|
 Заседание открыл Виктор Чумак, директор ГФЦ МЗ Украины. Говоря о проблемах обеспечения взаимозаменяемости лекарственных средств (ЛС), докладчик отметил, что представления о ЛС, принятые в Советском Союзе (согласно документу «Порядок организации работ по созданию и промышленному освоению новых лекарственных средств на предприятиях и организациях Министерства медицинской промышленности. Основные положения. ОМУ 64-33-81»), отличались от европейских, нашедших отражение в Директиве ЕС № 65/65 (табл. 1). По советским нормам инструкцию ЛС утверждали, когда технология производства ЛС еще не была разработана, а фармакокинетика — не изучена.
Заседание открыл Виктор Чумак, директор ГФЦ МЗ Украины. Говоря о проблемах обеспечения взаимозаменяемости лекарственных средств (ЛС), докладчик отметил, что представления о ЛС, принятые в Советском Союзе (согласно документу «Порядок организации работ по созданию и промышленному освоению новых лекарственных средств на предприятиях и организациях Министерства медицинской промышленности. Основные положения. ОМУ 64-33-81»), отличались от европейских, нашедших отражение в Директиве ЕС № 65/65 (табл. 1). По советским нормам инструкцию ЛС утверждали, когда технология производства ЛС еще не была разработана, а фармакокинетика — не изучена.
Таблица 1
Основные отличия советских представлений о ЛС от европейских
| – Поисковые исследования, включая доклиническое и клиническое изучение препарата (по сути молекулы) – Положительные результаты КИ служили основанием для научно-технической разработки нового ЛС (доработки или переработки лекарственной формы) – Научно-техническая разработка проводилась на основании утвержденного протокола Фармакологического комитета, рекомендовавшего новое ЛС к применению в медицинской практике, и планового задания на проведение данной работы |
Соответственно, другими были и принципы разработки ЛС (о чем на одной из конференций говорил ректор Национального фармацевтического университета Валентин Черных; табл. 2).
Таблица 2
Принципы разработки ЛС в СССР и мире
| Советский подход | Мировой подход |
| Препарат — нужная молекула в необходимом количестве, все остальное — выдумки | Препарат — нужная молекула определенной структуры, должна попасть в кровь: – в нужное время; – в определенной концентрации; – внужном месте организма |
Мы должны четко понимать, что зависит и что не зависит от производителя, — отметил В. Чумак. После поступления препарата в организм происходит высвобождение действующего вещества с последующим растворением последнего. Факторы, влияющие на два этих процесса, — и есть сфера ответственности компании. После перехода ЛС в растворенное состояние и начала всасывания все зависит от природы действующего вещества и особенностей больного. Таким образом, мы разделяем вклад производителя, от которого зависят процессы, происходящие с ЛС до его поступления в кровь (рис. 1), и дальнейшее взаимодействие препарата с организмом (модификация этого взаимодействия — задача, связанная с открытием (discovery, в отличие от development — разработки) ЛС — прим. ред.).
|
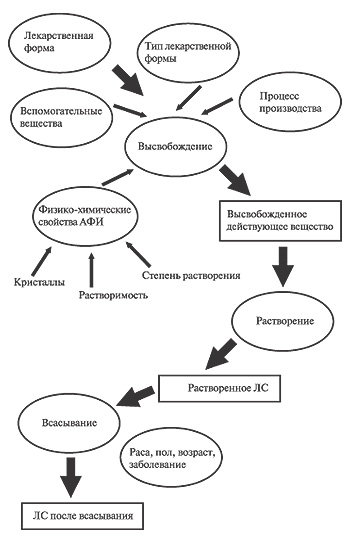
Сегодня в Украине сложилась ситуация, когда часть экспертизы регистрационных материалов, а именно то, что касается технологии получения ЛС, выполняется одним подразделением МЗ Украины (Государственной службой лекарственных средств и изделий медицинского назначения), а остальное — другим подразделением (ГФЦ). Таким образом, мы практически воссоздали советскую систему, описанную выше. Получается, мы зарегистрировали препарат, а что производит компания, мы не знаем, отметил В. Чумак. Поэтому когда о GMP говорят, что это «не евроремонт», обижаться не нужно. Цепочка надлежащих практик (GCP, GMP, GSP, GDP, GPP) призвана гарантировать воспроизведение качества ЛС от этапа его разработки до применения, то есть то, что ни во время клинических испытаний (КИ), ни во время производства, хранения, дистрибьюции, распространения через аптеки ничего в препарате не меняется. Вот это и есть идеология GMP. Как говорит В.А. Загорий, GMP — это не стандарт, это философия. Следование идеологии требует огромного объема знаний.
Итак, мы имеем дело либо с инновационным, либо с генерическим ЛС. К сожалению, наш научный потенциал, полученный в наследство от СССР, был сориентирован только на получение оригинальных препаратов. А что это такое? Это фармацевтическая разработка, доклинические испытания и КИ, производство по GMP. Примерно такой этапности мы придерживаемся. Когда Украина столкнулась с генериками, понадобилась другая идеология и методология. И что мы придумали? Фармацевтическую разработку, соответствующую нашим представлениям, краткие КИ и производство. В результате мы вышли из сферы доказуемости, потому что результаты таких КИ не могут служить доказательством воспроизведения качества препаратов. Если же организовывать КИ, обеспечивающие статистически значимые результаты, наши генерики по цене ничем не будут отличаться от оригинальных ЛС. Но это противоречит основному призванию генерических препаратов. Поэтому в приказ МЗ Украины от 17.04.2007 г. № 190 имплементирована особая идеология доказательства качества генериков, согласно 41-му отчету Экспертного комитета по спецификациям для ЛС ВОЗ (WHO Expert committee on specifications for pharmaceutical preparations) (TRS 937). То есть нормативная база создана. Осталось лишь одно — выполнять требования этих документов. Что не урегулировано? В следующем году будет издан список референтных препаратов.
Дискуссионный момент: нужно или нет выносить на публичное обсуждение нормативные документы, подобные приказу № 190? Его особенность — точное повторение международных норм, и если мы будем вносить изменения, эту норму не узнаем ни мы, ни ее зарубежные разработчики. Поэтому обсуждать нужно, что и на каком этапе можно требовать. Мы ничего нового не придумывали, а взяли те документы, которые были внедрены в США, в ЕС, рекомендованы ВОЗ. Если нам еще удастся внедрить систему стандартизации, чтобы расшифровать все составляющие этого документа в виде стандартов, мы обеспечим нормативными актами все необходимые аспекты обращения ЛС.
Зачем это нужно? В стране уже зарегистрировано огромное количество препаратов — 26 991 наименование, с учетом дозировок и упаковок. А доказано ли их качество с учетом современных международных норм? На украинском рынке 60% отечественных ЛС и 40% зарубежных не требуют проведения испытаний биоэквивалентности. Взаимозаменяемость подобных ЛС гарантируется фармакопейными спецификациями, что само по себе тоже непросто (табл. 3).
Таблица 3
Необходимые для подтверждения
фармацевтической эквивалентности параметры соответствия фармакопейным спецификациям
| Идентичность действующих веществ |
| Идентичность концентрации |
| Одна и та же лекарственная форма |
| Идентичность или сходство качественного и количественного состава вспомогательных веществ |
| Тот же путь введения |
| Соответствие спецификаций качества (профиль примесей) |
| Выполнение требований к производству, установленных законодательством Украины и международными нормами |
Что делать с остальными препаратами — в твердых лекарственных формах? Определению этого служит биофармацевтическая классификационная система (БКС), легализованная отечественным фармацевтическим законодательством (приказы МЗ Украины от 28.06.2005 г. № 426, от 01.03.2006 г. № 95, от 17.04.2007 г. № 190). Если препарат относится к I классу БКС, его приравнивают к растворам и доказывают биоэквивалентность путем проведения теста «растворение». В некоторых случаях то же относится и к препаратам II и III классов, и только относительно препаратов IV класса исследования биоэквивалентности проводят всегда.
Когда выбирают тот или иной метод оценки биоэквивалентности? Есть ряд особенностей (табл. 4).
Таблица 4
Критерии выбора метода оценки эквивалентности
| Фармакокинетические испытания проводят для препаратов системного действия, для которых возможно определить концентрацию действующего вещества в биологических жидкостях |
| Фармакодинамические испытания проводят для препаратов, для которых невозможно определить концентрацию действующего вещества в биологических жидкостях, но возможно оценить значимые фармакодинамические показатели, изменения которых вызваны ЛС |
| Сравнительные КИ проводят для препаратов, для которых невозможно определить фармакокинетические параметры или получить соответствующие фармакодинамические точки |
| Исследования in vitro проводят для препаратов в твердых лекарственных формах немедленного высвобождения системного действия |
Сравнительных исследований эквивалентности in vitro недостаточно для подтверждения эквивалентности ЛС в таких случаях:
– препарат предназначен для купирования неотложных состояний;
– препарат имеет форму таблеток для рассасывания в ротовой полости;
– препарат имеет вспомогательные вещества, влияющие на абсорбцию действующего вещества;
– препарат имеет узкий терапевтический диапазон («крутой» наклон зависимости доза/эффект);
– действующие вещества используются в другой полиморфной модификации, чем в референтном препарате;
– имеются документально подтвержденные доказательства проблем с биодоступностью (не связанные с растворением).
 Детально о современных регуляторных требованиях к исследованиям и регистрации генерических ЛС рассказала Ольга Баула, директор департамента фармацевтической деятельности ГФЦ МЗ Украины.
Детально о современных регуляторных требованиях к исследованиям и регистрации генерических ЛС рассказала Ольга Баула, директор департамента фармацевтической деятельности ГФЦ МЗ Украины.
«Украина заинтересована в инновационных препаратах; они должны выходить на фармацевтический рынок при обеспечении соответствующих гарантий качества со стороны государства, — отметила О. Баула. — Однако ни одна страна, даже с очень развитой фармпромышленностью, не оставляет сектор генериков». Наша задача — обеспечить, чтобы все последующие генерики выходили на рынок, будучи терапевтически эквивалентными. Ведь генерический препарат не проходит всесторонних исследований, но при этом все, что заложено в инновационном, должно быть в нем воспроизведено. На рынке могут присутствовать только взаимозаменяемые генерики. Для этого необходимо доказательство фармацевтической эквивалентности или альтернативности и биоэквивалентности.
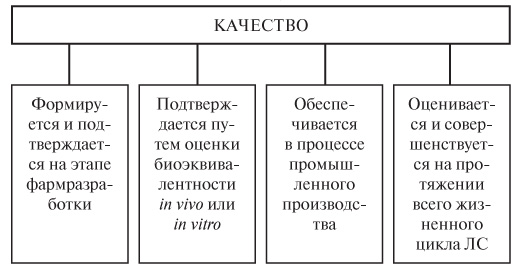
Этапы обеспечения качества включают формирование, подтверждение, обеспечение в процессе промышленного производства и оценку (рис. 2). Фармацевтическая разработка проводится всегда и для всех ЛС. На этом этапе качество должно быть сформировано, «встроено» в препарат и подтверждено результатами исследований. При помощи методов с высоким уровнем доказательности качество должно постоянно оцениваться и совершенствоваться на протяжении всего жизненного цикла препарата.
|
Современная концепция обеспечения качества в сфере разработки и производства ЛС Международной конференции по гармонизации технических требований к регистрации ЛС для человека (ICH) базируется на оценке рисков (рис. 3).
|
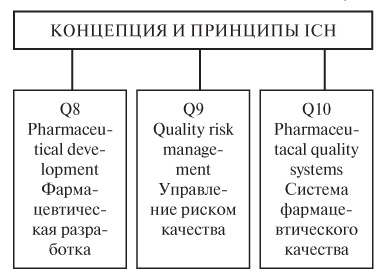
Три ее основополагающих документа — Q8, Q9, Q10 — сейчас внедряются в Европе. Отечественные производители и эксперты ГФЦ уже начали обсуждать их. Это важно, поскольку создание планируемой системы обеспечения качества позволит выводить на рынок препараты с запланированными параметрами качества.
Основополагающий документ ЛС — регистрационное досье. Согласно отечественному Руководству 42-01-2001 «Надлежащая производственная практика» владелец лицензии на производство обязан производить ЛС так, чтобы обеспечить их соответствие своему предназначению, требованиям регистрационного досье.
На протяжении всего жизненного цикла препарата владелец лицензии должен выпускать ЛС в соответствии с требованиями регистрационного досье, а не аналитико-нормативной документации, спецификаций и пр., что принято на постсоветском пространстве, отметила О. Баула. В России также пришли к этим же требованиям.
Структура регистрационного досье, утвержденная отечественными нормативными документами, на 100% гармонизирована с международными требованиями. Старый формат («4 части») ушел в прошлое. Теперь общепринятым считается досье в формате Общего технического документа. Используемый на протяжении 7 лет в других странах, этот формат показал свою уникальность, он позволяет проследить полную историю препарата и узнать об обязательствах компании относительно всех фаз жизненного цикла препарата.
Если подается заявка на генерический препарат, фармакологические и токсикологические сведения приводят согласно библиографическим данным. Клиническая документация в таких случаях описывает исследования по биодоступности и биоэквивалентности. «Мы настоятельно советуем переходить на этот формат, — подчеркнула докладчик. — На этих упомянутых выше обязательствах компании базируется все производство».
Основополагающий этап фармразработки — формирование качества. Компанией должна быть убедительно подтверждена либо фармацевтическая эквивалентность, либо альтернативность. В поле зрения разработчика и экспертов регуляторных органов должны быть следующие аспекты — действующие и вспомогательные вещества, лекарственная форма, которая, кстати, не усовершенствована, а воспроизведена.
Для генерика характерно, что все исследования на этапе фармацевтической разработки проводят путем сравнения с референтным препаратом. На сегодня его выбор должен быть законодательно внедрен на национальном уровне, и Украина, как отметила докладчик с надеждой, будет продолжать путь к интеграции в этом вопросе. ВОЗ разработала свои подходы к выбору инновационного препарата. В качестве источников информации о референтных препаратах О. Баула назвала следующие: ; ; ; ; .
Важная часть досье посвящена качеству действующих веществ. Ни европейская, ни украинская фармакопеи, как отметила О. Баула, не содержат монографий на готовые ЛС. Поэтому производитель должен разработать свои спецификации качества. Их основой должны стать аналогичные данные из доступного в любой аптеке сертификата качества референтного препарата. Если последний представлен на рынке США, Великобритании, задача производителя — выполнить требования национальных фармакопей этих стран.
Как только на наш рынок выходит генерический препарат со своим сертификатом, компания, представляющая на украинском рынке инновационный препарат, может сравнить свои спецификации с таковыми у «новичка». Эта информация не конфиденциальна, и таким образом легко составить представление о качестве ЛС.
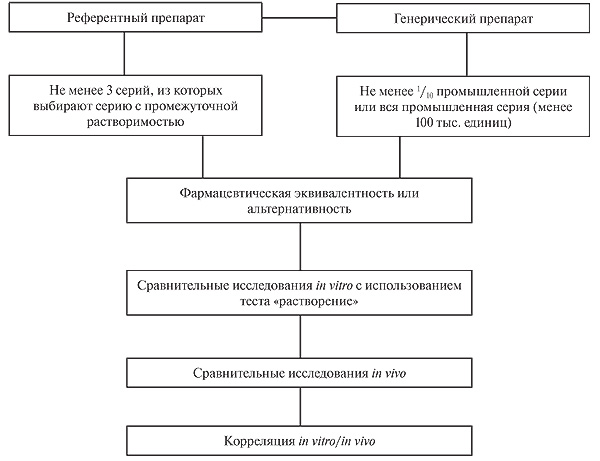
Фармразработка должна быть перенесена в серийное производство, что следует выполнять очень ответственно. В досье должен быть зафиксирован период, когда осуществляется перенос технологии производства. Если качество «встроено» в препарат, тогда понятны и риски на пути масштабизации производства. На стадии фармразработки необходимо предусмотреть пространство проектных параметров для постоянного усовершенствования. Для этого должны быть запрограммированы параметры, которыми можно управлять, отметила докладчик. Алгоритм доказательства эквивалентности показан на рис. 4. При этом, даже если для доказательства эквивалентности нельзя обойтись без сравнительных КИ, следует сначала провести сравнительные исследования in vitro, чтобы по возможности раньше выявить проблемы и устранить их до начала финансово затратной клинической части, которую в противном случае, вероятно, придется повторять.
|
Особое внимание слушателей О. Баула обратила на принципы доказательности исследований биоэквивалентности, сформулированные д-ром Лембитом Раго во время 1-й научно-практической конференции «Клинические испытания лекарственных средств в Украине» (см. «Еженедельник АПТЕКА» № 49 (570) от 18.12.2006 г.)
ИССЛЕДОВАНИЯ БИОЭКВИВАЛЕНТНОСТИ НИЧЕГО НЕ ЗНАЧАТ, если:
- имеются непостоянные производственные особенности как в составе продукта (качество активного фармацевтического ингредиента (АФИ), эксципиентов и пр.), так и в используемых процессах (GMP);
- исследуемый препарат — экспериментальный и его качество не спроецировано в масштабах промышленного производства;
- для изучения профилей растворения в ходе налаживания производственного процесса и последующих исследований биоэквивалентности использованы разные серии ЛС (не известны результаты теста «растворение» биосерий);
- отсутствуют регуляторные требования или контроль изменения качества в постмаркетиновый период.
 Вопросам качества действующих веществ посвятила свой доклад Таисия Герасимчук, эксперт отдела регистрации (перерегистрации) ЛС департамента фармацевтической деятельности ГФЦ МЗ Украины. Как отметила докладчик, критическими аспектами идентичности АФИ генерического и инновационного препаратов являются: химическая структура; полный профиль примесей (идентифицируемые и неидентифицируемые, в том числе отсутствующие в инновационном препарате, и их количество); физическая форма (морфология, размеры частиц, кристаллическая форма).
Вопросам качества действующих веществ посвятила свой доклад Таисия Герасимчук, эксперт отдела регистрации (перерегистрации) ЛС департамента фармацевтической деятельности ГФЦ МЗ Украины. Как отметила докладчик, критическими аспектами идентичности АФИ генерического и инновационного препаратов являются: химическая структура; полный профиль примесей (идентифицируемые и неидентифицируемые, в том числе отсутствующие в инновационном препарате, и их количество); физическая форма (морфология, размеры частиц, кристаллическая форма).
Производители, отметила Т. Герасимчук, должны хорошо разбираться в терминологии, в том числе, знать, что специфицированной примесью называют ту, которую индивидуально пересчитывают и ограничивают своим критерием в спецификации на АФИ. Она может быть идентифицированной (с определенной структурной формулой) и неидентифицированной. Неспецифицированной называют примесь, которую ограничивают при помощи общего критерия и отдельно не включают в спецификацию на АФИ. Квалификация примесей — процесс сбора и оценки данных, позволяющих установить безопасность индивидуальной примеси или данного профиля примесей на специфицированном уровне (уровнях). Порог квалификации — предел, выше которого примесь должна быть квалифицированной. Порог включения в отчет — предел, выше которого примесь следует включать в отчет.
Для подтверждения и выделения примесей, которые могут появляться в процессе хранения, проводят стрессовые исследования и определяют профиль примесей препарата сравнения, генерической субстанции и генерического препарата. На основании этих исследований можно классифицировать примеси, выявить те, которых нет в препарате сравнения, определить их уровень.
Разработчик генерического препарата обязан:
– гарантировать, что профили примесей генерика и препарата сравнения по сути одинаковы (а именно, все примеси в количестве выше порога идентификации и квалификации идентичны); содержание каждой из указанных примесей и сумма всех определяемых количественно примесей не превышают таковые для препарата сравнения на протяжении периода хранения обоих препаратов;
– найти и закупать АФИ у производителя, который детально его охарактеризовал с составлением соответствующего досье на АФИ;
– или стандартизировать профиль примесей в собственной спецификации на препарат и провести доклинические исследования и КИ безопасности, необходимые для регистрации препарата.
 Любовь Пилипчук, эксперт отдела регистрации (перерегистрации) ЛС департамента фармацевтической деятельности ГФЦ МЗ Украины, представила доклад о требованиях регистрационного досье к качеству готового ЛС. Основная цель регистрационного досье, как отметила докладчик, — установить уровень качества ЛС, предназначенного для размещения на рынке. Необходимые стандарты качества (спецификации) производитель предлагает и обосновывает, а компетентные и уполномоченные органы согласовывают, что является условием регистрации ЛС.
Любовь Пилипчук, эксперт отдела регистрации (перерегистрации) ЛС департамента фармацевтической деятельности ГФЦ МЗ Украины, представила доклад о требованиях регистрационного досье к качеству готового ЛС. Основная цель регистрационного досье, как отметила докладчик, — установить уровень качества ЛС, предназначенного для размещения на рынке. Необходимые стандарты качества (спецификации) производитель предлагает и обосновывает, а компетентные и уполномоченные органы согласовывают, что является условием регистрации ЛС.
В регистрационном досье в формате Общего технического документа спецификации приводят в разделах 3.2.S.4 «Контроль действующего вещества», 3.2.Р.4 «Контроль вспомогательных веществ» и 3.2.Р.5 «Контроль лекарственного средства». Различают два вида спецификаций готового ЛС: применяемые при выпуске и в период установленного срока хранения. Показатели качества и допустимые пределы значений спецификаций, применяемых в период хранения, должны быть установлены при изучении стабильности на основании: уровня содержания действующего вещества (эффективность); допустимого уровня содержания примесей (безопасность); постоянства фармакотехнологических характеристик (Руководство 42-3.2:2004. Спецификация: контрольные испытания и критерии приемлемости и ICH Q6A).
Спецификации на готовые ЛС при их выпуске должны предусматривать более жесткие параметры. Например, отклонение от количественного содержания действующего вещества не должно превышать ±5%, если нет других указаний. Если согласно предложенным спецификациям отклонение превышает 5%, необходимо это обосновать с помощью результатов экспериментальных исследований.
Л. Пилипчук подчеркнула, что раздел регистрационного досье «Анализ серий» должен содержать результаты, полученные относительно всех показателей качества, которые включены в спецификацию, применяемую при выпуске готового ЛС. А в сертификат качества готового ЛС производитель должен включить все показатели качества, соответствующие спецификациям при выпуске.
Уровень требований к параметрам спецификаций и методам контроля готового ЛС может быть выше требований украинской, европейской, британской, японской фармакопей, фармакопеи США, но не ниже. Параметры спецификаций генерических препаратов, не представленных в фармакопеях, должны быть такие же, как и у оригинального ЛС.
Источников подобной специфической информации — масса, нужно только уметь ими пользоваться. Если производитель хочет получить доступ сразу к очень большому объему информации, а не осуществлять поиск по отдельным источникам самостоятельно, он может воспользоваться платным доступом к определенным базам данных, к примеру названным О. Баулой: ; .
Дарья Полякова, фото Любови Столяр
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим