 Основным предназначением любой регуляторной системы в сфере обращения лекарственных средств является обеспечение допуска на рынок и дальнейшего обращения на нем исключительно качественных, эффективных и безопасных медикаментов по доступным для населения ценам. В данной публикации будут освещены первые три перечисленных аспекта рациональной лекарственный политики, вопросы же ценообразования, реимбурсации и прочие, которые обеспечивают экономическую доступность лекарственных средств, здесь рассматриваться не будут. Как было описано в наших ранних публикациях [1–3], эффективность, безопасность и качество лекарственных средств могут быть обеспечены только тогда, когда каждый этап «жизненного цикла» препарата осуществляется согласно соответствующей специфической системе качества, применяемой на этом этапе. Таким образом, обеспечение необходимых характеристик препарата является результатом эффективного функционирования глобальной системы качества, включающей отдельные специализированные системы качества (их универсальное название — GXP) на каждом из этапов его жизненного цикла. При этом задача регуляторной системы в сфере обращения лекарственных средств — осуществление контрольно-надзорных функций, ориентированных на контроль соответствия вышеуказанным системам качества и их соблюдения на всех этапах обращения лекарственных средств.
Основным предназначением любой регуляторной системы в сфере обращения лекарственных средств является обеспечение допуска на рынок и дальнейшего обращения на нем исключительно качественных, эффективных и безопасных медикаментов по доступным для населения ценам. В данной публикации будут освещены первые три перечисленных аспекта рациональной лекарственный политики, вопросы же ценообразования, реимбурсации и прочие, которые обеспечивают экономическую доступность лекарственных средств, здесь рассматриваться не будут. Как было описано в наших ранних публикациях [1–3], эффективность, безопасность и качество лекарственных средств могут быть обеспечены только тогда, когда каждый этап «жизненного цикла» препарата осуществляется согласно соответствующей специфической системе качества, применяемой на этом этапе. Таким образом, обеспечение необходимых характеристик препарата является результатом эффективного функционирования глобальной системы качества, включающей отдельные специализированные системы качества (их универсальное название — GXP) на каждом из этапов его жизненного цикла. При этом задача регуляторной системы в сфере обращения лекарственных средств — осуществление контрольно-надзорных функций, ориентированных на контроль соответствия вышеуказанным системам качества и их соблюдения на всех этапах обращения лекарственных средств.
В 2011 г. ВОЗ ввел понятие «строгий регуляторный орган» (stringent regulatory authority), которое в контексте обсуждаемых вопросов может восприниматься как «строгая регуляторная система» (СРС) [4]. Отнесение страны к категории «СРС» основано, по мнению ВОЗ, на строгом соблюдении и контроле выполнения в этой стране требований GLP, GCP и GMP. ВОЗ относит к странам СРС членов ICH [5], наблюдателей ІСН (Швейцария, Канада), страны, с которыми заключены соглашения о взаимном признании (Австралия, Исландия, Лихтенштейн, Норвегия). При этом ВОЗ отмечает, что данный список может быть продолжен. В отношении стандартов GMP и соответствующих систем инспектирования ВОЗ относит к странам СРС регуляторные органы, входящие в PIC/S (но только в части GMP!) [6].
Очевидно, что регуляторная система в сфере обращения лекарственных средств любой страны имеет ориентир для совершенствования — построение СРС, основанной на соблюдении и контроле выполнения требований основополагающих систем качества в сфере обращения лекарственных средств — надлежащих лабораторной, клинической и производственной практик. Однако, реформирование существующей регуляторной системы следует рассматривать комплексно, учитывая все сопутствующие факторы.
В данной публикации будут рассмотрены регуляторные процедуры, используемые в сфере обращения лекарственных средств, средства их реализации и нормативная основа для их осуществления, а также будут указаны наиболее серьезные проблемы, требующие первостепенного решения.
Регуляторные процедуры в сфере обращения лекарственных средств
Основными регуляторными процедурами в сфере обращения лекарственных средств являются лицензирование (производства, оптовой и розничной реализации), государственная регистрация и государственный контроль качества препаратов при их обращении. Не умаляя важности лицензирования оптовой и розничной реализации лекарственных средств, подчеркнем, что в контексте данной публикации мы остановимся не на всех видах лицензирования, а на лицензировании прежде всего производства лекарственных средств, которое требует гармонизации как соответствующих процедур, так и нормативной базы, о чем речь пойдет ниже. Чем эффективнее показанная на рис. 1 взаимосвязь процедур лицензирования, государственного контроля и государственной регистрации препаратов, тем ближе регуляторная система к СРС. Такая взаимосвязь проявляется в рамках всего регулирования сферы обращения лекарственных средств.
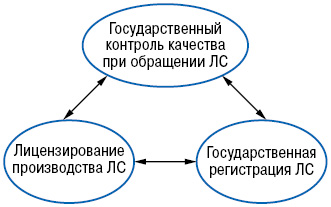
Приведем ряд примеров такой взаимосвязи. Регистрировать следует препараты, выпущенные только лицензированными производителями (а условием выдачи лицензии является, в свою очередь, соответствие GMP, о чем более подробно будет сказано ниже). В Украине с конца 2011 г. введена законодательная норма, предусматривающая регистрацию (перерегистрацию) только тех препаратов, которые произведены в условиях GMP. В отношении забракованных в рамках государственного контроля препаратов может приостанавливаться или аннулироваться как регистрация, так и лицензия на производство (последнее — в отношении отечественных, первое — в отношении как отечественных, так и зарубежных препаратов). Сообщение о непредвиденной побочной реакции, полученное в рамках системы фармаконадзора, может повлечь за собой ужесточение частоты и глубины государственного контроля в отношении данного препарата, опять же приостановление либо аннулирование лицензии или регистрации.
Приведенные примеры свидетельствуют о необходимости дальнейшего повышения эффективности взаимодействия регуляторных составляющих. Это еще более актуально для стран, где вышеуказанные регуляторные функции разделены между различными органами исполнительной власти и экспертными организациями. Вопросами государственного контроля и лицензирования производителей в Украине занимается Государственная служба Украины по лекарственным средствам (Гослекслужба), а государственной регистрацией — Министерство здравоохранения при экспертной оценке регистрационных материалов Государственным экспертным центром МЗ Украины (ГЭЦ). Отметим, что в ЕС и странах СРС эти функции, как правило, находятся в компетенции единого уполномоченного органа исполнительной власти (из стран ЕС только в Польше и Германии функции государственной регистрации лекарственных средств выполняет один уполномоченный орган, а государственного контроля, лицензирования и инспектирования производителей — другой). Такое сосредоточение функций в рамках одного уполномоченного органа исполнительной власти дает возможность повысить эффективность всей регуляторной системы в целом, в том числе путем обеспечения взаимосвязи и взаимодействия ее отдельных компонентов (см. рис. 1). Сегодня в Украине все более активно муссируется вопрос передачи функции регистрации лекарственных средств Гослекслужбе. При этом ссылки делаются на отчет 2008 г. миссии ВОЗ, Еврокомиссии и Агентства США по международному развитию. Справедливости ради отметим, что в этом отчете нет категоричной рекомендации о создании единого регуляторного органа в фармацевтической сфере. В нем отмечается целесообразность создания такого органа или налаживание более тесной координации между существующими органами. Для того чтобы не произвести эффект «слона в посудной лавке», необходимо тщательно взвесить последствия передачи Гослекслужбе функции регистрации лекарственных средств. При принятии решения о реорганизации системы контрольных и экспертных органов следует учитывать положение этих органов в системе органов исполнительной власти, в сфере здравоохранения, опыт работы экспертных органов в данной области, кадровый и научный потенциал, задействованный при проведении экспертиз, материальную базу, международные связи и др. Мы прекрасно понимаем, что государственные служащие Гослекслужбы никогда не смогут тщательно и профессионально провести самостоятельно экспертизу регистрационных досье, то есть все равно решения о госрегистрации будут приниматься на основании заключений экспертов/экспертной организации. В области экспертизы лекарственных средств авторитет ГЭЦ на сегодня бесспорен, а система регистрации в Украине хоть и не лишена серьезных недостатков, все же идет путем гармонизации с ЕС, поэтому отказаться от услуг этой организации при проведении регистрации препаратов практически невозможно, да и не нужно. В таком случае передача функций регистрации лекарственных средств от МЗ Украины Гослекслужбе выглядит как банальное «перетягивание одеяла на себя». Польза такого шага сомнительна, зато вред — очевиден. Такая пертурбация неизбежно парализует фармрынок (во всяком случае на неопределенное ближайшее время), что приведет к очередным социальным проблемам, с которыми мы уже столкнулись после введения НДС на лекарственные средства. Рациональным на данный момент представляется не революционный, а эволюционный путь усовершенствования регуляторной системы — обеспечение более эффективного взаимодействия в вышеуказанных вопросах и процедурах в треугольнике: МЗ — Гослекслужба — ГЭЦ путем издания внутренних приказов о таком взаимодействии при их неуклонном исполнении.
Средства реализации регуляторных процедур
Указанные выше регуляторные процедуры реализуются посредством определенных действий: лицензирование — с помощью инспектирования, государственный контроль качества — путем организации проведения лабораторного контроля, а государственная регистрация основана на экспертных заключениях. При этом перечисленные средства также должны быть связаны между собой. Так, в рамках государственной регистрации лекарственных средств могут потребоваться инспектирование производственной площадки либо информация о результатах такого инспектирования. Факт забраковки препарата в результате госконтроля может быть причиной проведения внепланового инспектирования производства, и, наоборот, результаты инспектирования могут служить основой ужесточения режима государственного контроля в отношении препарата или определенных лекарственных форм, или вообще всей продукции данного производителя. Примеры такого взаимодействия регуляторных мер можно продолжать и далее, но очевидно, что чем более тесное взаимодействие этих составляющих обеспечивается (рис. 2), тем выше эффективность регуляторной системы в целом, а значит, пациенту будут гарантированы эффективные, безопасные и качественные лекарственные средства.
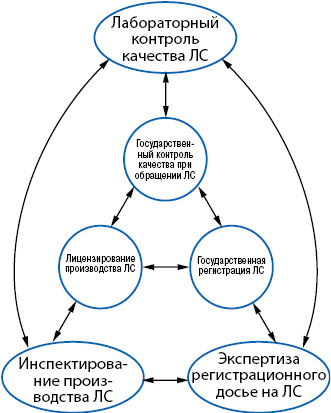
Важно, чтобы эти составляющие развивались по пути, проторенному ранее странами СРС, то есть, необходимо максимально гармонизировать средства осуществления регуляторных процедур. В Украине уже давно на высшем уровне принята стратегия гармонизации с законодательством ЕС, а Гослекслужба с 2011 г. стала полноправным членом PIC/S. Этому предшествовала большая и кропотливая работа по организации Инспектората GMP в 2000 г., его подготовку в 2000–2002 гг. в рамках проекта «Tacis», который финансировался Еврокомиссией. В дальнейшем была построена система качества инспектората, в лицензионные условия частично введены требования GMP, с 2003 г. начата процедура добровольной сертификации по GMP. С 2009 г. требования GMP вошли в обязательные лицензионные требования, соответственно были гармонизированы процедуры контроля за их соблюдением. На всех этапах вступления в PIC/S осуществлялось профессиональное сопровождение этого процесса. Путь перехода на GMP и присоединения к PIC/S был довольно тернистым, подробно об этом речь пойдет в следующей публикации. Отметим лишь, что люди, которые сейчас ожесточенно сражаются за звание «лучших евроинтеграторов», подключились к «марафону» внедрения GMP и вступлению в PIC/S уже на заключительной его «стометровке».
Рассмотрим подробнее состояние гармонизации, достигнутое при осуществлении регуляторных процедур в фармацевтической сфере в Украине. Система лабораторного контроля качества лекарственных средств традиционно у нас очень сильна. С одной стороны, это связано с наследием СССР, регуляторная система которого основывалась именно на лабораторном контроле. С другой — в последние годы в Украине были затрачены значительные финансовые и организационные ресурсы для усовершенствования материальной базы государственных контрольных лабораторий. ГП «Центральная лаборатория по анализу качества лекарств и медицинской продукции» принята в сеть официальных лабораторий по контролю медикаментов (OMCL), прошла аккредитацию на соответствие стандартам ISO 17025 со стороны Европейского директората по качеству лекарственных средств (EDQM) [7]. Кроме этого, она и лаборатория Фармацевтического анализа ГЭЦ получили одобрение со стороны ВОЗ и вошли в соответствующий список, подтверждающий техническую компетентность и независимость [8]. Заслуженным авторитетом пользуется лаборатория фармакопейного анализа ГП «Украинский научный центр качества лекарственных средств», которая, в числе прочих задач, стала экспериментальной базой подготовки и проведения туров профессионального тестирования для лабораторий контроля качества препаратов (в прошлом году был проведен 10-й юбилейный тур профессионального тестирования лабораторий при поддержке НФаУ и Гослекслужбы Украины). В Украине действует 6–7 уполномоченных лабораторий, имеющих техническую возможность провести тестирование препаратов любым фармакопейным методом, и сеть региональных лабораторий Гослекслужбы в соответствующих территориальных единицах Украины.
Следующий аспект — экспертиза в рамках государственной регистрации. Надлежащий уровень экспертизы обеспечивается тем, что в Украине есть компетентные эксперты и экспертные организации (ГЭЦ), определены нормативные требования к экспертам, порядку проведения экспертизы, ее срокам и пр. Таким образом, с точки зрения профессионального подхода к проведению экспертных оценок регистрационных материалов, внедренная в Украине система мало чем отличается от практики стран СРС (мы не акцентируем внимание на некоторых процедурных вопросах, а рассматриваем профессиональную составляющую экспертных работ). То есть, в организации и проведении экспертной оценки регистрационных материалов в Украине достигнут адекватный уровень по сравнению с практикой ЕС. Конечно, существуют проблемы конфликта интересов в рамках экспертной организации и отдельных экспертов, что отмечалось в 2008 г. в материалах отчета экспертов ВОЗ, Европейской комиссии и Агентства США по международному развитию (миссия была направлена на оценку регуляторной системы Украины в фармацевтической сфере). По мнению миссии, следует строго обеспечивать отсутствие такого конфликта на уровне штатных и внештатных экспертов ГЭЦ, чтобы не допустить лоббирования интересов той или иной фармкомпании. На уровне организации конфликт интересов заключается в том, что лаборатория биоэквивалентности ГЭЦ проводит исследования для субъектов рынка, которые затем подают материалы этих исследований в составе регистрационного досье и, естественно, успешно регистрируют свои препараты в Украине. Однако, во-первых, эти проблемы при желании могут быть достаточно легко преодолены административными мероприятиями, а, во-вторых, передача функций экспертизы либо переподчинение ГЭЦ Гослекслужбе проблему конфликта интересов не решают. В нынешних условиях нестабильного валютного курса любые «потрясения» фармсектора путем введения необоснованных новых регуляций или проведения ненужных реорганизаций неизбежно вызовут новый скачок цен и/или дефицит лекарственных препаратов, что категорически недопустимо.
Нормативная основа реализации регуляторных процедур
Все вышеперечисленные регуляторные процедуры и средства для их реализации не могут существовать без фундамента, каковым является совокупность нормативно-технических документов, изображенных на рис. 3. В основе системы лабораторного контроля качества лежит Государственная Фармакопея Украины (ГФУ), инспектирование производств должно быть основано на соблюдении требований GMP, а регистрационные материалы (и, соответственно, их экспертиза) — на формате Общего технического документа (CTD).
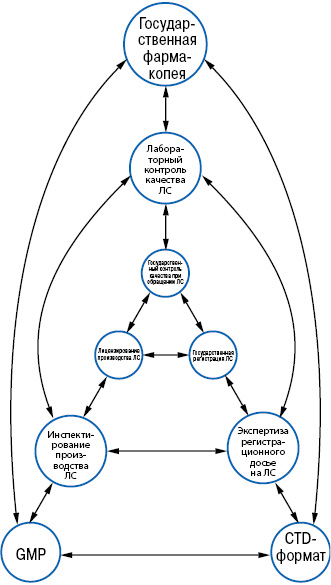
Очевидно, что указанные документы тесно связаны друг с другом. Базовым уровнем требований к качеству при регистрации лекарственных средств являются требования ГФУ, в CTD-формат включаются данные о соответствии производства требованиям GMP. Требования GMP ссылаются на фармакопейные методы стерилизации, регламентируют использование спецификаций со ссылкой на фармакопейные методы, оговаривают необходимость валидации аналитических методик и др. Принципиально важно построение или совершенствование регуляторной системы именно на фундаменте гармонизированных с ЕС нормативно-технических документов. Начнем с наличия нормативно-технического документа, то есть гармонизированного стандарта (руководства) GMP. Украина еще с 2001 г. полностью гармонизировала свое руководство GMP c GMP ЕС, после этого документ 6 раз переиздавался (только в 2011 г. — дважды) в связи с внесением изменений в GMP ЕС. Отдельно остановимся на характеристике состояния ГФУ. Не имея ни копейки бюджетного финансирования, коллектив специалистов Фармакопейного центра сделал в свое время прорыв в сфере отечественной фармацевтической стандартизации, подготовив первое издание ГФУ. В подготовке этого документа участвовали Национальный фармацевтический университет, ведущие институты НАН и АМН, вузы, предприятия и организации Украины. В 2001 г. в Украине была издана ГФУ, которая полностью гармонизирована с Европейской Фармакопеей, после этого изданы четыре Дополнения к первому изданию. С 1998 г. Украина стала наблюдателем Европейской Фармакопеи, а с 2009 г. — Фармакопейной конвенции США. В 2013 г. Украина стала 38-м членом Европейской Фармакопеи, присоединившись к Конвенции по разработке Европейской Фармакопеи, что закреплено на законодательном уровне Украины и ЕС. В настоящее время ведется активная подготовка второго издания ГФУ, первый том которого уже вышел в свет.
Последняя часть рассматриваемой нормативной основы регуляторных процедур связана с использованием CTD-формата в регистрационных досье и соответствующим проведением экспертизы этих досье. В Украине требование подачи регистрационных досье в таком формате применяют к впервые регистрируемым препаратам. К сожалению, для препаратов, которые проходят перерегистрацию, допускается и другой, «национальный», формат, не гармонизированный с CTD-форматом. В Украине пока есть серьезные проблемы с исследованием биоэквивалентности с позиций доказательной медицины. Если в нашей стране в год число проводимых исследований биоэквивалентности составляет примерно 5–7, то в соседней России в 2012 г. проведено 322 исследования биоэквивалентности, объективно характеризующих эффективность и безопасность регистрируемых генерических лекарственных средств!
Таким образом, в части нормативно-технических документов, поддерживающих всю регуляторную систему, существуют большие или меньшие проблемы, требующие систематической и целенаправленной работы для их решения.
Направления совершенствования регуляторной системы в сфере обращения лекарственных средств Украины
Учитывая проведенный выше анализ компонентов регуляторной системы в сфере обращения лекарственных средств, можно выделить блоки по трем направлениям дальнейшего совершенствования регуляторной системы Украины. При этом в каждом из блоков можно сформулировать следующие мероприятия для решения описанных проблем:
В сфере ГФУ:
- национальная часть ГФУ при введении GMP в обязательные лицензионные требования должна стать информационно-методической, а не требующей проведения дополнительных видов контроля;
- следует обеспечить дальнейшее развитие ГФУ, увеличение количества монографий на субстанции и частных монографий на готовые лекарственные формы;
- обеспечить дальнейшее развитие системы фармакопейных стандартных образцов.
В сфере GMP:
- регламентировать использование субстанций, произведенных только в условиях GMP;
- проводить инспектирование производства препаратов для клинических исследований в рамках плановых и внеплановых лицензионных инспекций производства лекарственных средств.
В сфере государственной регистрации лекарственных средств:
- не допускать регистрации (перерегистрации) генерических препаратов с уровнем качества ниже установленного в ГФУ (при отсутствии в ГФУ и других фармакопеях — не ниже уровня качества оригинального препарата);
- ввести обязательность доказательства терапевтической эквивалентности для генериков (биоэквивалентность);
- в рамках процедур государственной регистрации лекарственных средств предусмотреть инспектирование на соответствие GLP и GCP;
- исключить все формы регистрационных документов, кроме CTD-формата.
законодательное регулирование фармацевтического сектора
Евроинтеграционное направление развития фармотрасли Украины может быть обеспечено, безусловно, только при наличии соответствующих законодательных и подзаконных актов, регулирующих эту сферу. Стратегия интеграции Украины в Европейский Союз, включающая адаптацию украинского законодательства, была утверждена Верховной Радой Украины еще в 1998 г. В фармацевтической сфере Украины евроинтеграционные процессы зачастую опережали законодательные рамки. Так, Закон Украины «Про лікарські засоби», принятый в 1996 г., не определяет до настоящего времени такие основные понятия, как GMP, GLP, GCP, GDP, и соответствующие процедуры, по которым уже давно работает отрасль. Вместе с тем насущные требования развития фармсектора продиктовали необходимость внесения этих и других понятий и процедур в подзаконные акты (постановления Кабинета Министров Украины, приказы Минздрава, Руководства и пр.), оперативно регулирующие сферу обращения лекарственных средств. Очевидно, что ныне действующий Закон Украины «Про лікарські засоби», в который после его принятия внесены многочисленные правки, стал похож на «лоскутное одело», и на сегодня нуждается в кардинальном пересмотре, то есть, в подготовке и утверждении новой его редакции, полностью гармонизированной с законодательством ЕС. Такие попытки ранее предпринимались неоднократно. Так, в 2002 г. Верховна Рада Украины в первом чтении приняла Закон Украины «Про лікарські засоби» в новой редакции, учитывавшей все положения законодательства ЕС в фармацевтической сфере. Автором этого законопроекта был народный депутат Украины П. Порошенко. В 2010 г. проект новой редакции закона, учитывающий соответствующие положения законодательства ЕС, был подан народным депутатом П. Жебровским. К сожалению, ни один из указанных законопроектов так и не был принят в целом. В настоящее время под эгидой МЗ Украины созданы рабочие группы, в частности, по разработке новой редакции закона. При этом хотелось бы пожелать этим рабочим группам максимально использовать рациональные предыдущие наработки и опыт законотворческой работы, а также опираться на помощь не только ассоциаций, но и профессионалов ведущих вузов, НИИ, а также юристов, работающих в сфере фармацевтического права.
Кроме этого, необходимо помнить, что фармацевтический сектор Украины и так зарегулирован, и вводить новые регуляции, отсутствующие в ЕС (например «национальные регуляции» в виде лишения аптечной сети лицензии за «неправильное» ее название либо несоблюдение торговой наценки и пр.), категорически недопустимо.
Таким образом, изобретать ничего не надо, следует просто взять законодательные нормы ЕС, грамотно и профессионально их перевести и принять в Украине, тем более что они, по сути, у нас уже практически все имплементированы.
доктор фармацевтических наук, профессор
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим