Відкриваючи захід, директор ДП «УФІЯ» Олена Кричевська зазначила, що вступ Держлікслужби до Системи співробітництва фармацевтичних інспекцій (Pharmaceutical Inspection Cooperation Scheme — PIC/S) свідчить про визнання з боку цієї авторитетної міжнародної організації того, що в частині належної виробничої практики (Good Manufacturing Practice — GMP) в Україні досягнуто необхідного рівня і ступеня гармонізації. Вона відзначила, що численна представленість фахової спільноти на заході підтверджує важливість тематики конференції та загалом питань належної практики і гармонізації українського законодавства.
НОВЕЛИ ТА КЛЮЧОВІ ЗАСАДИ ПРОЕКТУ ПОРЯДКУ ЩОДО ПІДТВЕРДЖЕННЯ ВІДПОВІДНОСТІ УМОВ ВИРОБНИЦТВА ВИМОГАМ GMP
9 лютого 2018 р. проект Порядку розміщено на сайті Держлікслужби. «До 12 березня ми приймаємо ваші пропозиції, після опрацювання яких подаватимемо на затвердження МОЗ України», — зазначила Наталія Гудзь, перейшовши до презентації проекту Порядку.
Працюючи над проектом Порядку, Держлікслужба спільно з ДП «УФІЯ» ставили перед собою за мету:
- систематизувати положення чинного Порядку, затвердженого наказом МОЗ України від 27.12.2012 р. № 1130;
- дерегулювати в рамках Порядку процеси, що існують на сьогодні;
- встановити єдині підходи до виробництва, яке має підтверджену органами країн — членів PIC/S відповідність вимогам GMP.
Структура проекту Порядку чітко визначає 2 напрямки роботи Держлікслужби. Підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики буде здійснюватися 2 шляхами:
- шляхом визнання офіційних документів, що підтверджують відповідність реального стану виробництва лікарських засобів вимогам GMP (ЄС, PIC/S тощо), результатом чого стане виданий Держлікслужбою висновок про підтвердження відповідності умов виробництва лікарського засобу вимогам належної виробничої практики (далі — Висновок про підтвердження);
- шляхом здійснення Держлікслужбою процедури видачі сертифіката GMP.
Розробники проекту Порядку заклали в нього декілька основних так званих меседжів. Зокрема, поставлено мету врахувати майбутні двосторонні угоди про визнання результатів інспектувань. На даний час проводяться дії щодо їх укладення з регуляторними органами інших країн у сфері обігу лікарських засобів. У проекті Порядку міститься відповідне положення, яке визначає, що за умови наявності двосторонніх угод про визнання результатів інспектувань виробництв лікарських засобів між Держлікслужбою та регуляторним органом, який видав документ, що представлений для визнання, можливе автоматичне визнання з невідкладним внесенням реквізитів документів до бази даних Держлікслужби.
Проектом Порядку встановлено чіткі підстави, згідно з якими може бути зупинена дія Висновку про підтвердження та призначено позапланове інспектування. Зокрема, до таких підстав віднесено:
- невиконання заявником гарантійних зобов’язань;
- встановлення факту та/або отримання інформації щодо невідповідності виробництва лікарських засобів вимогам GMP;
- отримання інформації від регуляторних органів країн — членів PIC/S (у тому числі України) про рішення щодо заборон/вилучення з обігу серії/серій препаратів, у тому числі шляхом припинення дії реєстраційного посвідчення;
- наявність фактів виробництва та обігу неякісних/фальсифікованих лікарських засобів.
Для відновлення дії Висновку про підтвердження обов’язковим є інспектування.
Проектом Порядку запроваджується прозора методика розрахунку строків проведення інспектувань (тривалість інспектувань розраховується з урахуванням загальної матриці розрахунку строків проведення інспектувань, визначеної додатком 12 до проекту Порядку. Загальна кількість днів інспектування — це керівні значення, що включають необхідний час для проведення інспектування, а також відображають загальні часові витрати з урахуванням кількості інспекторів (наприклад, усього 10 днів інспектування дорівнює 2 інспекторам, що проводять інспектування протягом 5 днів, чи 4 інспекторам, що проводять інспектування протягом 2½ дня).
Розрахунок строків проведення інспектувань може бути скоригований з урахуванням наступних факторів:
- режим роботи підприємства;
- календарні особливості країни, у якій розташовано виробництво (національні та релігійні свята, офіційні заходи тощо);
- особливості транспортного сполучення;
- розташування окремих складових об’єкта, що перевіряється (у межах однієї території, одного населеного пункту, одного регіону);
- особливості аутсорсингової діяльності підприємства;
- кількість виробничих дільниць, лікарських форм, найменувань лікарських засобів, включених до інспектування;
- результати попередніх інспектувань.
Представляючи порядок отримання сертифіката GMP (рисунок), очільниця Держлікслужби звернула увагу на те, що проведення первинної експертизи проектом Порядку не передбачено.
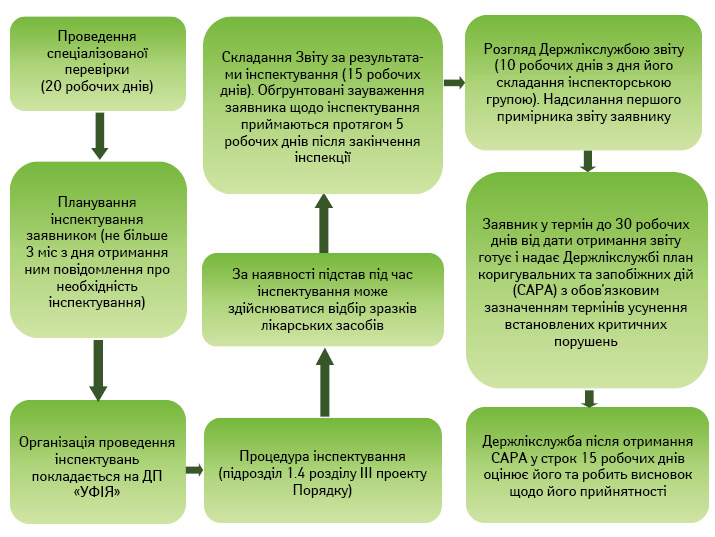
Рішення Держлікслужби про видачу сертифіката GMP прийматиметься на підставі позитивних результатів інспектування та, за наявності, позитивних результатів лабораторного аналізу і висновку Держлікслужби про прийнятність плану коригувальних та запобіжних дій. «Ще раз звертаю увагу: звіт за результатами інспектування надсилається вам. За результатами звіту виробник готує план коригувальних та запобіжних дій, який повертається Держлікслужбі. Держлікслужба оцінює його прийнятність та терміни усунення встановлених критичних порушень і за сукупністю позитивних результатів приймає рішення про видачу сертифіката GMP», — підкреслила Н. Гудзь.
«Не дивлячись на те що ми говорили про єдині підходи, для резидентів — українських виробників — регулятор залишив норми, які сьогодні закладені в Ліцензійних умовах, але це ні в якому разі не послаблює увагу державного регулятора до забезпечення вимог GMP вітчизняним виробником», — зауважила голова Держлікслужби.
Так, для резидентів сертифікат GMP може видаватися за результатами планової перевірки дотримання Ліцензійних умов провадження господарської діяльності з виробництва лікарських засобів, оптової та роздрібної торгівлі лікарськими засобами, імпорту лікарських засобів (крім активних фармацевтичних інгредієнтів) за відсутності в акті перевірки критичних порушень. Виробник протягом 15 робочих днів після проведення такої перевірки подає Держлікслужбі заяву щодо видачі сертифіката GMP.
Підставами для відмови у видачі сертифіката GMP проектом Порядку визначено наступні:
- негативні результати інспектування;
- негативні результати лабораторного аналізу (за наявності);
- ненадання заявником прийнятного САРА протягом 30 днів з дня отримання звіту;
- виявлення недостовірних відомостей у заяві або документах до неї;
- виявлення в ході інспектування підробки даних;
- наявність хоча б одного критичного порушення у звіті;
- неусунення більшості порушень, виявлених в ході попереднього інспектування;
- встановлення високого рейтингу ризику для виявлених порушень;
- відмова заявника надати за запит інспекторської групи необхідну інформацію, доступ до приміщень, обладнання, зон контролю, документів систем управління якістю (СУЯ) тощо — про що зазначається у звіті.
Проектом Порядку встановлено чіткі підстави, на основі яких може бути зупинена дія сертифіката GMP та призначене позапланове інспектування. Зокрема, такими визначено:
- ненадання заявником документальних підтверджень усунення суттєвих порушень згідно зі строками, зазначеними в плані коригувальних та запобіжних дій;
- невиконання гарантійних зобов’язань, що надаються заявником;
- встановлення факту та/або отримання інформації щодо невідповідності виробництва лікарських засобів вимогам GMP;
- отримання в період дії сертифіката GMP інформації від регуляторних органів (у тому числі України) про прийняті рішення щодо заборон та/або вилучення з обігу серій лікарського засобу, які вироблялися на даній дільниці, що пов’язані з якістю лікарського засобу, у тому числі шляхом припинення дії реєстраційних посвідчень;
- наявність фактів виробництва та обігу неякісних лікарських засобів.
Під час здійснення позапланового інспектування перевірці підлягатимуть лише ті питання, що стали підставою для зупинення дії сертифіката GMP, з вичерпним зазначенням цих підстав в організаційних документах для проведення інспектування.
Більш чітко проектом Порядку сформульовано розділ, присвячений процедурі внесення змін та переоформлення висновку про підтвердження або сертифіката GMP. Підставами для цього визначено наступні:
- зміна номера або строку дії ліцензії на виробництво лікарських засобів;
- зміни, пов’язані з додаванням поштового індексу, перейменуванням вулиць, селищ, місць провадження господарської діяльності з виробництва, які підтверджені відповідними документами міських, селищних рад або відповідних органів країн, де відбулися зміни;
- уточнення організаційної форми підприємства (наприклад «Лімітед» або «Лтд»);
- технічні помилки, які не змінюють зміст висновку про підтвердження або сертифіката GMP в цілому.
Н. Гудзь звернула увагу на те, що термін дії сертифіката GMP залишається незмінним і становить 3 роки від дати проведення інспектування.
Проект Порядку загалом містить 21 додаток. Одним з нових є останній 21-й додаток, який визначає методологію проведення розрахунку рекомендованого терміну наступного інспектування. Такий розрахунок здійснюватиметься на підставі попередньо визначеної складності процесів та критичності лікарських засобів; виявлених порушень та їх класифікації; визначеної категорії ризику виробничої дільниці.
«Ми завжди відкриті до конструктивної співпраці, тому ще раз наголошую, що ми будемо чекати ваших конструктивних пропозицій. Я б хотіла, щоб ви звернули увагу на серйозні процесні моменти даного проекту», — наголосила наостанок голова Держлікслужби, закликаючи учасників конференції активно брати участь у громадському обговоренні проекту Порядку.
Зауваження та пропозиції можна надсилати до 12 березня 2018 р. до Держлікслужби за адресою: м. Київ, 03115, просп. Перемоги, 120А, e-mail: [email protected]; виконавець — Віктор Владиславович Касьяненко (контактний телефон: +38 (044) 422-55-76; e-mail: [email protected]).
ІСТОРІЯ ТА ДІЯЛЬНІСТЬ ДП «УФІЯ»
З історією та діяльністю ДП «УФІЯ» учасників конференції детально ознайомила Олена Кричевська.
Так, у рамках проекту ЄС TACIS (Technical Assistance to the Commonwealth of Independent States) «Підтримка в реструктуризації фармацевтичної промисловості України» у жовтні 2001 р. створено ДП «Державний навчальний центр з належної виробничої/дистриб’юторської практики» (GMP/GDP Центр). Тоді вперше був створений інспекторат, а співробітники пройшли базове теоретичне й практичне навчання в частині інспектування у відповідності до вимог європейської спільноти.
У листопаді 2009 р. було створено ДП «УФІЯ» разом з філіями «Державний навчальний центр з належної виробничої/дистриб’юторської практики», «Медичний центр з оцінки відповідності» та Донецькою філією.
У 2011 р. відбулася одна зі знакових для України подій — за активної роботи GMP/GDP Центру Україна в обличчі Держлікслужби долучилася до PIC/S.
У 2017 р. було прийнято рішення реорганізувати філії ДП «УФІЯ», які не працювали. Держлікслужба затвердила нову редакцію статуту ДП «УФІЯ» — головного документа, яким керується підприємство під час своєї роботи. Усі види діяльності, які здійснював GMP/GDP Центр, а також його фахівці перейшли до ДП «УФІЯ».
Окрім статуту, у своїй роботі ДП «УФІЯ» також керується Настановою СТ-Н МОЗУ 42‑4.0:2016 «Лікарські засоби. Належна виробнича практика» і наказом МОЗ України від 27.12.2012 р. № 1130.
За словами О. Кричевської, процес гармонізації законодавства України у сфері виробництва та контролю якості лікарських засобів почався ще в 2000 р., на що вплинув проект TACIS.
Cистему управління якістю підприємства сертифіковано за міжнародним стандартом ISO 9001:2008, що підтверджується відповідним сертифікатом компанії «Бюро Верітас Сертифікейшн Україна» (Bureau Veritas Certification Holding SAS-UK Branch). «Це третій сертифікат, який підтверджує відповідність нашої системи управління якістю, яка щороку удосконалюється», — підкреслила директор ДП «УФІЯ», додавши, що щорічно система управління якістю підприємства проходить наглядові аудити.
Одним з важливих напрямків роботи ДП «УФІЯ» є проведення його, а також залученими спеціалістами навчальних семінарів для професіоналів фармацевтичної галузі.
Фахівці підприємства також безпосередньо задіяні в діяльності глобальної регуляторної системи. «Наші спеціалісти виступають у якості експертів в рамках PIC/S і щорічно беруть участь в засіданнях і різних заходах, які проводять регуляторні органи країн PIC/S», — наголосила О. Кричевська.
Активну участь фахівці ДП «УФІЯ» беруть у моніторингу, аналізі й підготовці пропозицій щодо гармонізації нормативних документів у розрізі належної виробничої та дистриб’юторської практики.
Окремо О. Кричевська зупинилася на питанні постійного професійного розвитку спеціалістів фармацевтичного сектору. «Ми проводимо як планові, так і корпоративні навчальні заходи з фокусом на практичні аспекти втілення набутих знань і навичок у сфері виробництва та контролю якості лікарських засобів. Новий напрямок нашої діяльності — проведення семінарів і тренінгів у сфері обігу медичних виробів», — зазначила доповідач.
Представляючи результати роботи підприємства за 2017 р., О. Кричевська повідомила, що штат ДП «УФІЯ» включає 8 висококваліфікованих експертів, які проходять постійне навчання, що є обов’язковою вимогою європейської спільноти.




У минулому році експерти підприємства пройшли в межах України навчання, що еквівалентно 271 год, а також навчання з підтвердження компетентності та спеціалізації поза Україною, що еквівалентно 104 год. «Навчання на таких семінарах дає можливість нашим експертам отримати рішення тих проблем, які вже виникали в європейської спільноти», — зауважила О. Кричевська, додавши, що такий підхід дозволяє попередити аналогічні проблеми в Україні.
У минулому році інспекторами підприємства проведено 377 різних експертиз, проінспектовано 57 виробничих дільниць в 10 країнах світу — Китаї, В’єтнамі, Індії, Пакистані, Туреччині, Білорусі, Болгарії, Сербії, Німеччині, Франції та на Кубі.
Говорячи про результати проведених експертиз, директор підприємства зазначила, що переважають позитивні результати. Це пов’язано з підвищенням рівня системи якості у виробників та певною стабілізацією на вітчизняному фармацевтичному ринку.
У 2017 р. до Держлікслужби звернулося 7 нових зарубіжних виробників лікарських засобів із заявами на видачу сертифіката GMP: 3 китайські виробники, 2 — турецькі та 4 — індійські. Проте, за словами О. Кричевської, не всі з них отримали сертифікат GMP. «Підготовка до сертифікації — процес дуже тривалий у часі та в матеріальному плані затратний, тому це вимагає великої роботи», — зауважила вона.
Наостанок О. Кричевська подякувала всім за партнерство в ході співпраці з ДП «УФІЯ», оскільки всі сторони процесу потребують впевненості в тому, що на українському ринку знаходяться дійсно якісні лікарські засоби.
НАЙБІЛЬШ ПОШИРЕНІ ПОРУШЕННЯ ВИМОГ GMP
Заступник директора з якості ДП «УФІЯ» Олексій Сухомлинов представив статистику результатів інспектувань за 2013–2017 рр., а також звернув увагу присутніх на найбільш часті порушення вимог GMP, що виявляли інспектори протягом 2017 р., надавши роз’яснення щодо того, як відбувається трактування того чи іншого зауваження.
Як уже було зазначено, у 2017 р. інспекторами ДП «УФІЯ» було проведено 57 інспекцій в 10 країнах. Результати 19% з них були негативними. При цьому за останні 5 років спостерігається тенденція до зменшення кількості негативних рішень за результатами інспектувань. «Це обумовлено тим, що на ринку залишаються більш-менш одні й ті ж самі виробники, до яких виїжджаємо на інспекцію не перший раз. Ще одна причина такої тенденції — заявники й виробники користуються можливістю отримати висновок про підтвердження на основі сертифіката, виданого в країнах — членах PIC/S», — зазначив О. Сухомлинов.
Доповідач навів приклади найбільш розповсюджених критичних невідповідностей, які найчастіше виявлялися експертами під час проведення інспектування у 2017 р.
Мова йде про невідповідність, яка стосується виробництва стерильних лікарських засобів, а саме не проведено валідацію стерилізуючої фільтрації, а встановлений час, необхідний для фільтрації відомого об’єму розчину та різниця тиску по різні сторони фільтру не ґрунтуються на відповідних валідаційних дослідженнях. Натомість така вимога відображена в п. 113 додатку 1 до Настанови СТ-Н МОЗУ 42‑4.0:2016 «Лікарські засоби. Належна виробнича практика».
Ще одне критичне порушення — не розроблена методика щодо перевірки флаконів з розчином для ін’єкцій, перевірки на цілісність після стадії закупорювання; під час виробництва не передбачена та не проводиться перевірка флаконів на цілісність. Це є порушенням п. 117 додатку 1 до Настанови СТ-Н МОЗУ 42‑4.0:2016.
Критичним порушенням також є те, що уповноважена особа підприємства дає дозвіл на постачання в Україну лікарських засобів без засвідчення їх відповідності вимогам реєстраційного досьє.
Розповсюдженим порушенням є й те, що під час здійснення вхідного контролю активних фармацевтичних інгредієнтів не передбачено проведення перевірки ідентичності вмісту кожного контейнера з сировиною, що є прямою вимогою Настанови СТ-Н МОЗУ 42‑4.0:2016. Непроведення такої процедури розцінюється інспекторами як критична невідповідність. «Перш за все це пов’язано з тим, що є ризик переплутування у виробника АФІ й в ємності може бути не та речовина, яка буде використовуватися для виготовлення готової лікарської форми», — зауважив О. Сухомлинов.
Серед найбільш частих суттєвих невідповідностей, виявлених у ході інспекцій, проведених у 2017 р., доповідач виокремив наступні:
1) не проводяться аудити виробників і дистриб’юторів діючих речовин для підтвердження дотримання ними відповідних вимог належної виробничої практики та належної практики дистрибуції. Таке порушення частіше пов’язано з тим, що не завжди виробники та дистриб’ютори вчасно поінформовані про зміни, які відбулися в Настанові й, відповідно, не виконують їх;
2) дезінфекційні розчини, які використовуються в приміщеннях класу «Б» зони «А», не випробовуються за показником «Стерильність», а відповідно, не виконується пряма вимога Настанови СТ-Н МОЗУ 42‑4.0:2016;
3) при валідації очищення обладнання межі стосовно кількості залишків препаратів, що переносяться, не ґрунтуються на токсикологічній оцінці. Така вимога міститься в останніх змінах до Настанови GMP ЄС й України відповідно;
4) необґрунтована відмова від проведення під час вхідного контролю випробувань якості сировини за певними показниками. «Наразі GMP дозволяє не проводити вхідний контроль сировини, якщо це належним чином обґрунтовано, тобто проведені аудити виробника чи лабораторії, яка здійснює такий контроль. Але таке рішення має прийматися на підставі якогось заключення чи результатів», — підкреслив доповідач;
5) ще одне суттєве порушення, яке достатньо часто зустрічається у звітах — відсутня поточна верифікація процесів протягом життєвого циклу лікарських засобів. Така вимога також передбачена останніми змінами до Настанов GMP ЄС й України, однак через несвоєчасну поінформованість про такі зміни підприємство порушує її;
6) не продемонстровано, що характер повітряних потоків у зоні класу «А» не становить ризику контамінації, що є порушенням п. 54 додатку 1 до Настанови СТ-Н МОЗУ 42‑4.0:2016.
Окрім проведення інспектувань, фахівці ДП «УФІЯ» здійснюють оцінку планів коригувальних та запобіжних дій. Основними невідповідностями, які виявляють інспектори під час аналізу таких планів, є незазначення запобіжних заходів. Також не зазначаються причини тих чи інших виявлених невідповідностей. Є випадки, коли невиконання вимог GMP намагаються обґрунтувати застосуванням аналізів ризиків. «Таке буває, коли на зауваження підприємство робить наукову роботу з обґрунтуванням, чому GMP помиляється», — зазначив О. Сухомлинов, додавши, що ніякий аналіз ризиків не може підмінити собою пряму норму GMP. Ще одне порушення — строки усунення невідповідностей або не встановлено, або вони перевищують 1 рік після інспектування. Помилкою є й те, що документальне підтвердження не в повній мірі демонструє весь обсяг виконаних заходів. «Є такі випадки, коли виконані дії представлені в плані, а документального підтвердження немає, чи його обмаль», — зауважив доповідач.
Користуючись нагодою, учасники конференції змогли поставити питання доповідачам, які виникають у заявників під час отримання висновків про підтвердження та сертифікатів GMP.
фото Сергія Бека
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим