|
Мы уже неоднократно обращались в своих публикациях (см. «Еженедельник АПТЕКА» № 10 (531) от 13 марта 2006 г.; № 27 (548) от 10 июля 2006 г.) к вопросу об украинских и международных нормах регистрации лекарственных средств (ЛС). Понимание того, как они применяются, совершенно необходимо для оценки перспектив их использования в нашей стране.
СТРАТЕГИЧЕСКИЙ БАЛАНС
Законы, согласно которым разрешение на маркетинг ЛС начали выдавать только после доказательства эффективности и безопасности, в США и странах Европейского экономического сообщества появились в один и тот же период, отмеченный «талидомидовой драмой» — в 1962 и 1965 гг. соответственно. За четыре десятилетия, прошедшие с тех пор, в фармацевтическом законодательстве произошли значительные изменения, ведущие к гармонизации и повышению эффективности регуляторной системы.
Принятой в 1962 г. поправкой к федеральному закону о пищевых продуктах и лекарственных средствах (Federal Food and Drugs Act) введено требование доказательства эффективности препарата перед выдачей разрешения на маркетинг; ранее она производилась только на основании безопасности. Для доказательства эффективности и безопасности от компаний стали требовать проведения клинических исследований с участием людей и представления этих результатов в Управление по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) вместе с заявкой на получение разрешения на маркетинг (new drug application — NDA). От заявителей, обращающихся за получением разрешения на маркетинг генерических версий препаратов, одобренных на основании поправки 1962 г., требовали проведения других исследований эффективности и безопасности, которые неизбежно повторяли проведенные инновационными компаниями. Они не являлись необходимыми, были связаны с потерей времени и, более того, были неэтичными, поскольку включали больных, получавших плацебо. Именно так FDA оценивало ситуацию, когда более простая процедура выдачи разрешения на маркетинг генерических версий оригинальных ЛС с истекшей патентной защитой еще не действовала (H.R. Rep. No. 98-857(I) (1984)). По оценкам FDA, к 1984 г. таких оригинальных препаратов, не имевших генерических версий, насчитывалось около 150 (там же).
Другим фактором, замедляющим процесс выведения на рынок генерических ЛС, было то, что компании не могли начать процесс получения разрешения на маркетинг, прежде чем истечет срок патентной защиты оригинального препарата (Roche Products, Inc. v. Bolar Pharmaceutical Co., 733 F.2d 858 (Fed. Cir. 1984)). Такое применение патентного права отсрочивало одобрение генериков и задерживало их выведение на рынок. В то же время инновационные компании нередко сталкивались с тем, что срок патентной защиты истекал вскоре после того, как инновационный препарат получал разрешение на маркетинг.
|
Принятие закона о ценовой конкуренции на лекарственные средства и восстановление срока действия патентов, который иначе называют поправкой Ваксмена — Хетча к федеральному закону о пищевых продуктах и лекарственных средствах (The Drug Price Competition and Patent Term Restoration Act of 1984 (Pub. L. No. 98-417) (the Hatch-Waxman Amendments to the Federal Food, Drug and Cosmetic Act), преследовало две цели. Во-первых, компаниям разрешили предпринимать необходимые для получения разрешения на маркетинг генерического препарата действия до истечения срока действия патентной защиты оригинального. Во-вторых, генерическим компаниям при подаче заявки позволили полагаться на полученные при разработке оригинального ЛС данные о его безопасности и эффективности. Это не означает, что им предоставили доступ к конфиденциальной информации, просто ее стали использовать регуляторные органы при выдаче разрешения на маркетинг. Для того чтобы в некоторой степени компенсировать инновационным компаниям ускорившийся выход генериков, предусмотрено продление действия патентов в некоторых случаях (см. ниже).
Таким образом, поправка Ваксмена — Хетча призвана поддерживать баланс между быстрым выведением на рынок генериков и сохранением заинтересованности инновационных компаний в создании новых ЛС. Одновременно с заявкой на получение разрешения на маркетинг компания-оригинатор представляет в FDA список патентов, имеющих отношение к тому или иному ЛС, которые публикуются в издании «Получившие разрешение на маркетинг лекарственные средства с оценками терапевтической эквивалентности» («Approved Drug Products with Therapeutic Equivalence Evaluations») — базе данных, которую принято называть «Оранжевой книгой» («Orange Book»). Это издание общедоступно в электронной форме (). Информация о генерических препаратах и патентах в нем обновляется ежедневно. Список патентов, относящихся к определенному NDA, можно увидеть, щелкнув мышкой на номере заявки (первая колонка).
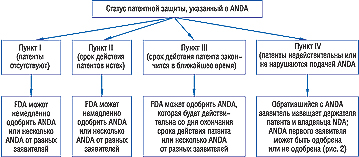
Сокращенная заявка о получении разрешения на маркетинг (abbreviated new drug application — ANDA), которая используется согласно статье 505(j) поправки Ваксмена — Хетча, должна включать информацию о патентах, защищающих права на соответствующий NDA препарат. Этот раздел заявки должен содержать одно из четырех утверждений, которые для удобства обозначают как пункт (paragraph) I, II, III, IV. Обозначают они следующее: (I) в FDA не подается никакой патентной информации о продукте, который является предметом ANDA; (II) срок действия патента закончился; (III) срок действия патента не истек, но закончится вскоре, и ANDA вступит в силу в этот момент; (IV) патент недействителен или не будет нарушен генерическим препаратом, которого касается ANDA.
Если заявитель апеллирует к п. I или II и соблюдены все остальные требования, FDA может одобрить ANDA немедленно, если к п. III — в день истечения срока действия патента (рис. 1).
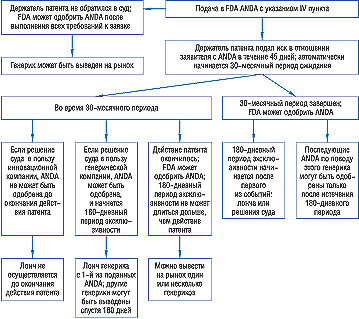
Утверждение, касающееся IV пункта, влечет за собой два важных последствия. Первое — 30-месячный период ожидания, предоставляемый компании-оригинатору. Заявитель, подающий ANDA с указанием IV пункта, должен предоставить как держателю патента, так и владельцу NDA детальное описание как правовых, так и фактических оснований для вывода подателя ANDA о том, что патент недействителен или не нарушается. В ответ на это держатель патента (обычно это компания-оригинатор) в 45-дневный срок может подать судебный иск, после чего наступает 30-месячная отсрочка. Если держатель патента в течение 45 дней после получения сведений от подателя ANDA об использовании IV пункта не обратится в суд, процесс одобрения может продолжаться, и FDA может выдать разрешение на маркетинг. Подача судебного иска задерживает одобрение ANDA до одного из трех, наступившего раньше других, событий: (1) истечение срока действия патента; (2) окончательное решение суда* о недействительности патента или ненарушении патентных прав; (3) истечение 30-месячного срока после подачи ANDA.
О маркетинговой эксклюзивности и защите данных мы подробно писали в предыдущем номере (см. «Еженедельник АПТЕКА» № 33 (554) от 28.08.2006 г.). В США ANDA не может быть объектом регуляторных действий, если не истекли хотя бы 4 года из 5-летнего срока маркетинговой эксклюзивности, предоставляемой владельцу NDA (21 CFR314.101). 5 лет — срок эксклюзивности для препарата, содержащего новый активный фармацевтический ингредиент; 3 года — других ЛС, одобренных на основании NDA; 7 лет — ЛС для лечения редких заболеваний (orphan drugs — препараты-сироты). Если проведены клинические испытания в педиатрии, предоставляется дополнительная эксклюзивность сроком на 6 мес.
Второе важное положение статьи 505(j) поправки Ваксмена — Хетча, касающееся IV пункта, предназначено, как сказано в соответствующем руководстве, для поощрения производителей генерических ЛС оспаривать патенты, которые могут быть недействительными или не распространять свое действие на генерик, по поводу которого подается ANDA. Генерические компании, обратившиеся в FDA с ANDA, содержащей пояснения по IV пункту, получают 180-дневный период эксклюзивности. Первый заявитель, представивший такую ANDA, имеет право на эту привилегию, являющуюся экономическим стимулом для компаний, и в течение этого периода FDA не может одобрить другую ANDA на такой же генерический продукт. Доказывать, что действие патента не относится к ее генерическому препарату или создавать продукт с измененными характеристиками, чтобы «обойти» патентные права компании-оригинатора — непростая задача, выполнение которой возможно только при наличии мощных стимулов, каким и является маркетинговая эксклюзивность. Началом 180-дневного периода считается выведение генерика на рынок или решение суда в пользу генерической компании, в зависимости от того, что наступит раньше (рис. 2).
Так в США организована взаимосвязь выдачи разрешения на маркетинг генерических препаратов и статусом патентной защиты оригинальных (patent-registration linkage). FDA не берет на себя функции владельца патента в обеспечении его прав, а только участвует в информировании о наличии патентной защиты (при помощи «Оранжевой книги») и намерениях третьей стороны ее оспорить. Отстаивать свои права предоставляется самому владельцу патента, который волен решать возникший спор через суд. В Европе, как будет указано ниже, взаимосвязь патентов и разрешения на маркетинг не применяется. Но европейские уполномоченные органы, как указано в пресс-релизе Европейской ассоциации производителей генерических препаратов (European Generic Medicines Association — EGA), в настоящее время испытывают растущее давление, что EGA расценивает как одну из серьезнейших угроз для отрасли в ее стремлении повышать доступность препаратов для пациентов. Это давление осуществляется инновационными компаниями непосредственно или через американских торговых представителей в европейских странах. По мнению EGA, такая практика замедлит выведение на рынок генериков (, пресс-релиз от 2 февраля 2006 г.).
«Замедляя распространение достижений технического прогресса, патенты создают условия для того, чтобы распространялись более значимые достижения» (Robinson J., 1956) — в этой фразе — суть и смысл патентной защиты, которая обеспечивает возврат инвестиций, осуществленных держателем патента, позволяя ограничить конкуренцию в случае коммерциализации изобретения. Это становится стимулом для значимых инноваций. На получение патента в США уходит в среднем 3,4 года, а в области биотехнологий — 4,4 года. Основанием для его выдачи являются новизна, польза и неочевидность. Если патент выдает Управление по патентам и торговым маркам США (United States Patent and Trademark Office — USPTO), изобретатель получает 20-летнюю монополию (со дня подачи патента; со дня выдачи — 17 лет) в обмен на разглашение информации, которая является предметом интеллектуальной собственности. В соответствии с законом о патентах (Patent Act) USPTO продлевает срок действия патента «день за день», если в процессе рассмотрения потеряно время не по вине заявителя. Патентная защита также может быть продолжена в соответствии с поправкой Ваксмена — Хетча, но не более чем на 5 лет, при этом действие патента не должно продолжаться более 14 лет с момента его получения ().
Прорывать патентную защиту Большой Фармы удается только компаниям — лидерам генерического сектора рынка, которых называют молодыми волками. Подобно инновационным, эти компании имеют свои продуктопроводы генерических препаратов, находящихся на разных стадиях готовности к лончу. Это требует постоянного анализа портфеля патентов компаний-оригинаторов, что позволяет определить «слабые места» или «обходные пути», которыми можно воспользоваться при разработке генериков. Участвовать в судебных процессах, отстаивая право вывести на рынок генерик, может позволить себе далеко не каждая компания. Сообщается, что в среднем услуги адвокатов в таких случаях обходятся в 2 млн дол. США (Antolin S., Boyd W., 2005).
Анализ, мониторинг и применение информации о патентах требует участия консультантов со специальной подготовкой. Ведь патент на молекулу действующего вещества — только верхушка айсберга защиты оригинального ЛС. Интересные факты о препаратах и интеллектуальной собственности на примере оригинального препарата аторвастатина сообщают специалисты «GenericsWeb Pty Ltd» — австралийской фирмы по патентному анализу и поиску (). На рынок ЕС (в Великобритании) Lipitor впервые был выведен в 1996 г., и срок действия патента на активный фармацевтический ингредиент (АФИ) истекает в 2011 г. Существует около 230 семейств (!) патентов, касающихся препаратов аторвастатина, а именно молекулы действующего вещества, состава препаратов, процесса производства, применения и пр. Только в 80% из них упоминается аторвастатин. Держателей соответствующих патентов насчитывается более 120, часть из них — генерические компании, которые разрабатывают препарат аторвастатина таким образом, чтобы не посягать на интеллектуальную собственность Большой Фармы. Среди них лидируют «Biocon» (АФИ), «Teva» (АФИ и препарат), «Lek». Основной упор в разработке делается на использовании аморфного, а не кристаллического аторвастатина, причем активность получения патентов в последние 5–6 лет существенно возросла. То есть понятие «разработка генерических препаратов» в условиях соблюдения прав интеллектуальной собственности — это не что-то символическое и абстрактное. Получая патенты, определенным образом касающиеся оригинальных препаратов, уже через 2–3 года после их выведения на рынок лидеры генерической отрасли вносят свой вклад в интеллектуальной сфере.
Генерический препарат, вышедший на рынок первым, в США становится предметом 50–80% всех назначений уже спустя 10 нед после выведения на рынок. Доля рынка, которую занимает генерик-пионер, в денежном выражении составляет примерно 57,6%. При этом розничная цена препарата ниже, чем оригинального примерно на 40%. С появлением новых генериков первый лишь немного теряет свою долю в назначениях (Fernandez D.S., Huie J.T., 2003; Milanese R.S., 2005). С учетом этих экономических соображений выведение своего генерика на рынок первым с использованием периода маркетинговой эксклюзивности является очень желанной целью для компаний. В последнее время подача ANDA с указанием IV пункта весьма участилась: если в первые годы после вступления в силу поправки Ваксмена — Хетча их доля составляла только 2%, то в 1998–2000 гг. около 20% заявок были такими (FTC, 2002). Согласно последнему отчету Федеральной комиссии по торговле США (Federal Trade Commission — FTC), в котором собрана такая статистика, с 1 января 1992 г. по 1 января 2001 г. в FDA были поданы АNDA с указанием IV пункта, касающиеся 104 NDA. Инновационные компании сочли необходимым отстаивать свое право на патентную защиту 75% оригинальных ЛС, то есть подали иск в суд против производителей, намеревавшихся первыми вывести генерики на рынок. В подавляющем большинстве оставшихся 25% случаев АNDA были одобрены в среднем через 24 мес и 2 нед после подачи. Из принятых по состоянию на 1 июня 2002 г. судебных решений 73% — в пользу генерических компаний. Хотя справедливости ради следует отметить, что 30% дел на тот момент еще находились в «подвешенном» состоянии.
ПОРЯДОК РЕГИСТРАЦИИ В ЕС
Существующая европейская система регистрации ЛС включает 4 основных процедуры: централизованную, взаимного признания, децентрализованную и на национальной основе.
Основной целью централизованной процедуры, разработанной в 1993 г., является совершенствование процесса экспертизы регистрационных документов и обеспечение быстрого доступа ЛС на рынки стран-членов. Ее появление имеет первостепенную важность для генерических компаний, которые получили возможность подавать единую для всех стран-членов краткую характеристику препарата (Summary of product characteristics — SPC) и проект инструкции. Ранее, поскольку регуляторное одобрение инновационных препаратов производилось в каждой стране отдельно, их регистрационные данные (к примеру, доза, способ применения, противопоказания и т.п.) могли отличаться в разных странах. В связи с этим Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA)** выявляло значительные отличия в SPC поданных на регистрацию генериков и референтных препаратов, что препятствовало выдаче разрешения на маркетинг (Almeida S., 2006).
|
||||
| **Согласно новому фармацевтическому законодательству децентрализованный орган ЕС, ранее носивший название Европейское агентство по оценке лекарственных средств (European Medicines Evaluation Agency — EMEA), называется Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA), однако акроним ЕМЕА остался без изменений. |
Централизованную процедуру согласно Регламенту 726/2004/EC (пояснение этих положений дается в соответствующем руководстве) в обязательном порядке следует применять ко всем продуктам, перечисленным ниже, с ноября 2005 г.:
1) ЛС, разработанные с применением следующих биотехнологических процессов:
- рекомбинантных ДНК-технологий;
- контролируемой экспрессии генов, кодирующих биологически активные белки прокариот и эукариот, включая трансформированные клетки млекопитающих;
- методов гибридомы и моноклональных антител;
2) ЛС для применения в ветеринарии;
3) ЛС, содержащие новый АФИ, который к моменту вступления в силу регламента не был одобрен в сообществе, показанный для лечения одного из следующих заболеваний:
- приобретенный синдром иммунодефицита;
- онкологические заболевания;
- нейродегенеративные расстройства;
- сахарный диабет;
… и с мая 2008 г.:
4) ЛС, классифицированные как препараты-сироты.
Также с 1993 г. существует процедура взаимного одобрения, предусмотренная для того, чтобы заявитель мог получить разрешение на маркетинг сразу в нескольких странах-членах, если к моменту подачи заявки препарат одобрен на национальном уровне. В 2004 г. разработана еще и децентрализованная процедура, при помощи которой можно получить разрешение на маркетинг в нескольких странах, не имея национального одобрения.
Появилась возможность ускорить получение одобрения по централизованной процедуре: если продукт представляет большую потенциальную ценность, Консультативный научный комитет (Committee for Medicinal Products for Human Use) EMEA должен предоставить заключение в пределах не 210, как обычно, а 150 дней (Регламент 726/2004/ЕС; п. 33 преамбулы и ст. 6(3)).
Важно, что создается новый гармонизированный подход в обеспечении защиты данных и маркетинговой защиты. Впервые такая мера, действующая в ЕС совершенно отдельно от патентного законодательства, была введена в 1987 г. Свое дальнейшее развитие она получила со вступлением в силу нового фармацевтического законодательства. Согласно ст. 14(11) Регламента 726/2004/ ЕС и ст. 10.1. Директивы 2004/27/ЕС, регуляторные органы могут полагаться на данные преклинических и клинических испытаний референтного препарата при одобрении генерика, только если референтный был зарегистрирован (согласно требованиям нового законодательства) не менее 8 лет тому назад. Это так называемый период эксклюзивности данных. Вывести же на рынок такой генерический препарат можно спустя не менее 10 лет после выдачи разрешения на маркетинг референтного. Десятилетний период маркетинговой эксклюзивности может быть продолжен максимум до 11 лет, если во время первых 8 из этих 10 лет было одобрено одно или больше показаний к применению, что во время предшествующей научной оценки было расценено как источник серьезных преимуществ по сравнению с существующими терапевтическими возможностями. Директивой 2004/27/ЕС положена легальная основа используемого во многих других странах так называемого регуляторного исключения или условия «Bolar» (по названию компании, которая добивалась такой возможности через суд). В соответствии с ним проведение необходимых для разработки генерика клинических и других исследований не считается противоречащим действию патентов или дополнительных защитных сертификатов референтного препарата.
Важное дополнение: согласно ст. 10.2(b) Директивы 2004/27/ЕС различные лекарственные формы немедленного высвобождения для перорального применения должны считаться одной и той же формой, и в случае расширения линии дополнительная маркетинговая эксклюзивность не предоставляется.
Европейские разработчики генериков лишены такого полезного и удобного в использовании ресурса, как «Оранжевая книга» в США. Общеевропейский регистр (ec.europa.eu/enterprise/pharmaceuticals/register/index.htm) содержит данные, да и то только самые основные, без указаний референтного препарата, без сведений о патентной защите и терапевтической эквивалентности, только о зарегистрированных по централизованной процедуре препаратах. Часть информации можно получить, пользуясь ресурсами национальных регуляторных агентств (heads.medagencies.org/index.html). С целью повысить эффективность процесса регистрации и улучшить обмен информацией о продукте (краткая характеристика препарата, листок-вкладыш и инструкция) между всеми участниками (ЕМЕА, национальными регуляторными органами и заявителями) была создана система управления информацией о продукте (product management information — PIM) (pim.emea.eu.int). В июне 2006 г. в PIM поступил первый реальный, не тренировочный, пакет документов. В дальнейшем планируется расширить сферу действия системы с обеспечения только централизованной на децентрализованную и процедуру взаимного признания.
Только Директивой 2004/27/ЕС в европейское законодательство введены термины «референтный препарат» и «генерический препарат». Ранее, а именно в Директиве 2003/63/ЕС, появилось используемое в формате общего технического документа определение по сути аналогичного лекарственного средства. Директива предоставляет больше свободы производителям генериков, потому что согласно ст. 10.1 не требуется, чтобы референтный препарат был зарегистрирован в той стране, где подают заявку на генерик. То есть то, что инновационная компания не посчитала рынок Эстонии или Мальты, к примеру, интересным для своих препаратов, не должно становиться препятствием для одобрения их генерических версий. n
*Суд, выносящий финальное решение, после которого апелляции невозможны (21 CFR 314.107(e)(1) (1999) — если ANDA подавалась до марта 2000 г. Если позднее — окружной суд (FDA, Guidance for Industry, Court Decisions., 2000).
Продолжение следует
Дарья Полякова
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим