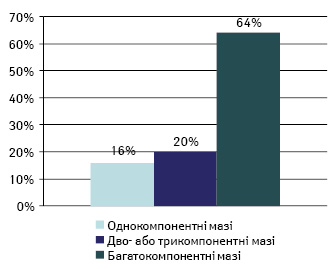
Насамперед зауважимо, що в структурі аптечного виробництва переважають розчини для зовнішнього застосування. Мазі нерідко займають 2-ге місце за обсягами виготовлення серед інших екстемпоральних лікарських форм; водночас серед мазей левову частку займають багатокомпонентні препарати (Коритнюк Р.С., 2007; рисунок).
Забезпечення якості готового продукту гарантується контролем якості на всіх етапах його виробництва. У даному випадку оптимальним є проведення валідації технологічного процесу виготовлення, особливо для ліків, які готують про запас. Важливо в такому разі визначити критичні стадії виготовлення препарату, які можуть вплинути на якість готового препарату (Савченко Л.П., Георгіянц В.А., 2013). Що стосується мазей, можна виділити 4 основні стадії їх виготовлення, які здатні напряму впливати на якість кінцевого продукту (Савченко Л.П., 2016):
- зважування та відмірювання інгредієнтів;
- розчинення та диспергування;
- емульгування та змішування;
- пакування.
У Національному фармацевтичному університеті (НФаУ) під керівництвом професора Вікторії Георгіянц, завідувача кафедри фармацевтичної хімії, почесного професора НФаУ, відмінника освіти України, — більше 10 років тому створено робочу групу, головним завданням якої є стандартизація препаратів аптечного виготовлення. Вчені НФаУ спільно з науковцями ДП «Український науковий фармакопейний центр якості лікарських засобів» (директор — професор Олександр Гризодуб) займаються розробкою стандартів контролю якості різноманітних екстемпоральних лікарських форм. Проводяться дослідження з розробки та валідації методик контролю якості активних інгредієнтів розчинів, суспензій, очних крапель, м’яких лікарських форм, порошків. Крім цього, основним завданням досліджень робочої групи є вивчення стабільності лікарських форм, які готуються про запас. У рамках проведення досліджень ведеться робота з провідними виробничими аптеками України.
* * *
Коли йдеться про серійне виробництво ліків, — як на зарубіжних, так і на вітчизняних фармацевтичних підприємствах з кожним роком приділяється все більше уваги практиці валідації технологічного процесу — вона є основною з ключових умов впровадження вимог належної виробничої практики (GMP), виконання стандартів якої є обов’язковим для реєстрації лікарських препаратів у всьому світі та в Україні (Савченко Л.П., 2013). Важливість проведення валідації технологічного процесу важко переоцінити (Swarbrick J., 2007; Frederick J. et al., 2008): проведення валідації процесу виробництва дозволяє провести оцінку ризиків для якості готового продукту; при проведенні валідації технологічного процесу відповідно до концепції GMP, контроль якості переноситься з контролю якості готового продукту на контроль якості самого процесу виробництва — у такий спосіб якість «вбудовується» в продукт через суворо достеменне дотримання стадійної змістовності процесу. Складові процесу випуску серії в реалізацію включають: 1) перевірку виробництва, а також проведених випробувань серії; 2) сертифікацію Уповноваженою особою серії готового лікарського засобу, яка означає, що серія вироблена із дотриманням стандартів GMP і відповідає вимогам реєстраційного досьє (так званий випуск серії за характеристикою якості); 3) передачу в товарний запас та/або експорт серії готової продукції (Підпружников Ю.В., 2016). Докладніше щодо нагальних питань забезпечення якості лікарських засобів у серійному виробництві — див. «Щотижневик АПТЕКА» № 38 (859) від 1 жовтня 2012 р., № 8 (1079) від 6 березня 2017 р., № 35 (1106) від 11 вересня 2017 р., а також монографію «Система качества и надлежащие практики в фармации» під науковою редакцією академіка НАН України Валентина Черних та професора Юрія Підпружникова.
* * *
Що ж стосується аптечного виготовлення лікарських препаратів, то на сьогодні в Україні воно, як відомо, регулюється наказом МОЗ України від 17.10.2012 р. № 812 «Про затвердження Правил виробництва (виготовлення) та контролю якості лікарських засобів в аптеках» — і, звісно ж, Державною Фармакопеєю України другого видання (ДФУ 2.0), яка містить удосконалені фармакопейні статті стосовно екстемпоральної рецептури). ДФУ 2.0 охоплює:
- нестерильні лікарські засоби, виготовлені в аптеках;
- розрахунки при виготовленні лікарських засобів в аптеках;
- м’які лікарські засоби, виготовлені в аптеках;
- порошки, виготовлені в аптеках;
- супозиторії та песарії, виготовлені в аптеках.
Наказ МОЗ України від 17.10.2012 р. № 812 передбачає, що
- виготовлення серій лікарських засобів, внутрішньоаптечної заготовки та лікарських засобів про запас в аптеках повинно здійснюватися згідно з технологічними інструкціями;
- контроль якості лікарських засобів, що виготовляються серіями, повинен охоплювати всі стадії їх виготовлення і має здійснюватися за всіма показниками якості.
Крім того, наказом МОЗ України від 01.07.2015 р. № 398 затверджено дві настанови: «Вимоги до виготовлення нестерильних лікарських засобів в умовах аптек» та «Вимоги до виготовлення стерильних та асептичних лікарських засобів в умовах аптек».
Також підготовлено методичні рекомендації під редакцією професорів НФаУ, лауреатів Державної премії України в галузі науки і техніки Олександра Тихонова і Тетяни Ярних. Вказане методичне джерело складається з двох частин: в частині 1 представлено перелік лікарських речовин із зазначенням їх фізико-хімічних, фармакологічних властивостей, несумісностей, обґрунтовано спосіб введення цих речовин до складу різних лікарських форм та наведено номери прописів, до складу яких входять дані субстанції, представлено посилання на офіцинальні прописи лікарських препаратів; в частині 2 узагальнено та систематизовано за медичним призначенням екстемпоральну рецептуру (442 прописи, які наведені з оптимальним варіантом технології (з паспортами письмового контролю), оформленням до відпуску та із зазначенням умов зберігання); окрім цього, до рекомендацій включено предметний покажчик за захворюваннями та лікарськими формами.
У преамбулі до зазначених методичних рекомендацій автори підкреслюють, що перевагою екстемпоральних лікарських засобів є індивідуалізація медичної допомоги для кожного хворого, можливість підбору найраціональнішого співвідношення інгредієнтів, широкий вибір доз (від мінімальної до максимально переносимої), врахування генетичних, вікових, статевих та інших особливостей організму людини. Адже збереження та розвиток індивідуально орієнтованого підходу до потреб пацієнта у більшості прогресивних країн світу розглядається як пріоритет, — а нині вже й в Україні до багатьох суб’єктів господарювання приходить розуміння важливості розвитку концепції «персональних лікарських засобів».






У цьому контексті та за таких умов саме екстемпоральне виготовлення ліків набуває нового звучання і на сьогодні позиціонується як розробка ліків для потреб кожного конкретного пацієнта. Понад те: як один з реальних напрямів підвищення ефективності фармакотерапії різних захворювань, екстемпоральне виготовлення поряд з фармакотерапевтичними перевагами також має багато інших важливих фармакоекономічних і фармацевтичних особливостей. Водночас систематизована інформація про склад, технологію та оформлення до відпуску екстемпоральних прописів, на жаль, недостатньо представлена в сучасній літературі, — і це не дозволяє повною мірою використовувати переваги і значний потенціал екстемпоральних препаратів лікарем та фармацевтом для надання якомога якіснішої фармацевтичної допомоги пацієнтові.
* * *
Тут, виключно в інтересах історіографічної справедливості, зробимо лишень одну історичну ремарку-ремінісценцію і від себе нагадаємо, що інформаційну розбудову в Україні цієї триєдиної ідеології, —
- фармація: не продаж ліків, — але фармацевтична допомога;
- аптека: не торгова точка, — але заклад охорони здоров’я;
- пацієнт: не «істота безсловесна», — але зміст процесу та носій комплайєнсу, а тому — ключовий член медичної команди у повноправному тріумвіраті з лікарем та фармацевтом; —
пояснення та поширення цієї стрункої парадигми охорони здоров’я наше видання розпочинало за духом і літерою ВООЗ та каноном FIP ще дуже задовго до першого натяку на позитив появи у фармацевтичному секторі України паростків фахово-громадських утворень, які задекларують підтримку цього триєдиного принципу і згодом під цим гаслом ставатимуть під знамена фармацевтичних міжнародно-федеративних інституцій професійно-громадського ґатунку.
Це ж стосується й належної аптечної практики загалом і як такої.
Адже нове — це добре забуте старе.
А важливе — це з користю згадуване забуте.
Однак, повернімося знову —
- у часі: з глибин десятиріч — до дня сьогоднішнього;
- у площині: від концептуальних декларацій — до реальної аптечної практики;
- у векторі: від фахово-громадського сектору — до міждержавної регуляторної інстанції, зі згадки про яку ми почали аж від заголовку цієї статті.
А саме — повернімося знову до PIC/S (офіційним членом якої наша країна стала ще на зорі поточної декади) — як до мірила та законодавця якості ліків у прогресивних країнах світу, — адже саме її чинне трактування належної практики аптечного виробництва ліків є для нас зараз предметно-актуальним: оскільки на території країни-члена PIC/S — за відповідність вимогам-критеріям саме PIC/S — і відповідь тримати…
* * *
Отже — чинний міжнародний регуляторний канон якості.
Невеликий за обсягом, не перенавантажений подробицями й насичений за контентом Додаток 2 до «Посібника PIC/S з належної практики виготовлення лікарських препаратів у закладах охорони здоров’я» (PIC/S Guide to Good Practices for the Preparation of Medicinal Products in Healthcare Establishments, PE 010-4) має таку назву: «Керівні вказівки щодо стандартів, обов’язкових для виконання при приготуванні нестерильних розчинів, кремів та мазей» (Annex 2. Guidelines on the standards required for the preparation of non-sterile liquids, creams and ointments).
У стислій вступній частині до Додатку 2 зазначається, що він є доповненням до основної частини Посібника з належної практики виготовлення лікарських препаратів у закладах охорони здоров’я та визначає загальні правила підготовки нестерильних розчинів, кремів та мазей, — однак у разі виготовлення препарату в окремій ємності (окремому контейнері) для негайного застосування деякі з наведених у документі вимог можуть бути пом’якшені/скорочені.
Принципово важлива й беззастережна для врахування обставина полягає в тому, що під час приготування розчинів, кремів та мазей вони можуть бути особливо чутливими до мікробного та іншого забруднення, тому необхідно вживати спеціальних заходів для запобігання будь-якому забрудненню. Звідси випливає низка принципових вимог до приміщень та обладнання:
- з метою захисту препарату від забруднення рекомендовано використовувати закриті системи для переробки та транспортування;
- виробничі зони, де виставлено продукт або відкриті чисті ємності (контейнери), зазвичай повинні ефективно вентилюватися фільтрованим повітрям;
- виробничі площі не повинні використовуватися для інших видів діяльності.
Заходи, спрямовані на зниження ризику забруднення, повинні включати:
- використання спеціального одягу та покриття для волосся;
- при використанні відкритих процедур рекомендується використовувати місцеву фільтрацію повітря та рукавички;
- оперативне очищення використаного обладнання;
- після промивання обладнання, яке контактувало з продуктом, необхідне його виполіскування відповідним сортом води (підійде очищена вода або вода для ін’єкцій чи зрошень у флаконах, якщо вона використовується протягом 24 год після відкривання ємності);
- необхідно переконатися у видаленні залишків чистильних та санітарних речовин (наприклад гіпохлоритів);
- після обробки слід перевірити, чи є обладнання чистим і сухим;
- ретельне зберігання очищеного обладнання;
- усі матеріали, що знаходяться у виробничій зоні, повинні бути чистими;
- перед використанням найбільш значущі поверхні слід дезінфікувати спиртом;
- має здійснюватися перевірка контейнерів та кришок, щоб вони були чистими та сухими перед використанням;
- ємність (контейнер) для готових препаратів не повинна використовуватися повторно;
- засоби для прибирання не повинні залишати волокна, підлягають щоденному очищенню при повторному використанні, не застосовуються для прибирання інших зон;
- якщо в одній виробничій зоні проводиться більше однієї технологічної процедури, має існувати адекватна сегрегація, щоб запобігти перехресній контамінації й перемішуванню (необхідно провести оцінку ризику);
- використання спеціалізованого обладнання рекомендується для сильнодіючих речовин, пеніцилінів, цефалоспоринів, сенсибілізуючих сполук, цитотоксичних засобів, ектопаразитицидів та інших речовин, які є високо небезпечними або такими, що важко піддаються елімінації (такі матеріали повинні бути ідентифіковані, після чого необхідно провести відповідну оцінку ризику).
Баки, контейнери, трубопроводи та насоси повинні мати таку конструкцію та встановлені таким чином, щоб їх можна було легко чистити й, за необхідності, дезінфікувати. Зокрема, конструкція обладнання повинна містити мінімум «мертвого простору» або ділянок, де може відбуватися накопичення залишків, що сприяє мікробній проліферації.
Характерно, що розробники документа рекомендують, за можливості, уникати використання скляної апаратури, пропонуючи в якості матеріалу першого вибору для деталей, що контактують з продуктом, високоякісну нержавіючу сталь. Якщо ж скляне обладнання все ж застосовується, слід ретельно відстежувати його пошкодження як до, так і після використання.
У настанові загострюється увага на необхідності визначення та суворого контролю за хімічною та мікробіологічною якістю води, що застосовується у виробництві екстемпорального препарату. Це повинно здійснюватися згідно з положеннями «Вказівок щодо якості води для фармацевтичного використання» (), виданих Європейською агенцією з лікарських засобів (тоді ще з їх оцінки — European Agency for the Evaluation of Medicinal Products (EMEA), а з 2010 р. лаконічніше: — EMА), а також відповідно до фармакопейних вимог. (Як здавна відомо — без води й ні туди, й ні сюди: тож в одній з майбутніх публікацій ми докладніше торкнемося духу й літери, глибше оглянемо зміст та структуру згаданого важливого європейського настановчого документа — прим. авт.).
При використанні відповідної сертифікованої стерильної бутильованої води для ін’єкцій або зрошення у проведенні мікробіологічних та хімічних аналітичних досліджень немає прямої необхідності. Для рутинного моніторингу водних систем періодично необхідно фіксувати (як правило, щотижня) загальні випробування — такі як біологічне навантаження (загальна кількість живих одиниць аеробних мікроорганізмів), провідність, загальний органічний вуглець або дотичні показники. Специфічний хімічний аналіз можна проводити рідше (зазвичай 1 раз на 3 міс). Задля уникнення ризику мікробної проліферації, необхідно постійно дбати про утримання водних систем. А після кожної хімічної дезінфекції водопровідної системи слід дотримуватися процедури перевірки промивки, щоб забезпечити ефективне видалення дезінфектанту.
Зазначається, що в зонах, де зберігаються препарати чи чисті ємності (контейнери), не повинні знаходитися матеріали, які можуть залишати волокна або інші забруднюючі речовини (наприклад картон або необроблена деревина).
Під час наповнення слід приділяти значну увагу і вживати заходів для підтримки однорідності сумішей, суспензій та ін. Процеси змішування та наповнення можуть потребувати валідації, а час і швидкість перебігу змішування повинні бути задокументовані. Щоб забезпечити збереження однорідності, слід дотримуватися особливої ретельності на початку процесу наповнення, після зупинок і в кінці процесу.
За будь-яких рівних обставин готовий препарат рекомендується упаковувати якнайшвидше (в той же день). Однак якщо готовий продукт не буде упакований негайно, слід зазначити максимальний термін зберігання з умовами зберігання й чітко їх дотримуватися.
Рамки проведення фізичних, хімічних та мікробіологічних досліджень якості повинні визначатися на підставі оцінки ризику (оцінка ризиків вкрай важлива для демонстрації відповідності, тому в подальших публікаціях цей блок з основного тексту посібника буде окремо розглянуто нами докладніше — прим. авт.). Але, за можливості, слід візуально перевіряти зразки готового продукту перед його випуском.
І, — цілком логічно насамкінець, — щодо термінів закінчення придатності (використання до певної дати): вони повинні бути встановлені та обґрунтовані для невідкритого препарату, — натомість може знадобитися рекомендувати дату закінчення терміну застосування з відліком від того моменту, коли упаковку (контейнер) було відкрито.
* * *
Під завісу даної пілотної статті авторського циклу публікацій про аптечне виготовлення ліків нагадаємо про той приємний факт, що нещодавно затверджено та з 16 березня 2018 р. введено в дію Доповнення № 2 до ДФУ 2.0, яке включає, зокрема, 5 національних монографій на лікарські засоби (концентровані розчини), виготовлені в аптеках.
Розробка таких монографій здійснюється фахівцями ДП «Український науковий фармакопейний центр якості лікарських засобів» та Державної служби України з лікарських засобів та контролю за наркотиками за наукової підтримки провідних вчених НФаУ. Цими спільними зусиллями відкривається новий напрямок розвитку ДФУ, який дозволяє залучити препарати аптечного виготовлення до загального фармакопейного процесу стандартизації якості ліків. А Україну — підштовхує до членства у клубі прогресивних країн, які піклуються про власне аптечне виробництво ліків та приділяють належну увагу забезпеченню й контролю його якості.
Далі буде…
фото автора
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим