Національні регуляторні органи (НРО) займаються видачею дозволів на маркетинг, постмаркетинговими заходами, експертизою клінічних випробувань, наданням наукових рекомендацій та організацією інспекцій (у зоні своєї відповідальності). При цьому хоча НРО намагаються уникнути дублювання оцінок та об’єднати зусилля та досвід (ЕМА, 2020), зберігається чимало національних правил. Так, з 1995 р. існують три різні шляхи для отримання дозволу на маркетинг продукту в державах — членах Європейського Союзу: централізована, децентралізована, взаємного визнання та чисто національна процедура. Порядок надання макетів упаковок, а також зразків упаковок та самих препаратів має свої особливості в різних країнах. Це ж стосується і першого з 5 модулів реєстраційного досьє, додаткових даних та способів надання документів (оригінали, копії або нотаріально засвідчені).
Централізована процедура, що має правову основу в Регламенті ЄС № 726/2004, є обов’язковою для біологічних, орфанних, що містять нову діючу речовину, та деяких інших лікарських засобів. Європейський комісійний дозвіл на маркетинг, що видається при цьому, дійсний у всіх країнах — членах ЄС.
Процедура взаємного визнання (MRP) реалізується в країнах ЄС з опорою на Директиву 2001/83/EC для лікарських засобів, які вже мають реєстраційне посвідчення в одній державі-члені.
Децентралізована процедура (DCP) є ще одним шляхом реєстрації лікарського засобу в ЄС, який був введений пізніше Директивою 2004/27/EC і застосовується у випадку, якщо на момент подання заявки в жодній державі-члені дозволу на маркетинг препарату ще немає. При цьому решта НРО визнають оцінку першого з них як референтну.
Як зазначено у ст. 8 Директиви 2001/83/ЕС, для отримання дозволу на маркетинг необхідно подати відповідну заявку. Її слід супроводити рядом документів, серед яких — стисла характеристика продукту (summary of product characteristics — SPC), а також один або кілька зразків або макетів (mock-ups) вторинної та первинної упаковки лікарського засобу разом із листком-вкладишем.
«Макет» (mock-up) — це повнокольоровий площинний ескіз художнього оформлення, представлений таким чином, що після розрізання та складання, де це необхідно, виходить копія як вторинної, так і внутрішньої упаковки так, щоб чітко прочитувався текст, орієнтований у всіх трьох напрямках.
«Зразок» (specimen) — це екземпляр фактичної вторинної та первинної упаковки та листка-вкладиша (тобто в товарному вигляді).
Маркуванню та листку-вкладишу присвячений цілий розділ Директиви — V. Після викладення (ст. 54–59) вимог до їх змісту у ст. 60 зазначено, що країни-члени не можуть забороняти або перешкоджати розміщенню на ринку лікарських засобів на підставах, пов’язаних з маркуванням або листком-вкладишем, якщо вони відповідають вимогам розділу V.
У ст. 61 знову йдеться про те, що при зверненні за отриманням дозволу на маркетинг в НРО повинні (shall — традиційна для інструкцій форма для вказання необхідності) бути надані один або кілька зразків або макетів (mock-ups) вторинної та первинної упаковки лікарського засобу разом з листком-вкладишем. Відповідно до п. 2 ст. 61 компетентний орган повинен відмовити у видачі дозволу на маркетинг, якщо маркування або листок-вкладиш не відповідають розділу V або відомостям, наведеним у SPC.
Якщо зміни маркування або листка-вкладиша в аспектах, що покриваються положеннями розділу V, не відповідають SPC, їх слід надати до органів, уповноважених на видачу дозволу на маркетинг. Якщо компетентні органи не будуть заперечувати пропоновані зміни протягом 90 днів після подання запиту, заявник може ввести зміну в дію (ст. 61, п. 3).
Відповідно до ст. 64 якщо положення розділу V не дотримуються, а повідомлення, направлене заінтересованій особі, не має ефекту, НРО країн-членів можуть призупинити дію дозволу на маркетинг доти, доки маркування та листок-вкладиш відповідного лікарського засобу не будуть виправлені для забезпечення відповідності розділу V.
Централізована процедура
«Коли я повинен подавати макети та/або зразки?» — таку назву має один із розділів (3.1.6.) постійно оновлюваного збірника рекомендацій для тих, хто використовує централізовану процедуру (тут і далі — за ЕМА, 2021). У відповіді використовують слово «must»: «Макети та зразки вторинної та первинної упаковки разом з листком-вкладишем ПОВИННІ бути представлені на розгляд EMA заявником/власником реєстраційного посвідчення ДО КОМЕРЦІАЛІЗАЦІЇ лікарського засобу. При цьому висувають такі вимоги:
(1) до макета: на момент подання заявки один повнорозмірний макет англійською мовою та один багатомовний кольоровий повнорозмірний макет («найгірший варіант») вторинної та первинної упаковки кожної лікарської форми в кожному типі контейнера з найменшою кількістю в упаковці повинні бути включені до Модуля 1.3.2 заявки. Можуть бути включені (за бажанням) також макети листка-вкладиша. Далі вказані терміни, в які заявник вирішує з ЕМА питання та адресу електронної пошти для зв’язку з ЕМА: [email protected];
(2) до зразків: не пізніше ніж через 15 робочих днів до маркетингу необхідно надати ЕМА один набір відповідних зразків вторинної та первинної упаковки, а також листок-вкладиш для кожного різновиду за силою дії, концентрацією та формою випуску з використанням Форми подачі зразків (див. EMEA/305821/2006):
- перед першим маркетингом у ЄС,
- перед першим маркетингом у вигляді багатомовної упаковки (з більшою кількістю мов, ніж раніше).
На завершальному етапі оцінки EMA виконає загальну перевірку з точки зору зручності читання протягом 15 робочих днів і перевірить, чи були належним чином реалізовані будь-які попередні коментарі до макетів/зразків. Заявника буде поінформовано про результати перевірки.
«Чи повинен я подавати зразки (продукту, samples) разом із заявкою?» — так називається ще один із розділів (3.1.8). «Зразки для випробувань пропонованого лікарського засобу на момент подання заявки не потрібні, — зазначено у відповідь. — Однак Комітет з лікарських засобів для застосування у людини (Committee for Medicinal Products for Human Use — CHMP) може запросити тестування зразків лікарського засобу та/або його інгредієнтів під час оцінки заявки відповідно до положень статті 7(b) Регламенту (ЄС) № 726/2004».
Особливості процедур, реалізованих НРО
Координаційна група із взаємного визнання та децентралізованих процедур (Co-ordination Group for Mutual Recognition and Decentralised Procedures — Human — CMDh) вперше опублікувала у 2012 р., а потім ще неодноразово уточнювала зведені дані про вимоги НРО щодо надання макетів, зразків упаковок та препаратів у межах MRP/DCP (CMDh (a), 2020).
CMDh тут посилається на вимогу ст. 8 Директиви 2001/83/ЕС щодо обов’язковості надання макетів упаковок та листків-вкладишів (див. вище) і зазначає, що країни-члени можуть вимагати надання зразків упаковок у їх товарному вигляді для перевірки відповідності розділу V Директиви.
Вимоги більшості країн-членів до надання зразків лікарських засобів передбачають їх доступність на будь-який запит органів влади, але супроводжувати ними кожну заявку необов’язково. Однак частина НРО передбачає особливі положення, що стосуються діючих та допоміжних речовин, а також готових лікарських засобів (табл. 1):
| Чехія | Німеччина | Естонія | Угорщина | Мальта | Польща | Швеція | Словаччина | Словенія | |
| Готові ліки | * | * | * | * | * | * | * | * | * |
| Усі субстанції | – | – | * | * | з/з | * | – | * | * |
| Нефармакопейні субстанції | – | – | – | – | з/з | * | – | * | * |
| Допоміжні речовини | – | – | – | – | з/з | * | – | * | * |
1. Чехія: до заявки (принаймні до винесення рішення щодо неї) повинен бути доданий один зразок лікарського засобу в первинній упаковці кожного виду, можливо — без остаточного маркування; перед розміщенням лікарського засобу на ринку вимагається один зразок лікарського засобу в остаточному товарному вигляді; в обґрунтованих випадках від подання зразка можна відмовитися.
2. Німеччина: у кількості, достатній для проведення повного аналізу та перевірки методів контролю, що використовуються виробником; для всіх лікарських препаратів, щодо яких компетентний Інститут Пауля Ерліха (Paul-Ehrlich-Institut), — сироватки, вакцини, алергени, препарати крові, генної терапії, соматичних та ксеногенних клітин та продукти тканинної інженерії), зразки повинні надаватися одночасно з поданням досьє.
3. Естонія: слід надавати готовий лікарський засіб, якщо країна є референтною для регуляторної процедури (Reference Member State — RMS), референтні матеріали, основні домішки, продукти деградації та неактивні речовини слід надавати за запитом та в кількості, достатній для проведення повного аналізу та перевірки методів контролю, що використовуються виробником.
4. Угорщина: зразки лікарського препарату, активної речовини та комерційно недоступних референтних матеріалів повинні бути надані на запит уповноваженого органу; при цьому кількість наданих зразків має давати можливість провести три повні аналізи.
5. Мальта: зразки повинні бути включені при подачі заявки, якщо Mальта є RMS.
6. Польща: повинні бути представлені на запит компетентного органу в кількості та в строки, зазначені в запиті (зазвичай має бути достатнім для проведення повного аналізу та перевірки методів контролю, що використовуються виробником).
7. Швеція: до заявки повинен бути доданий зразок лікарського засобу в первинній упаковці кожного виду, призначеного для реалізації; зразок може бути представлений без маркування. Якщо вимірювальний пристрій (мірний посуд) постачається разом із препаратом, необхідно також надати відповідний зразок.
8. Словаччина: зразки препарату в товарному вигляді, референтні речовини, основні продукти деградації та основні домішки повинні бути надані на запит НРО в кількості, достатній для проведення трьох повних аналізів (але якщо Словаччина — RMS).
9. Словенія: зразки повинні бути представлені за запитом НРО в кількості та в строки, зазначені в запиті (у принципі кількість матеріалів має бути достатньою для проведення повного аналізу та перевірки методів контролю виробника).
Як оформляють зміни в маркуванні?
Буває, що лікарський засіб, який отримав дозвіл на маркетинг, у тій чи іншій країні-члені, представлятиме інший місцевий представник, або виникає потреба узгодити/уточнити формулювання листків-вкладишів, змінити позначення номера партії тощо. Для подібних випадків, не пов’язаних зі змінами SPC, статтею 61(3) Директиви 2001/83/ЕС передбачено так зване повідомлення (61(3) Notification) (див. вище).
Щоб повідомлення 61(3) було дійсним:
- зміна (-и) повинна торкатися лише Додатка IIIA (маркування) та/або IIIB (PL), без змін у SPC та/або Додатка II;
- зміни слід вносити у версії всіма мовами.
Під дію повідомлення 61(3) не підпадають:
- зміни в SPC або додатку II,
- зміни, які стосуються маркування лише деякими мовами,
- зміни у спільному компонуванні, дизайні, зручності читання маркування та/або товарного знака без зміни тексту; у такому разі необхідність розгляду EMA пропонованих змін із наданням зразків слід обговорити із Сектором медичної інформації EMA ([email protected]). Після подання ЕМА поінформує власника дозволу на маркетинг протягом 90 днів про те, прийняті запропоновані зміни чи ні.
Вимоги країн-членів при внесенні змін CMDh розглядає відносно певних типів згідно з настановою (Guidelines, 2013) (CMDh (b)).
B.II.a.1. Зміна або додавання елементів оформлення, включаючи заміну або додавання фарб, що використовуються для маркування продукції.
B.II.a.2. Зміна форми або розмірів лікарської форми.
B.II.a.3. Зміни у складі (допоміжних речовин) готового продукту.
B.II.e.1. Зміна первинної упаковки готового продукту.
B.II.e.4. Зміна форми або розмірів контейнера/закупорювання (первинної упаковки).
B.IV.1. Заміна засобу дозування або доставки лікарського засобу до місця призначення.
Зразки повинні бути доступні в більшості країн ЄС за будь-яким запитом НРО в межах конкретних термінів та кількості, а в деяких — і з додатковими умовами, наприклад, надавати зразки разом із заявкою (табл. 2).
| Тип змін | Порядок надання зразків | ||
| За запитом | Разом із заявкою | Тільки нових контейнерів/дозувань | |
| B.II.a.1 | Усі, крім Чехії, Франції та Словаччини | Франція (по 3 зразки), Чехія | Словаччина (окрім B.II.a.3 і B.IV.1) |
| B.II.a.2 | |||
| B.II.a.3 | |||
| B.II.e.4 | |||
| B.IV.1 | |||
| B.II.e.1 | Болгарія, Кіпр, Іспанія, Фінляндія, Франція, Угорщина, Ірландія, Латвія, Мальта, Нідерланди, Польща | Чехія | – |
«Що робити, якщо я хочу подати кілька заявок одночасно/зробити дублікати вже схвалених заявок на той самий лікарський засіб?», — на це запитання (1.11) ЕМА відповідає наступним чином. У EMA регулярно звертаються заявники, які бажають одночасно або послідовно отримати більше одного реєстраційного посвідчення на конкретний лікарський препарат (тобто з однаковим якісним та кількісним складом активної речовини та однією й тією самою лікарською формою) під різними найменуваннями (invented names). Зокрема, до цього вдаються, виводячи на ринок авторизовані генерики (прим. ред.).
Відповідно до статті 82(1) Регламенту (ЄС) № 726/2004, Європейська комісія може дозволити заявникам подавати більше однієї заявки за наявності об’єктивних, що піддаються перевірці, причин стосовно громадської охорони здоров’я або у зв’язку зі спільним маркетингом. При цьому заявників попросять пояснити та обґрунтувати мотиви подання кількох/дублікатів заявок та їх наміри щодо використання реєстраційного посвідчення.
Відповідно до спеціально узгодженої процедури заявники повинні приблизно за 4 міс до передбачуваної дати подання повідомити Європейську комісію про мотиви подання кількох заявок та надати необхідні пояснення та обґрунтування з копією для EMA. Аналогічні положення знаходимо в нормативних вимогах НРО.
Реєстри та практика компаній
 На відміну від США, де діє інша регуляторна практика, в ЄС зареєструвати препарати під власним брендом можуть дозволити собі лише дуже великі оператори. При цьому дозволи за централізованою процедурою практично відсутні. Найбільші аптечні мережі схильні отримувати дозволи за іншими процедурами — децентралізованим або взаємним визнанням. Наприклад, один із лідерів східноєвропейського роздрібного сектору «Dr. Max Group», що перебуває у володінні «Penta Investments», в одній лише Польщі, згідно з офіційним реєстром, оформив 22 дозволи на маркетинг лікарських засобів.
На відміну від США, де діє інша регуляторна практика, в ЄС зареєструвати препарати під власним брендом можуть дозволити собі лише дуже великі оператори. При цьому дозволи за централізованою процедурою практично відсутні. Найбільші аптечні мережі схильні отримувати дозволи за іншими процедурами — децентралізованим або взаємним визнанням. Наприклад, один із лідерів східноєвропейського роздрібного сектору «Dr. Max Group», що перебуває у володінні «Penta Investments», в одній лише Польщі, згідно з офіційним реєстром, оформив 22 дозволи на маркетинг лікарських засобів.
Оптові компанії нерідко вважають за краще об’єднуватися в одну організацію з виробниками, як, наприклад, найбільша в Польщі група NEUCA, до складу якої входить Synoptis Pharma. У портфелі останньої — 146 дозволів у країні.
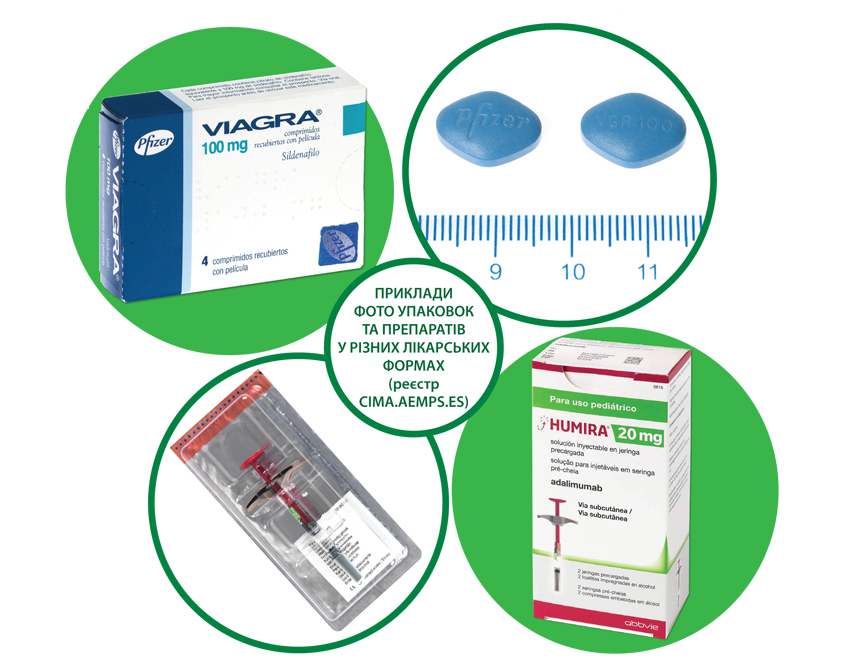
Оскільки одному дозволу на маркетинг в ЄС відповідає один препарат з форматом упаковки (включаючи логотипи, стиль та колірну гаму оформлення), закріпленим у регуляторному досьє, особливої необхідності включати макети та фото упаковок до громадських реєстрів препаратів немає. З усіх національних реєстрів лікарських засобів, що отримали дозволи на маркетинг, фото упаковок і ліків у готових лікарських формах розміщують лише в одному, іспанському (рисунок).
Крім того, Іспанське агентство з лікарських засобів та продуктів санітарії (Agencia Espanola de Medicamentos y Productos Sanitarios) розмістило на відповідному сайті інформацію, а також інфографіку з розшифровкою всіх графічних елементів, які можна побачити на макетах упаковок лікарських засобів. Складність сучасного маркування робить цей крок цілком виправданим, що полегшує життя як працівникам фармації, так і пацієнтам.
Список використаної літератури
- Mock-ups, specimens and samples — New application. CMDh (а) December 2020, CMDh/260/2012, Rev.7. http://www.hma.eu/fileadmin/dateien/Human_Medicines/CMD_h_/procedural_guidance/Application_for_MA/ CMDh_260_2012_Rev07_2020_12_clean_-_ Mock-ups_specimen_and_samples_New_ application.pdf.
- Mock-ups, specimens and samples — Variation. CMDh (b) December 2020, CMDh/261/ 2012, Rev.4. http://www.hma.eu/fileadmin/dateien/Human_ Medicines/CMD_h_/procedural_guidance/Variations/CMDh_261_2012_Rev_4_2020_12_clean_-_Mock-ups_specimens_and_samples_ Variation.pdf.
- European medicines agencies network strategy to 2025 Protecting public health at a time of rapid change gencies network strategy to 2025 (EMA/85501/2020). European Medicines Agency and Heads of Medicines Agencies, 2020. http://www.ema.europa.eu/ en/documents/report/european-union-medicines-agencies-network-strategy-2025-protecting-public-health-time- rapid-change_en.pdf.
- European Medicines Agency pre-authorisation procedural advice for users of the centralised procedure (EMA/821278/2015). Human Medicines Division, 20 December 2021. http://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/european-medicines-agency-pre-authorisation-procedural-advice-users-centralised-procedure_en-0.pdf.
- European Medicines Agency post-authorisation procedural advice for users of the centralised procedure (EMEA-H-19984/03 Rev. 96). Last updated: 22/12/2021.
- Guidelines on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products and on the documentation to be submitted pursuant to those procedures (2013/C 223/01). Official Journal of the European Union, 2.8.2013.
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим