как и в отношении продуктов из группы А.
Когда на локальном рынке доступен лидер рынка, но инновационный продукт не может быть идентифицирован или получен со своего первого рынка, лидирующий на рынке продукт может быть выбран в качестве ПС, если установлены его качество, эффективность и безопасность. В другом случае используют второго лидера рынка или компендиальный подход (как в случаях со списком В). Важно, что ВОЗ, как и регуляторные органы разных стран, тесно сотрудничает с предприятиями отрасли в составлении списка РП.
Итак, ВОЗ рекомендует стремиться использовать в качестве ПС прежде всего инновационный продукт. Только при невозможности идентифицировать или получить его, следует воспользоваться лидирующим на рынке препаратом. Может быть, необходимость включать в список РП в нашей стране лидирующие на локальном рынке препараты, в том числе генерики, — только временная, и компании в основном не будут выпускать копии копий? Последнее, скорее всего, вообще искажает суть такого явления, как разрешение при регистрации полагаться на данные о доказанной эффективности и безопасности препарата. Да, ВОЗ не считает обязательным условием регистрацию ПС на основании полного досье, но, думается, недоступность инновационных препаратов или невозможность идентифицировать их производителей при сегодняшнем развитии коммуникаций — проблема специфическая и вряд ли является неразрешимой для нашей страны. Так что производители, стремящиеся соблюдать современные стандарты с учетом возможности выведения своих препаратов на внешние рынки, должны стремиться учитывать требования самого высокого уровня, которые выдвигают и отечественные регуляторные органы. Как можно заметить из обзора зарубежной ситуации, в разных странах подходы к выбору РП отличаются, и в нашей стране сейчас ведется активная работа по согласованию соответствующих критериев и списка.
Появление такого инструмента очень важно для прогресса отечественной фармацевтической отрасли. n
1Трехзначное обозначение (АВ1, АВ2, АВ3 и т.п.) может применяться только в ситуациях, когда существует два и более РП с одинаковой силой действия (дозировкой). Например, в качестве РП нифедипина с продленным высвобождением действующего вещества указаны Adalat® CC (Miles), Procardia XL® (Pfizer). Между собой эти препараты не биоэквивалентны. Генерические препараты с установленной биоэквивалентностью Adalat® CC или Procardia XL® будут обозначены АВ1 или АВ2, соответственно.
Дарья Полякова
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим