Руководство ВОЗ для органов по регулированию лекарственных средств. 1999 г.
IV. РАССМОТРЕНИЕ ЗАЯВОК О ПОЛУЧЕНИИ РАЗРЕШЕНИЙ НА МАРКЕТИНГ МНОГОИСТОЧНИКОВЫХ (ГЕНЕРИЧЕСКИХ) ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
А. Сфера применения
Рекомендации, приведенные в настоящем руководстве, относятся к многоисточниковым фармацевтическим продуктам, содержащим хорошо известные лекарственные средства, для которых не требуется представления комплекса токсикологических и клинических данных, необходимых в случае новых химических соединений. Многие принципы применимы и к другим группам лекарственных средств (таким как сложные биологические препараты, «альтернативные» препараты и лекарственные средства для использования в ветеринарии), хотя в деталях они могут и отличаться. Уже существует отдельное руководство в отношении лекарственных препаратов растительного происхождения (Guidelines for the assessment of herbal medicines. In: WHO Expert Committee on Specifications for Pharmaceutical Preparations. Thirty-fourth report. Geneva. World Health Organization, 1996: 178-184 (WHO Technical Report Series. No. 863). Повторно опубликовано в: Quality assurance of pharmaceuticals: a compendium of guidelines and related materials. Vol. 1. Geneva, World Health Organization, 1997: 31-37). Во многих странах обращение препаратов, предназначенных для использования в ветеринарии, регулируется другим агентством.
Настоящие детализированные рекомендации применимы к антибиотикам и субстанциям, которые, несмотря на биологическое происхождение, обладают низкой молекулярной массой и могут быть выделены в виде чистых веществ (например, очищенные стероиды и алкалоиды).
С другой стороны, данные рекомендации не относятся к более сложным биологическим веществам бoльшей молекулярной массы, чистоту, активность и состав которых нельзя легко и надежно определить с помощью химического или физико-химического анализа. Вещества этой группы включают вакцины, препараты крови, модифицированные ткани животного происхождения, гормоны с высокой молекулярной массой, аллергены, продукты генной инженерии или других новейших биотехнологий производства.
Владельцы новых лицензий редко, если это вообще случается, могут получить разрешение на производство биологических препаратов второй группы лишь на основании результатов исследований in vitro с препаратом сравнения. Препараты различных торговых марок могут иметь одинаковые показания к применению (например, противококлюшная вакцина), но эффективность и безопасность каждого из них должны быть продемонстрированы независимо друг от друга. Они даже могут иметь различные схемы дозирования. Данные о концентрациях «активных ингредиентов» в плазме крови обычно также мало информативны, поскольку неясно, проводилось ли измерение одного и того же вещества. Поэтому эти препараты нельзя утвердить без данных о безопасности и эффективности; следовательно, они не являются предметом настоящего руководства. Для этой группы препаратов не применима типовая форма заявки (которая будет рассмотрена в Приложении 6).
В настоящее время ВОЗ разрабатывает руководство по оценке вакцин и выдаче разрешений на их маркетинг.
Данное руководство охватывает как отпускаемые по рецепту, так и безрецептурные («over-the-counter» или OTC) лекарственные препараты. Считается, что безрецептурные препараты несут в себе меньший риск для потребителей, чем рецептурные. Таким образом, в условиях ограниченных ресурсов, при выделении средств для оценки качественных аспектов заявок ОРЛС могут отдать приоритет препаратам, отпускаемым по рецептам. С другой стороны, контроль за информацией о безрецептурных лекарственных средствах и их промоционными материалами может оказаться не менее важным — для содействия их рациональному использованию и сведения к минимуму затрат пациентов на неэффективное или недостаточно эффективное лечение.
В настоящем руководстве не рассматривается проблема фальсифицированных лекарственных средств, так как ВОЗ рассматривает их отдельно (The dangers of counterfeit and substandard active pharmaceutical ingredients. WHO drug information, 1997,11(3): 123-127). Еще одно руководство, содержащее сведения и рекомендации по борьбе с фальсифицированными лекарственными средствами, находится в процессе подготовки (Manual on combatting counterfeit drugs. Geneva, World Health Organization, 1998 (Draft)) (Уже опубликовано под названием: Counterfeit drugs: guidelines for the development of measures to combat counterfeit drugs. Geneva, World Health Organization, 1999. WHO/EDM/QSM/99.1 — Прим. ред.).
В. Предварительные решения при выборе вариантов проведения домаркетинговой оценки
Минимум обязательных требований, которые должны соблюдаться в отношении признанных препаратов, включает:
-
Воспроизводимое и удовлетворительное качество;
-
Взаимозаменяемость с фармацевтическими эквивалентами (будет подробнее рассмотрено в Приложении 3), представленными на том же рынке;
-
Точную и полезную для местного потребителя информацию о препарате.
Комплекс этих требований призван гарантировать качество, безопасность и эффективность.
При проведении оценки признанных препаратов ОРЛС может:
-
Подготовить свой собственный отчет;
-
Полагаться на отчеты об оценке, подготовленные регуляторными органами других стран;
-
Полагаться на решения, принятые регуляторными органами других стран;
-
Использовать различные сочетания вышеприведенных подходов.
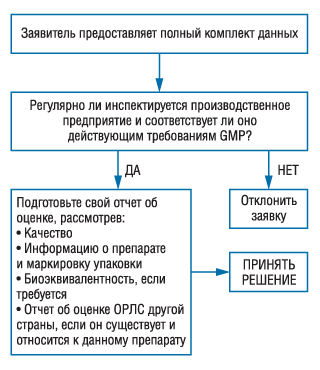
Типовая форма заявки (будет представлена в Приложении 6) предназначена для облегчения процедуры подачи данных — независимо от требуемого вида оценки — и должна быть заполнена во всех случаях. Отчет или решение регуляторного органа другой страны не всегда можно получить, особенно если речь идет о препаратах, разработанных и произведенных на местном уровне. В этих случаях ОРЛС должен провести собственную оценку. Точно так же, если существует сертификат, соответствующий установленному ВОЗ образцу, но в нем нет подтверждения на наличие разрешения на маркетинг в стране, выдавшей сертификат, ОРЛС должен провести собственную оценку данных. В этих случаях вышеуказанный сертификат предоставляет информацию о систематической инспекции линии производства данного препарата на соответствие требованиям GMP.
|
Каждый из вышеуказанных вариантов подробнее рассматривается ниже:
1. Подготовка отчета об оценке. ОРЛС может подготовить собственный отчет об оценке или прокомментировать какой-либо экспертный отчет, представленный заявителем (если этот отчет соответствует предъявляемым требованиям). Отчет об оценке нужен как для представления экспертному совету, так и для будущих ссылок в случае апелляции, принятия последующих регуляторных мер или дальнейших заявок о внесении изменений в уже полученное разрешение.
Отчет, как правило, должен содержать:
-
Краткий обзор данных, представленных в заявке;
-
Аргументацию любого несогласия с предложениями заявителя, например в отношении предлагаемого срока хранения, спецификаций готового препарата или содержания информации о препарате;
-
Резюме и оценку информации о взаимозаменяемости, сопровождаемые рекомендациями и аргументами.
Если ОРЛС считает некоторые из предложений заявителя неприемлемыми, ему направляется письмо и/или факс. Такие письма иногда называют «уведомлениями о недостатке информации», «запросом дополнительной информации» или другим сходным образом. Форма их составления должна соответствовать положениям действующего законодательства.
 ОРЛС имеет полное право подготовить замечания к экспертному отчету и использовать эти два документа в качестве собственного доклада об оценке. Специалист, проводящий оценку, должен помнить, однако, что его функция состоит в проведении независимого изучения препарата и представленных данных. Поэтому утверждения экспертов заявителя должны быть проверены путем сопоставления с имеющимися данными. Если экспертный отчет выполнен недостаточно качественно или содержит однобокие заключения, лицо, проводящее оценку, имеет право не использовать его.
ОРЛС имеет полное право подготовить замечания к экспертному отчету и использовать эти два документа в качестве собственного доклада об оценке. Специалист, проводящий оценку, должен помнить, однако, что его функция состоит в проведении независимого изучения препарата и представленных данных. Поэтому утверждения экспертов заявителя должны быть проверены путем сопоставления с имеющимися данными. Если экспертный отчет выполнен недостаточно качественно или содержит однобокие заключения, лицо, проводящее оценку, имеет право не использовать его.
2. Использование отчета об оценке, подготовленного органом другой страны. Если заявитель подтверждает, что данные, представленные в обеих странах, одинаковы, то, получив отчет об оценке другого национального органа, ОРЛС может подготовить и направить заявителю (при необходимости) только «уведомление о недостатке информации». Если некоторые рекомендации, сделанные в отчете другой страны, представляются неуместными, полезно отметить причины возражений как для будущих справок, так и на случай обжалования.
3. Использование решения, принятого органом другой страны. Рекомендуется, чтобы во всех случаях предъявлялся сертификат образца, установленного Системой ВОЗ по сертификации качества фармацевтической продукции для международной торговли. Он должен содержать информацию об утвержденном препарате и гарантии заявителя о том, что препарат, который будет поставляться, по всем аспектам производства и качества идентичен препарату, который использует страна-экспортер (типовая форма заявки предусматривает предоставление таких гарантий). Необходимо также определить, является ли информация, предложенная для препарата, приемлемой для импортирующей страны. Этот вопрос подробно рассматривается ниже.
С. Оценка данных о качестве
Независимо от уровня, на котором ОРЛС намерен самостоятельно проводить оценку данных, вся техническая информация о качестве должна быть представлена заявителем, чтобы в спорных случаях иметь возможность полностью исследовать данную серию препарата, например в лаборатории по контролю качества. Типовая форма заявки требует представления информации подобного рода.
Информация о качестве, которая может понадобиться в будущем, включает:
-
Методику проведения исследований — для использования при анализе образцов препарата.
-
Данные о природе примесей, полученные в ходе исследования стабильности, а также методы их определения и количественной оценки — для использования при обнаружении примесей в процессе испытаний. Важно знать, являются ли эти примеси продуктами распада ингредиентов или загрязняющими веществами, попавшими извне, а также определить, могут ли они в обнаруженном количестве оказывать вредное воздействие.
-
Участки производства активных ингредиентов или промежуточных веществ (например, гранулята) — на случай, если определенный производственный участок будет закрыт из-за несоответствия требованиям GMP.
-
Данные о стабильности — в целях сравнения, если какая-либо компания заявит о намерении продлить срок хранения препарата.
-
Маркировку контейнеров (упаковок) — для сравнения в процессе исследований, проводимых лабораторией по контролю качества.
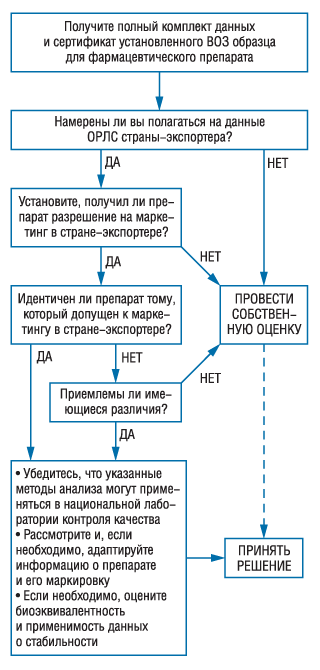
После получения данных о качестве, процедура оценки зависит от того, какой препарат оценивается — импортный или разработанный и произведенный в данной стране, и в какой степени ОРЛС намерен полагаться на решения, принятые органом другой страны.
После того как ОРЛС принципиально определил, будет ли он полагаться на подобные решения (и, если да, то при каких обстоятельствах), далее он должен действовать по установленной схеме, за исключением особых случаев. Важно, чтобы решения были логически последовательными. К категории «особых случаев» могут быть отнесены компании-заявители, ранее замеченные в маркетинге субстандартной продукции.
ОРЛС должен применять единые стандарты качества, безопасности и эффективности как к импортируемым, так и к отечественным препаратам.
Отечественные препараты
Если препарат был разработан и произведен в данной стране, ОРЛС должен сам провести оценку всего комплекта представленных данных. Он также должен удостовериться, что отечественный производственный участок имеет действительную лицензию, которая соответствует стандартам GMP. Прежде чем выдать разрешение рекомендуется провести испытания образцов в лаборатории по контролю качества ОРЛС или в другой лаборатории, работающей по контракту с ОРЛС.
Если препарат был разработан в другой стране (например, транснациональной компанией), но производится в данной стране, то может быть использован отчет ОРЛС страны-разработчика. ОРЛС этой страны может выбирать, полагаться ли ему на отчет другой страны или на решение, принятое зарубежным ОРЛС, либо проводить собственную оценку. Однако в этих обстоятельствах нельзя требовать сертификат установленного ВОЗ образца.
Импортные препараты
Если ОРЛС намерен принять свое собственное решение, он должен в первую очередь выяснить возможность получения отчета, составленного национальным органом другой страны. Если это возможно, то решение в отношении качества препарата может быть принято вскоре после получения отчета. Если такого отчета нет или получить его не представляется возможным, ОРЛС должен провести собственную оценку и принять решение на основании ее результатов. В любом случае рекомендуется не предоставлять разрешение на маркетинг до получения сертификата установленного ВОЗ образца.
Если этот сертификат включает разрешение на маркетинг препарата в стране-экспортере, то заявителю достаточно либо предоставить гарантии того, что препарат, на который выдан сертификат, во всех отношениях идентичен препарату, реализуемому в стране-экспортере, либо указать и обосновать имеющиеся между ними различия. Форма типовой заявки, которая будет представлена в Приложении 6, требует предоставления таких гарантий заявителем. Следует принимать во внимание влияние климата на стабильность препарата, а также местные условия хранения, например наличие кондиционируемых складских помещений, в которых поддерживается средняя кинетическая температура (Amidon GL, Robinson JR, Williams RL, eds. Scientific foundations for regulating drug product quality control. Alexandria, VA, AAPS Press, 1997). Если какая-либо стадия производственного процесса (например расфасовка) осуществляется в данной стране, то местный производственный участок должен иметь действительную лицензию и соответствовать требованиям GMP. Наконец, должна быть рассмотрена взаимозаменяемость препарата с фармацевтическими эквивалентами на местном рынке (см. ниже) даже в том случае, если ОРЛС намерен полагаться на сертификат установленного ВОЗ образца.
За исключением случаев, когда подобный сертификат содержит разрешение на маркетинг препарата в стране-экспортере или указанные в нем различия являются несущественными и обоснованными, ОРЛС обязан проводить свою собственную оценку данных так же, как и для препаратов, разработанных и произведенных в данной стране.
Сертификат установленного ВОЗ образца должен сопровождаться копией информации о препарате, утвержденной в стране-изготовителе (раздел 2А.5 типового сертификата; представлен в Дополнении 1 к Приложению 2). Ее необходимо сравнить с информацией о препарате, предлагаемой для данной страны, и оценить с точки зрения возможности применения в местных условиях, например в отношении показаний.
Факт получения разрешения на маркетинг в третьих странах в какой-то мере может гарантировать качество препарата. Однако, если разрешение на маркетинг препарата в третьих странах и повышает доверие к нему, заявитель должен предоставить гарантии того, что серии импортированного препарата являются по крайней мере такими же по качеству и, пока не будут представлены новые данные, они будут производиться на тех же предприятиях и из того же исходного сырья, включая сырье активных ингредиентов. Это нельзя считать само собой разумеющимся. Выдача разрешений на маркетинг исключительно на основе разрешений на маркетинг в других странах, то есть без сертификата установленного ВОЗ образца, не рекомендуется.
Отбор образцов и инспекция GMP
Во всех случаях, когда ОРЛС полагается на решения, принятые органами других стран, или сомневается в данных, представленных заявителем, — прежде чем препарат будет допущен на рынок, отбираются образцы препарата для анализа в собственной лаборатории контроля качества ОРЛС или в других лабораториях, работающих на контрактной основе.
Если речь идет о местном производстве, то все участки, задействованные в выпуске препарата, должны иметь действительные сертификаты о соответствии нормам GMP. Если представленные заявителем данные вызывают у ОРЛС сомнение, то проводится инспекция на соответствие нормам GMP условий производства конкретного препарата, и, если необходимо, дополняется инспекцией всего предприятия.
Рекомендации, представленные в двух предыдущих разделах («Предварительные решения при выборе вариантов домаркетинговой оценки» и «Оценка данных о качестве») представлены в виде «деревьев» для принятия решений (рис. 1, 2). n
|
|
Продолжение в следующих номерах «Еженедельника АПТЕКА»
*Продолжение. Начало см. «Еженедельник АПТЕКА» № 30 (451) от 9 августа, № 31 (452) от 16 августа, № 32 (453) от 23 августа, № 36 (457) от 20 сентября 2004 г.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим