 Фальсифікація лікарських засобів є глобальною проблемою, особливо в країнах із низьким та середнім рівнем доходів. Актуальність цієї теми для населення інколи призводить до спроб маніпуляцій політиків (задля привертання уваги та підвищення рейтингу у виборців) і чиновників (для отримання додаткових фінансувань та повноважень). У цій статті наведено огляд розвитку рекомендацій ВООЗ щодо боротьби з підробленими ліками та процесів створення, розвитку і ослаблення національної системи боротьби з фальсифікованими лікарськими засобами в Україні. Завдяки створенню цієї системи, масштаб фальсифікації препаратів в Україні є відносно незначним і становить значно менше 1% від їх кількості на ринку. У зв’язку з цим рекомендовано підтримувати й покращувати систему протидії фальсифікації ліків, що існує на сьогодні, а не витрачати обмежені ресурси на передчасні й сумнівні, з точки зору досяжності очікуваних результатів, нові масштабні й високовитратні проєкти, на кшталт 2D-кодування упаковок. На наш погляд, зараз у системі охорони здоров’я України та її фармацевтичному секторі існує ряд більш важливих і актуальних невирішених проблем, які потребують уваги й зусиль політиків, чиновників та регуляторів (покращення економічної доступності і раціональне використання ліків, поновлення системи виписування рецептів, зменшення кількості лікарських помилок тощо).
Фальсифікація лікарських засобів є глобальною проблемою, особливо в країнах із низьким та середнім рівнем доходів. Актуальність цієї теми для населення інколи призводить до спроб маніпуляцій політиків (задля привертання уваги та підвищення рейтингу у виборців) і чиновників (для отримання додаткових фінансувань та повноважень). У цій статті наведено огляд розвитку рекомендацій ВООЗ щодо боротьби з підробленими ліками та процесів створення, розвитку і ослаблення національної системи боротьби з фальсифікованими лікарськими засобами в Україні. Завдяки створенню цієї системи, масштаб фальсифікації препаратів в Україні є відносно незначним і становить значно менше 1% від їх кількості на ринку. У зв’язку з цим рекомендовано підтримувати й покращувати систему протидії фальсифікації ліків, що існує на сьогодні, а не витрачати обмежені ресурси на передчасні й сумнівні, з точки зору досяжності очікуваних результатів, нові масштабні й високовитратні проєкти, на кшталт 2D-кодування упаковок. На наш погляд, зараз у системі охорони здоров’я України та її фармацевтичному секторі існує ряд більш важливих і актуальних невирішених проблем, які потребують уваги й зусиль політиків, чиновників та регуляторів (покращення економічної доступності і раціональне використання ліків, поновлення системи виписування рецептів, зменшення кількості лікарських помилок тощо).1. Глобальна проблема країн з низьким та середнім рівнем доходів. Рекомендації ВООЗ щодо її подолання
Учітесь, читайте,
І чужому научайтесь…
Тарас Шевченко
Фальсифіковані лікарські засоби є нелегальними і небезпечними продуктами, оскільки вони можуть не відповідати базовим вимогам до лікарських засобів — щодо їх ефективності, безпеки та якості. Вони можуть бути неефективними (не містити активних компонентів або містити їх у невідповідній кількості, або не бути біоеквівалентними), бути небезпечними (містити неприпустиму кількість токсичних домішок або незадекларовані активні фармацевтичні інгредієнти (АФІ) з іншим небезпечним ефектом) та/або неякісними (оскільки вони не виробляються відповідно вимог належної виробничої практики (Good Manufacturing Practice — GMP), то немає впевненості щодо постійності їх складу та властивостей), навіть якщо зразки фальсифікованих лікарських засобів формально відповідають фармакопейним вимогам.
Фальсифіковані лікарські засоби менше поширені у розвинених країнах з високим рівнем доходів, де створено жорстку регуляторну систему контролю фармацевтичного ринку і національні регуляторні органи (НРО) застосовують ефективний комплекс заходів для запобігання й боротьби з фальсифікованими лікарськими засобами, з урахуванням ситуації на ринку, свого досвіду і значних наявних ресурсів. У результаті кількість випадків виявлення фальсифікованих лікарських засобів у цих країнах дуже невелика, за оцінками самих НРО, — значно менше 1%.
У країнах, що розвиваються (із середнім та низьким рівнем доходів), проблема фальсифікованих ліків є більш гострою у зв’язку з недостатньо жорсткими регуляторними вимогами і слабкою спроможністю НРО.
У зв’язку з цим ВООЗ у другій половині ХХ ст. активно розробляла рекомендації для таких країн, для створення і посилення ефективності їх НРО і забезпечення населення якісними лікарськими засобами.
Перші спеціальні рекомендації ВООЗ щодо боротьби з фальсифікованими ліками були викладені у 1999 р. в документі «Counterfeit drugs: guidelines for the development of measures to combat counterfeit drugs» (WHO/EDM/QSM/99.1).
У цих рекомендаціях було представлено огляд проблем і факторів, що спричиняють появу фальсифікованих лікарських засобів, а також запропоновано підходи до розробки національних стратегій та комплексу специфічних заходів для боротьби з фальсифікованими ліками (у тому числі щодо організації досліджень, інспекції підозрюваних фальсифікованих препаратів, скринінгу потенційно підроблених продуктів, навчання фахівців тощо).
У цьому документі вперше було рекомендовано використовувати визначення «фальсифіковані лікарські засоби (Counterfeit drugs) — це лікарські засоби, які навмисно неправильно промарковані щодо їх ідентичності та/або назви виробника. Фальсифікованими можуть бути як оригінальні, так і відтворені препарати; вони можуть містити інгредієнти у відповідному або невідповідному складі, можуть бути без діючих речовин, з недостатньою їх кількістю або в підробленій упаковці».
Важливість цього визначення була зумовлена тим, що до цього в законодавстві різних країн використовувалися дуже різні визначення фальсифікованих лікарських засобів, що часто викривляло реальну картину щодо рівня наявності цих продуктів у різних країнах. Наприклад, у нормативних документах деяких африканських країн до фальсифікованих ліків відносили також субстандартні, незареєстровані, контрабандні препарати, що дозволяло чиновникам цих держав маніпулювати цифрами в 30–50–70% фальсифікованих лікарських засобів на ринку і вимагати від своїх урядів додаткових повноважень і ресурсів, а від міжнародних організацій — фінансової й технічної підтримки.
Але у 2000-х роках у міжнародній експертній спільноті теза щодо фальсифікованих лікарських засобів як найбільш небезпечної загрози для життя і здоров’я пацієнтів у країнах, що розвиваються, почала змінюватися. На основі різноманітної інформації експерти дійшли висновку, що кількість субстандартних лікарських засобів у країнах, що розвиваються, є значно більшою (на порядок), а небезпека від таких препаратів є не меншою, ніж від фальсифікованих лікарських засобів.
Крім того, найбільш резонансні й масштабні випадки смерті й госпіталізації людей у 1990–2015 рр. у країнах, що розвиваються, були спричинені не фальсифікованими лікарськими засобами, а субстандартними зареєстрованими препаратами, виробленими легальними виробниками із відхиленнями від вимог GMP:
- 1995–1996 рр. — близько 80 дітей померло на Гаїті від гострої ниркової та печінкової недостатності після застосування сиропу від кашлю, для виробництва якого використовували гліцерин, забруднений діетиленгликолем;
- 2011 р. — більше 200 пацієнтів померло і 850 було госпіталізовано в Пакистані після прийому кардіологічного препарату, забрудненого протималярійним АФІ, який помилково було використано виробником замість інертної допоміжної речовини;
- 2012–2013 рр. — у Пакистані померло близько 60 дорослих наркоманів, які у великих кількостях споживали сироп проти кашлю з АФІ декстрометорфан, який було помилково замінено виробником на інший стереоізомер — левометорфан, з тією ж самою хімічною формулою, але в кілька разів активніший;
- 2013 р. у Парагваї було госпіталізовано 44 дитини після застосування сиропу проти кашлю з помилково використаним левометорфаном замість декстрометорфану (тієї ж серії АФІ, що спричинила загибель наркоманів у Пакистані);
- 2014 р. у Демократичній Республіці Конго десятки людей постраждали після прийому таблеток парацетамолу, забруднених великими кількостями фенобарбіталу.
Через серйозність проблеми в 2013 р. ВООЗ впровадила Систему глобального епіднагляду та моніторингу (Global Surveillance and Monitoring System — GSMS), яка стала основним міжнародним інструментом у боротьбі з субстандартними та фальсифікованими лікарськими засобами. Переліки повідомлень ВООЗ про фальсифіковані ліки можна знайти на . Ще одним з елементів GSMS стала програма навчання для співробітників НРО щодо визначення та повідомлення про лікарські засоби, що не відповідають стандартам. Зі збільшенням числа фахівців, що пройшли це навчання, спостерігалося зростання кількості повідомлень, наданих через GSMS.
Через 5 років, у травні 2017 р., Всесвітня асамблея охорони здоров’я своїм рішенням в документі WHA70.23, на основі аналізу та узагальнення інформації в повідомленнях, отриманих ВООЗ, рекомендувала використовувати об’єднане визначення — «субстандартні та фальсифіковані лікарські засоби» («Substandard and falsified medical products» — SFMP), поставивши на перше місце субстандартні лікарські засоби як найбільш небезпечні. Також у документі WHA70.23 було наведено нові визначення для 3 категорій небезпечних ліків:
- субстандартні лікарські засоби (Substandard medical products; також названі «такими, що не відповідають специфікації») — зареєстровані лікарські засоби, які не відповідають стандартам якості та/або специфікаціям;
- незареєстровані/неліцензовані лікарські засоби (Unregistered/unlicensed medical products) — лікарські засоби, які не пройшли оцінку та/або не мають дозволу НРО для ринків, на яких вони реалізуються/розповсюджуються або використовуються, відповідно до регуляторних вимог національного або регіонального законодавства;
- фальсифіковані лікарські засоби (Falsified medical products) — лікарські засоби, в яких навмисно/по-шахрайськи неправильно вказано їх назву, склад або виробника.
Але таке об’єднання в одну категорію субстандартних і фальсифікованих лікарських засобів заклало певну суперечність. Субстандартні ліки є зареєстрованими продуктами, випуск і використання яких можна зменшити впровадженням GMP, посиленням реєстраційних і ліцензійних вимог. Щодо фальсифікованих лікарських засобів, то вони є нелегальними продуктами, виробництво і розповсюдження яких здійснюється поза правовим полем, і для боротьби з ними слід застосовувати інші механізми, ніж для субстандартних ліків. На рис. 1 показано всі рекомендовані функції НРО для протидії субстандартним і фальсифікованим лікарським засобам, червоним пунктиром виділено сегменти функціоналу НРО, які застосовують для протидії лише фальсифікованим лікам.
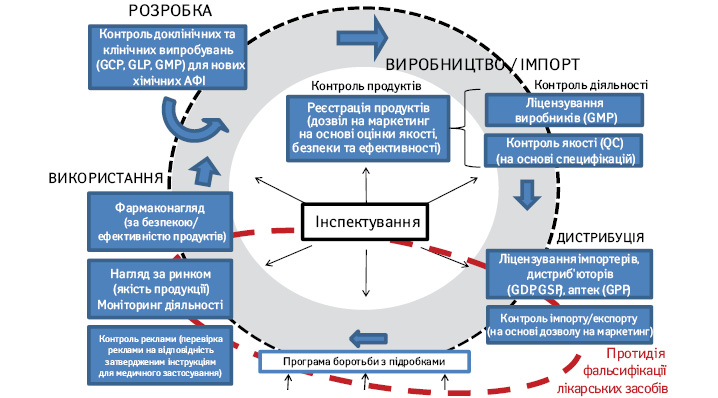
Для планування адекватної протидії цим двом групам небезпечних продуктів НРО важливо розуміти їх кількість і співвідношення на ринках. Від цього буде залежати, які напрямки роботи НРО мають стати пріоритетними і які ресурси слід задіяти для зменшення загроз для пацієнтів. Також НРО слід визначитися з цільовими кількісними індикаторами — який може бути прийнятний рівень субстандартних лікарських засобів та фальсифікованих лікарських засобів з точки зору співвідношення загрози для пацієнтів і ресурсів, необхідних для зменшення їх кількості.
ВООЗ неодноразово підкреслювала, що реальний обсяг і масштаб проблеми субстандартних і фальсифікованих лікарських засобів у різних країнах і регіонах важко оцінити у зв’язку з дуже розрізненими та фрагментарними даними.
В опублікованій у липні 2017 р. доповіді ВООЗ «» вперше були представлені об’єднані результати метааналізу даних зі 100 публікацій з надійних джерел за 10 років (2007–2016 рр.) щодо результатів лабораторного контролю різними методами, в основному високоефективної рідинної хроматографії та з використанням наборів MiniLabTM, більше 48 тис. зразків лікарських засобів, відібраних у 88 країнах з різним рівнем доходу.
З них лише 178 зразків були відібрані у 13 країнах з високим рівнем доходу, тому статистично було неможливо використати ці дані для екстраполяції з метою оцінки кількості субстандартних та фальсифікованих лікарських засобів на цих ринках.
Але до огляду були також включені публікації з результатами контролю більше 11 тис. зразків, відібраних у 19 країнах з низьким рівнем доходів, і близько 37 тис. зразків — з 56 країн із середнім рівнем доходів. При цьому для цих груп країн відсотки перевірених зразків, які не відповідали вимогам специфікацій (тобто були субстандартними і фальсифікованими), становили відповідно 10,6 та 10,5%.
Для оцінки ситуації із субстандартними і фальсифікованими ліками в окремих країнах ВООЗ рекомендує брати до уваги й інші джерела інформації, у першу чергу — статистичні дані НРО цих держав.
Ще однією важливою публікацією ВООЗ 2017 р. щодо субстандартних та фальсифікованих лікарських засобів стали рекомендації «». У цих оновлених у порівнянні з WHO/EDM/QSM/99.1 рекомендаціях були враховані рішення щодо об’єднання в одну категорію субстандартних і фальсифікованих лікарських засобів, нові статистичні дані ВООЗ і оцінка нових сучасних факторів, що спричиняють розповсюдження таких ліків.
Так, у цій публікації було зазначено, що субстандартні та фальсифіковані лікарські засоби найбільш часто виявлялися за наявності в країнах комбінації 3 основних передумов:
- обмежений доступ до економічно доступних, якісних, безпечних та ефективних лікарських засобів, зокрема необхідність купувати ліки самими пацієнтами за власний кошт;
- низькі стандарти нагляду за фармацевтичним ринком — від слабких етичних практик у закладах охорони здоров’я і точках продажу лікарських засобів до корупції в публічному і приватному секторах;
- обмежені засоби і технічна спроможність НРО забезпечити дотримання належних практик виробництва, контролю якості і дистрибуції лікарських засобів.
Серед інших проблем країн з низьким і середнім рівнем доходів автори публікації відзначили низький рівень розуміння загрози субстандартних і фальсифікованих лікарських засобів у багатьох країнах, навіть серед професіоналів охорони здоров’я; недостатньо сильні регуляторні системи або невиконання регуляторних вимог, що створює вакуум у законодавстві і використовується злочинцями; зростання рівня онлайн-продажу ліків, що створює значною мірою нерегульований та невидимий ринок. Крім того, автори відмічають, що ризики появи в обігу субстандартних і фальсифікованих лікарських засобів суттєво зростають у кризових ситуаціях і регіонах (військові конфлікти, стихійні лиха тощо), коли урядовий контроль за системою постачання ліків ослаблюється або взагалі втрачається.
Для протидії поширенню субстандартних і фальсифікованих лікарських засобів у вищевказаних рекомендаціях пропонується розробити національні стратегії дій у 3 напрямках — «запобігати, виявляти та реагувати»:
1) слід запобігати потраплянню субстандартних і фальсифікованих лікарських засобів до пацієнтів шляхом створення системи швидкого вилучення цих продуктів з аптек та лікарень. Також важливо проводити широку інформаційну компанію для підвищення рівня знань і розуміння як пацієнтами, так і медичним персоналом загроз субстандартних і фальсифікованих лікарських засобів. Треба забезпечувати цілісність каналів постачання, закривши можливість потрапляння в систему таких препаратів. І, нарешті, слід створювати сильну нормативну систему, щоб співробітники поліції і митниці також мали необхідні інформацію та засоби для захисту населення від субстандартних і фальсифікованих лікарських засобів;
2) слід виявляти субстандартні і фальсифіковані лікарські засоби. Це вимагає інвестицій у посилення контролю на кордонах, покращення системи повідомлень про такі ліки, більш розумне інспектування та збільшення доступу до лабораторій та обладнання для польового скринінгу зразків лікарських засобів;
3) слід реагувати. Має бути створена система сповіщення та відкликання виявлених субстандартних і фальсифікованих лікарських засобів, посилюватися регуляторна система (див. рис. 1), а юридичні процедури мають ставати більш прозорими.
Для посилення ефективності вказаних дій має існувати сильна політична воля щодо протидії субстандартним і фальсифікованим лікарським засобам, і всі зацікавлені сторони мають працювати разом. Проблема субстандартних і фальсифікованих лікарських засобів є проблемою не тільки системи охорони здоров’я, вона потребує залучення регуляторів, правоохоронців, митників, логістів та інших зацікавлених партнерів.
У боротьбі із субстандартними та фальсифікованими лікарськими засобами необхідна активна участь політичних лідерів, які б транслювати політику в конкретні дії шляхом залучення необхідних людських і фінансових ресурсів. Такі залучення не є витратами, вони мають розглядатися як інвестиції для захисту бізнесу та ринку, а також цілісності систем охорони здоров’я.
У 2016 р. в ЄС було опубліковано настанову з вимогами до нанесення елементів безпеки на пакувальні матеріали деяких лікарських засобів (Regulation (EU) 2016/161). Нею було передбачено, що маркування 2D-кодами буде обов’язковим тільки для рецептурних лікарських засобів (за винятком більшості інфузійних розчинів тощо) і 1 безрецептурного препарату. З 09.02.2019 р. в ЄС почала працювати система серіалізації та контролю першого розкриття упаковки як додатковий до існуючої в ЄС жорсткої регуляторної системи метод боротьби з фальсифікацією ліків. Вказана настанова встановила чіткі вимоги до унікальних ідентифікаційних номерів, що мають бути нанесені на пакувальні матеріали, до нанесення та якості друку 2D-кодів та системи верифікації елементів безпеки лікарських засобів.
Також у ЄС була створена Європейська організація з верифікації лікарських засобів (European Medicines Verification Organisation — EMVO), яка відповідає за збір та зберігання інформації щодо продуктів та елементів безпеки, а також містить платформи для перевірки автентичності лікарських засобів кінцевими та проміжними операторами ланцюга поставки — лікарнями, аптеками, дистриб’юторами тощо. EMVO є недержавною неприбутковою організацією, заснованою європейськими професійними асоціаціями аптек, лікарень, дистриб’юторів та виробників ліків. Вона фінансується за рахунок членських внесків учасників системи. Роль держав та союзних регуляторних органів ЄС полягає у нагляді за роботою EMVO та затвердженні відповідних регуляторних документів.
Впровадження системи і дотримання термінів проєкту в ЄС відбувалося непросто. За першими підсумками 2019 р. в країнах ЄС кількість технічних збоїв системи верифікації сягала близько 6%, і на цьому фоні було неможливо визначити кількість виявлених фальсифікованих лікарських засобів. Також поки що не опубліковано дані щодо підвищення собівартості однієї упаковки препаратів після впровадження такої системи в ЄС з урахуванням інвестицій у додаткове виробниче обладнання і інформаційну інфраструктуру, членських внесків до EMVO тощо.
Таким чином, поки що не можна зробити висновки стосовно того, чи досягали країни ЄС поставлених цілей у цьому проєкті і якою була вартість такого впровадження для виробників ліків та інших операторів ринку.
Відносно успішним був проєкт з впровадження системи 2D-кодування упаковок лікарських засобів у Туреччині, який було розпочато у 2009 р. Запровадження такої системи в масштабах країни потребувало кардинального удосконалення IT-інфраструктури (наявність комп’ютерів і точок доступу до інтернету), істотних фінансових витрат від виробників (встановлення додаткового обладнання, модернізація пакувальних ліній, встановлення систем track&trace), від дистриб’юторів та аптечних мереж (програмне забезпечення, сканери, навчання персоналу).
Інший результат від впровадження подібної системи показує Російська Федерація (РФ). Керівники НРО Росії заявляли, що кількість виявлених фальсифікованих лікарських засобів у РФ у 2011 р. до рівня менше 0,1–0,2%, а у 2017 р. — .
Але, незважаючи на такі мізерні кількості фальсифікованих лікарських засобів, Уряд РФ 14.12.2018 р. прийняв постанови № 1556, № 1557, № 1558 про створення системи моніторингу руху лікарських засобів. Для ліків, вироблених або ввезених у РФ після 1 липня 2020 р. стало обов’язковим нанесення 2D-кодів на упаковки та приймання і відпуск їх дистриб’юторами та аптеками лише після отримання підтвердження від системи. Ця вимога, на відміну від вимог ЄС, стосується всіх лікарських засобів (у тому числі безрецептурних), і не тільки вторинної, але й групової упаковки.
Зі збільшенням у мережі кількості лікарських засобів з 2D-кодами система почала давати збої, що призводило до кількагодинних і кількаденних затримок із прийманням і відпуском лікарських засобів дистриб’юторами та аптеками. У розпал другої хвилі епідемії COVID-19, наприкінці вересня і в жовтні 2020 р. у РФ спостерігався колапс дистриб’юторської і аптечної мережі, коли днями і тижнями не було можливості зареєструвати такі операції в системі і, відповідно, надати продукцію споживачам. У зв’язку з цим уряд РФ був вимушений постановою від 02.11.2020 р. № 17790 дозволити до 01.02.2021 р. спростити процедуру і дозволити операторам ринку приймати і відпускати лікарські засоби, не чекаючи дозволу системи.
2. Добре забуте старе. Створення і розвиток системи боротьби з фальсифікованими лікарськими засобами в Україні у 1991–2005 рр.
…І свого не цурайтесь
Тарас Шевченко
Після проголошення незалежності в 1991 р. в Україні почалися швидкі зміни в політичній та економічній сферах. У 1992–1993 рр. розпад централізованої системи постачання лікарських засобів, особливо оптової ланки, розрив виробничих зв’язків у межах колишнього Радянського Союзу, різке падіння економіки призвели до ситуації, коли полиці аптек в Україні стали майже порожніми. Лібералізація вимог до реєстрації лікарських засобів, їх виробництва та імпорту, дозволила в 1994–1995 рр. наповнити країну ліками, збільшити їх асортимент, однак при цьому на ринку значно зросла кількість препаратів низької якості та ефективності.
У зв’язку з цим в Україні виникла потреба реформування системи регулювання лікарських засобів з урахуванням змін на фармацевтичному ринку та світового досвіду.
Відповідно, в 1991–1992 рр. в Україні почалося створення власних НРО в галузі лікарських засобів. У той час українські спеціалісти ще не мали власного досвіду, а знання щодо досвіду інших країн і рекомендацій ВООЗ були обмеженими, тому в Україні були створені і почали працювати регуляторні та експертні органи за аналогією з такими в СРСР:
- Державний комітет України з медичної та мікробіологічної промисловості (Держкоммедбіопром) — у 1991 р. З розвитком ринкової економіки Держкоммедбіопром поступово втрачав функції управління і перетворювався на НРО з функціями ліцензування, контролю і створення нормативного поля у фармацевтичній галузі;
- Фармакологічний Комітет МОЗ України — у 1992 р. До 1997 р. він виконував функції державної реєстрації лікарських засобів. Після 1997 р. він став виконувати функції експертної організації, на основі рекомендацій якої вже МОЗ приймало рішення про реєстрацію ліків в Україні;
- Фармакопейний Комітет МОЗ України — у 1992 р. Після 1996 р. він був виведений з процедури реєстрації лікарських засобів і зосередився на підготовці Державної Фармакопеї України, атестації фармакопейних стандартних зразків та інших завданнях стандартизації;
- Державна інспекція з контролю якості лікарських засобів МОЗ України (Держлікінспекція) — у 1992 р.;
- Комітет з питань імунобіологічних препаратів МОЗ України — у 1994 р. У 2000 р. він був ліквідований з передачею його функцій іншим НРО.
Таким чином, у 1991–1994 рр. в Україні замість єдиного НРО (як це є сьогодні у більшості країн) були створені кілька НРО (як це було в СРСР), які почали свою історію розвитку і взаємодії, а інколи — конкуренції і суперництва за вплив, повноваження і ресурси. У певні періоди часу це створювало і до цього часу створює проблеми не тільки для їх функціонування, але й для інших учасників фармацевтичного ринку.
Основні етапи створення, перейменування, реорганізації та/або об’єднання НРО України показано на рис. 2. Зеленим фоном позначені організації, які мали/мають повноваження приймати рішення щодо допуску/заборони лікарських засобів та/або компаній; жовтим фоном — експертні установи, які надають рекомендації щодо таких дій.
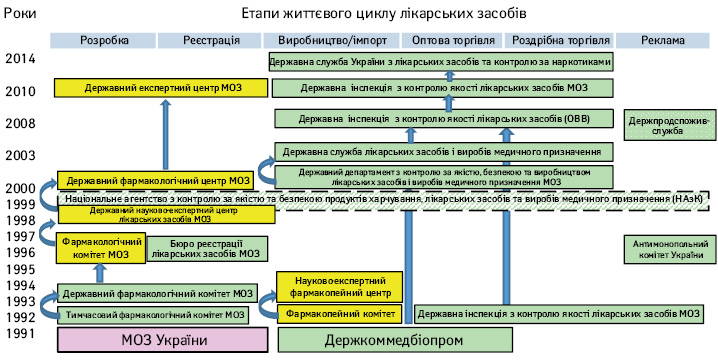
Суперечливим епізодом в історії НРО України було створення 01.02.1999 р. Національного агентства з контролю за якістю та безпекою продуктів харчування, лікарських засобів та виробів медичного призначення (), керівником якого було призначено колишнього міністра А.М. Сердюка. Але МОЗ було категорично проти таких змін. Протягом року тривало двовладдя і бюрократичне протистояння між МОЗ і НАзК, яке завершилося перемогою МОЗ.
Важливим моментом розвитку вказаних НРО було створення в їх структурі або у підпорядкуванні лабораторій з контролю лікарських засобів і оснащення їх сучасним аналітичним обладнанням. Зокрема, наказом МОЗ від 05.01.1993 р. № 3 на базі УНДІФТ МОЗ була створена Державна лабораторія з аналізу якості лікарських засобів у статусі робочого органу Держлікінспекції. У 1998 р. вона була реорганізована в самостійну Центральну лабораторію з аналізу якості лікарських засобів МОЗ (Центральна лабораторія).
У перші роки незалежності спрощені вимоги до реєстрації, виробництва й імпорту ліків, відсутність на той час нормативних вимог до фармацевтичної розробки, відповідності GMP, підтвердження біоеквівалентності призвели до появи на ринку України великої кількості нових лікарських засобів українських виробників і ліків з країн зі слабкими регуляторними вимогами: Індії, Пакистану тощо. І перші результати державного контролю в 1993–1995 рр. показали, що від 5 до 10% перевірених зразків цих груп лікарських засобів не відповідали вимогам специфікацій. Почалася практика вилучення з ринку України неякісних лікарських засобів приписами Держлікінспекції. Так, у 1994 р. за результатами аналізів Державної лабораторії з аналізу якості лікарських засобів було вилучено з ринку більше 30 серій неякісних лікарських засобів однієї з індійських компаній на суму близько 300 тис. дол. США.
Спочатку новостворені НРО України у своїй роботі використовували нормативні документи і підходи колишнього СРСР, а після накопичення власного досвіду й ознайомлення з регуляторною практикою інших країн і рекомендацій ВООЗ стала зрозумілою необхідність створення нових нормативних документів, які б сприяли підвищенню ефективності, безпеки та якості лікарських засобів.
Велике значення для регулювання фармацевтичного ринку мало прийняття у 1996 р. Закону України № 123/96-ВР «». У ст.ст. 14–17 цього Закону, які готувалися за допомогою фахівців Управління з контролю за харчовими продуктами і лікарськими засобами (Food and Drug Administration — FDA) США, були визначені статус, структура Держлікінспекції, джерела її фінансування і права її посадових осіб. Але в остаточний текст ст. 14 цього Закону за ініціативою впливового голови Держкоммедбіопрому Ю.П. Спіженка було введено положення про те, що державний контроль за додержанням умов виробництва лікарських засобів здійснюють Держкоммедбіопром та уповноважені ним державні органи. Це призвело до дублювання функцій і подальших проблем у взаємодії Держлікінспекції і Держкоммедбіопрому, які тривали до моменту об’єднання цих двох НРО у 2008 р.
У 1997–1999 рр. була створена система Держлікінспекції — 27 держлікінспекцій в АР Крим, областях, містах Києві і Севастополі, а також самостійна Центральна лабораторія. У цілому в системі Держлікінспекції в 2005 р. працювало близько 600 співробітників.
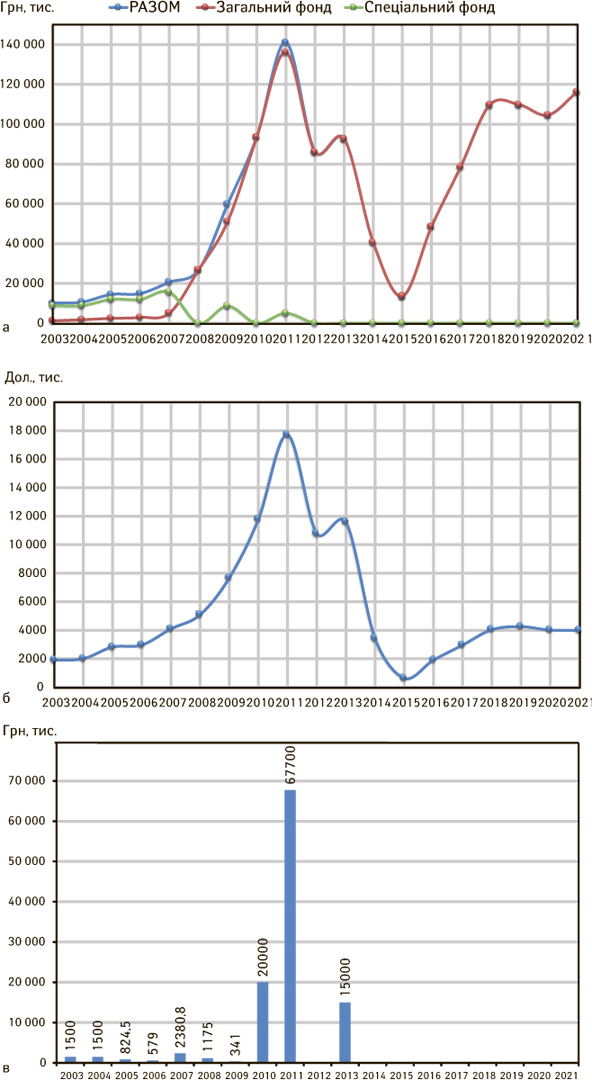
Запорукою для стійкого розвитку і стабільної роботи будь-якого НРО є достатнє і стабільне фінансування. За середньорічної потреби близько 10 млн грн (1,87 млн дол. в перерахунку), у 2000–2004 рр. фактичне фінансування з державного бюджету Держлікінспекції становило від 0,7 до 1,7 млн грн (рис. 3 а–в). З інших джерел фінансування, за рахунок тренінгів, семінарів, проведення лабораторного контролю зразків тощо в цей період залучалося у кілька разів більше коштів, від 5,3 млн грн до 10 млн грн на рік.
Важливим результатом роботи Держлікінспекції і Центральної лабораторії стало створення в Україні, згідно з рекомендаціями ВООЗ, дворівневої системи лабораторій з контролю якості лікарських засобів. Наказами Держлікінспекції від 08.09.98 р. № 01-Л та № 02-Л були визначені уніфіковані правила роботи в лабораторіях держлікінспекцій згідно з рекомендаціями належної лабораторної практики (Good Laboratory Practic —GLР) ВООЗ та порядок акредитації лабораторій у системі державного контролю якості лікарських засобів. Протягом 1998–2000 рр. було проведено навчання персоналу, придбано нове обладнання, створено систему забезпечення якості робіт і проведено акредитацію лабораторій.
Для оцінки і удосконалення рівня роботи лабораторій територіальних держлікінспекцій у 2000 р. була започаткована Програма професійного тестування (ППТ), участь у якій стала обов’язковою для всіх акредитованих лабораторій (табл. 1) і добровільною — для лабораторій фармацевтичних підприємств України.
| Таблиця 1 | Типи і кількість лабораторій, які брали участь у Програмі професійного тестування в Україні у 2001–2019 рр.* |
| Лабораторії | Раунди Програми професійного тестування/рік | ||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | ||||
| 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2012 | 2011 | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 | 2019 | |
| Територіальні ДІ/ДЛС | 30 | 30 | 30 | 30 | 27 | 14 | 26 | 21 | 11 | 18 | 4 | 1 | 1 | – | 2 | 6 | |||
| Уповноважені ДІ/ДЛС | 5 | 5 | 8 | 7 | 6 | 9 | 7 | 7 | 8 | 7 | 9 | 8 | 7 | 8 | 5 | 5 | |||
| Фармацевтичних підприємств | – | 10 | 12 | 11 | 20 | 21 | 26 | 34 | 32 | 29 | 27 | 28 | 33 | 30 | 41 | 37 | |||
| Зарубіжні | – | – | 6 | 11 | 6 | – | – | 7 | 8 | 10 | 10 | 5 | – | 3 | 4 | 3 | |||
| Усього: | 35 | 45 | 56 | 60 | 59 | 44 | 59 | 69 | 59 | 64 | 50 | 42 | 41 | 41 | 52 | 51 | |||
*За даними ДП «Український науковий фармакопейний центр якості лікарських засобів»
У кожному регіоні працювала лабораторія першого рівня, здатна простими методами перевіряти велику кількість зразків лікарських засобів, які відбирали інспектори під час перевірок аптек і дистриб’юторів. А на другому рівні Центральна лабораторія і ще кілька акредитованих лабораторій проводили контроль зразків більш складними методами, які потребували дороговартісного обладнання та висококваліфікованого персоналу.
На жаль, у 2002–2003 рр. внаслідок недостатньої кваліфікації співробітників і використання застарілого обладнання спостерігалася помітна кількість помилково негативних результатів контролю зразків лікарських засобів у територіальних лабораторіях та безпідставні зупинки обігу окремих серій ліків у регіонах. Це призводило до необхідності повторного контролю цих зразків у Центральній лабораторії для підтвердження або відміни таких рішень. Ситуація поступово стабілізувалася після навчання і набуття досвіду фахівцями лабораторій першого рівня, оснащення їх сучасним аналітичним обладнанням, впровадження системи забезпечення якості отриманих результатів.
У вересні 1999 р. в Україні було офіційно зафіксовано перший випадок виявлення фальсифікованих лікарських засобів. Зразки препарату азитроміцину тригідрату, капсули по 250 мг серії 215107 з маркуванням виробника «Пліва», Хорватія були вилучені з аптеки м. Київ співробітниками УДСБЕЗ Головного управлiння Міністерства внутрішніх справ (МВС) України у м. Києві і передані в Держлікінспекцію в м. Київ. Лабораторія цієї інспекції встановила, що середня маса вмісту капсул (370 мг) була завищеною у порівнянні з вимогами аналітичної нормативної документації фірми-виробника (304–336 мг). Додаткові дослідження, проведені в Центральній лабораторії, підтвердили, що перевірений зразок фальсифікованих ліків не містив АФІ азитроміцину, натомість він містив невідому речовину з інтенсивним поглинанням в УФ-ділянці спектру при довжині хвилі 262 нм. На підставі цієї інформації Держлікінспекція видала перший припис № 14Д/21-158 про заборону реалізації і вилучення з обігу вказаної серії фальсифікованих ліків.
Ще одну серію фальсифікованих лікарських засобів з такою ж діючою речовиною, капсули по 250 мг, серії 107117, з аналогічними ознаками фальсифікації у жовтні 1999 р. виявила Держлікінспекція в Харківській обл.
Стало зрозуміло, що проблема фальсифікованих ліків є актуальною і для України. Тому перед українськими регуляторами постало нове завдання — створення системи протидії обігу таких препаратів, для чого були використані щойно опубліковані рекомендації ВООЗ 1999 р. «Counterfeit drugs: guidelines for the development of measures to combat counterfeit drugs» (WHO/EDM/QSM/99.1).
Спеціальні заходи для боротьби з фальсифікованими лікарськими засобами, рекомендовані ВООЗ, були впроваджені в тексти нормативних документів, що вказані нижче.
В Інструкцію про порядок контролю якості лікарських засобів під час оптової та роздрібної торгівлі, затверджену Наказом МОЗ України від 30.10.2001 р. № 436, вперше в Україні було введено визначення фальсифікованих лікарських засобів (як у документі ВООЗ WHO/EDM/QSM/99.1). Також принципово новим положенням цієї Інструкції стала вимога щодо введення в аптеках і оптових компаніях ключового елементу GDP — посад Уповноважених осіб (УО) з якості, на яких було покладено обов’язки зі здійснення вхідного контролю якості лікарських засобів під час оптової та роздрібної торгівлі, надання дозволу на продаж серій ліків тощо. Для відслідковування шляхів постачання лікарських засобів в інструкцію була введена вимога вказувати номери серій і кількість упаковок кожної серії препарату в накладних. Це дало можливість покращити відстеження лікарських засобів і ефективність вилучення з обігу субстандартних і фальсифікованих ліків при їх виявленні.
Ще одним базовим документом системи забезпечення якості лікарських засобів і боротьби із субстандартними та фальсифікованими лікарськими засобами став Порядок заборони (зупинення) та вилучення з обігу лікарських засобів на території України, затверджений Наказом МОЗ України від 12.12.2001 р. № 497, з наступними змінами та доповненнями. Цей порядок визначив підстави для вилучення з обігу субстандартних та фальсифікованих лікарських засобів, а також терміни і обов’язки аптек і дистриб’юторів щодо виконання приписів і розпоряджень Держлікінспекції. У результаті впровадження цієї системи небезпечні продукти швидко вилучалися з аптек і лікарень та суттєво знижувався ризик їх потрапляння до пацієнтів.
Важливою подією для фармацевтичної галузі у 2003 р. стало затвердження Постановою КМУ від 17.07.2003 р. № 1075 першої Програми боротьби з виробництвом та розповсюдженням фальсифікованих лікарських засобів на 2003–2008 рр. Ця Програма заклала основи для координації дій МОЗ, МВС, Державної митної служби та Служби безпеки України (СБУ) у боротьбі з фальсифікованими ліками.
Правила зберігання та проведення контролю якості лікарських засобів у лікувально-профілактичних закладах, затверджені Наказом МОЗ України від 16.12.2003 р. № 584, встановили вимоги до зберігання ліків у лікувально-профілактичних закладах, започаткували введення в них УО, які мали контролювати відсутність у надходженнях лікарських засобів вилучених з обігу приписами і розпорядженнями Держлікінспекції.
Для запобігання повторному потраплянню в мережі постачання заборонених субстандартних і фальсифікованих лікарських засобів, Наказом МОЗ України від 08.07.2004 р. № 349 були внесені зміни до Правил проведення утилізації та знищення неякісних лікарських засобів, якими було визначено, що фальсифіковані лікарські засоби вилучаються з обігу лише шляхом знищення.
Порядок здійснення державного контролю якості лікарських засобів, що ввозяться в Україну, вперше було затверджено на останньому засіданні КМУ під керівництвом М.Я. Азарова в січні 2004 р., рішення якого не було опубліковано новим післяреволюційним Урядом. І вже після другої спроби цей було затверджено постановою КМУ від 14.09.2005 р. № 902.
У червні 2003 р. голова Державної служби України з лікарських засобів та контролю за наркотиками (Держлікслужба) М.Ф. Пасічник подав офіційну заяву про її вступ до Системи співробітництва фармацевтичних інспекцій (Pharmaceutical Inspection Co-operation Scheme — PIC/S). А 2004 р. були прийняті стратегічні постанови КМУ № 1419 щодо обов’язкового впровадження вимог GхP в Україні з 2009 р. та № 1570 щодо заборони торгівлі субстандартними та фальсифікованими лікарськими засобами в аптеках.
Вищевказані нормативно-правові акти, затверджені у 2001–2005 рр., заклали нормативну основу системи державного контролю лікарських засобів під час їх виробництва, ввезення в Україну, оптової та роздрібної реалізації та використання у медичних закладах, а також виявлення, вилучення з обігу та знищення субстандартних і фальсифікованих лікарських засобів. Закладені в них підходи і принципи продовжують і сьогодні використовуватися в практиці регулювання лікарських засобів в Україні.
Одним з найважливіших елементів успішної боротьби з фальсифікованими лікарськими засобами була освітня й інформаційна кампанія Державної інспекції з контролю якості лікарських засобів (Держлікінспекція). Першим завданням цієї кампанії було ознайомлення фармацевтичних і медичних працівників з проблемою і небезпекою фальсифікованих лікарських засобів.
Для цього співробітниками Центральної лабораторії у 2000 р. були перекладені українською мовою та опубліковані в «Щотижневику Аптека» рекомендації ВООЗ WHO/EDM/QSM/99.1.
Також у червні 2000 р. Держлікінспекція опублікувала брошуру «Візуальні методи виявлення підроблених або субстандартних лікарських засобів», у якій співробітникам аптек та дистриб’юторам були надані рекомендації щодо проведення візуального контролю лікарських засобів і дій з вилучення з обігу небезпечних продуктів (рис. 4). На останній сторінці цієї брошури були наведені контакти Держлікінспекції, Центральної лабораторії та всіх 27 територіальних держлікінспекцій. 5 тис. копій брошури за допомогою «Щотижневика Аптека» були безоплатно передані аптекам, дистриб’юторам і територіальним органам Держлікінспекції. А у 2002 р. була видана і розповсюджена друга редакція цієї брошури зі збільшеним накладом у 10 тис. копій.

Держлікінспекція у 2000–2005 рр. започаткувала проведення регулярних семінарів для керівників та співробітників територіальних органів Держлікінспекції, виробників, імпортерів, дистриб’юторів лікарських засобів та аптек. Для інформування пацієнтів про небезпеку фальсифікованих лікарських засобів та рекомендації щодо зниження цих ризиків співробітниками державної інспекції були організовані періодичні публікації у центральних і місцевих газетах та інтерв’ю на радіо і телебаченні.
Завдяки такій інформаційній кампанії медичні працівники і пацієнти України отримували професійну інформацію щодо фальсифікованих лікарських засобів, а Держлікінспекція стала беззаперечним лідером і визнаним експертом з цих питань, у тому числі для МОЗ, МВС, СБУ та митниці.
Виявлення субстандартних і фальсифікованих лікарських засобів здійснювали УО з якості аптек і дистриб’юторів, інспектори під час планових і позапланових перевірок, Держлікінспекцією проводилася перевірка повідомлень про підозрілі лікарські засоби з будь-яких інших джерел (пацієнти, лікарі, система фармаконагляду, МВС, СБУ, митниця тощо). Відібрані зразки ліків співробітники Держлікінспекції направляли на контроль до акредитованих лабораторій. У разі невідповідності зразків вимогам специфікацій Держлікінспекція приймала рішення про вилучення субстандартних і фальсифікованих лікарських засобів з обігу.
Держлікінспекцією була створена система швидкого інформування всіх суб’єктів фармацевтичного ринку про виявлені субстандартні і фальсифіковані лікарські засоби з приписами про їх вилучення з обігу. У цих приписах вказувалися показники, за якими субстандартні і фальсифіковані ліки не відповідали вимогам специфікацій, а для фальсифікованих препаратів — ознаки фальсифікації, за якими можна було відрізнити фальсифіковані від справжніх лікарських засобів. Приписи про вилучення з обігу субстандартних і фальсифікованих лікарських засобів терміново надсилалися всім виробникам, дистриб’юторам і аптекам та публікувалися в професійній пресі, зокрема у «Щотижневику Аптека». За невиконання приписів Держлікінспекції щодо вилучення субстандартних і фальсифікованих лікарських засобів аптеки і дистриб’ютори могли бути оштрафовані або позбавлені ліцензії.
Інформація про назви і серії виявлених субстандартних і фальсифікованих лікарських засобів використовувалася інспекторами та УО для цілеспрямованої перевірки інших серій таких препаратів. Оскільки виробники субстандартних і фальсифікованих ліків, як правило, виробляли більше, ніж одну серію невідповідної продукції одного найменування, то такий підхід дозволяв швидше виявляти і вилучати з обігу небезпечні продукти. Відповідно, у період 2001–2005 рр. Держлікінспекція щорічно виявляла і вилучала з обігу 33–89 серій і 18–23 найменувань фальсифікованих лікарських засобів (табл. 2)
| Таблиця 2 | Кількість виявлених і вилучених з обігу серій і найменувань фальсифікованих лікарських засобів в Україні у 2001–2020 рр. та кримінальних справ за ст. 321-1 Кримінального кодексу України у 2013–2018 рр.** |
| Роки | 2000 | 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 | 2019 | 2020 |
| Серій фальсифікованих лікарських засобів** | 17 | 40 | 62 | 33 | 49 | 89 | 57 | 41 | 21 | 46 | 69 | 34 | 62 | 66 | 117 | 29 | 59 | 16 | 9 | 59 | 4 |
| Найменувань фальсифікованих лікарських засобів** | 4 | 22 | 20 | 18 | 21 | 23 | 25 | 23 | 12 | 28 | 27 | 15 | 39 | 40 | 63 | 20 | 22 | 14 | 7 | 18 | 3 |
| Відсоток фальсифікованих лікарських засобів на ринку (упаковки)** | 0,38 | 0,33 | 0,20 | 0,34 | 0,08 | 0,09 | 0,19 | 0,05 | 0,03 | 0,04 | |||||||||||
| Зареєстровано кримінальних справ*** | 26 | 23 | 40 | ||||||||||||||||||
| Повідомлено про підозру*** | 5 | 3 | 2 | ||||||||||||||||||
| Подано до суду з обвинувальним вироком*** | 3 | 3 | 1 | ||||||||||||||||||
| Припинено провадження*** | 8 | 9 | 7 | ||||||||||||||||||
| Рішення не прийнято*** | 24 | 20 | 39 | ||||||||||||||||||
| Винесено вироки*** | 6 | 3 | 6 | 5 | 3 | 2 | |||||||||||||||
**С.О. Лебедь. Історичні аспекти та сучасний стан фальсифікації лікарських засобів в Україні та світі, Рівне, Волинські обереги, 2018. — 320 с.; дані Держлікслужби: www.dls.gov.ua.
***Arkusha L.I., Hloviuk I.V., Zavalniuk S.V. PROBLEMS OF COUNTERFEITING MEDICAL PRODUCTS IN UKRAINE//Wiadomosci lekarskie (Warsaw, Poland: 1960) 72 (5 cz 2), 1131–1135.
Для розрахунку кількості субстандартних і фальсифікованих лікарських засобів на ринку України використовували співвідношення кількості зразків виявлених субстандартних і фальсифікованих ліків до загальної кількості проконтрольованих зразків. У 2003–2004 рр. в лабораторіях територіальних органів Держлікінспекції за допомогою простих методів щорічно перевірялося більше 120 тис. зразків лікарських засобів. Із них було визнано субстандартними 3,2% від перевірених зразків (у тому числі 2,5% для імпортних і 3,5% для українських препаратів), а фальсифікованими — 0,35%. Тобто кількість виявлених субстандартних ліків в Україні в цей період на порядок перевищувала кількість виявлених фальсифікованих препаратів — як і в інших країнах, згаданих у дослідженнях у першому розділі цієї статті.
З урахуванням того, що зразки лікарських засобів під час перевірок аптек відбиралися не у випадковий спосіб, а цілеспрямовано (з урахуванням інформації про інші серії субстандартних і фальсифікованих лікарських засобів), цей відсоток виявлених фальсифікованих ліків міг бути вищим за їх реальну кількість на ринку. При використанні іншого способу розрахунку, коли кількість виявлених і вилучених з ринку упаковок фальсифікованих лікарських засобів порівнювалася з кількістю упаковок всіх препаратів на ринку, результат становив 0,013% фальсифікованих лікарських засобів (131 тис. упаковок фальсифікованих лікарських засобів, виявлених у 2003 р. порівнювалася з 1 млрд упаковок на ринку). З урахуванням результатів, розрахованих за цими двома підходами, Держлікінспекція оцінювала реальну кількість фальсифікованих лікарських засобів в Україні в цей період як «менше, ніж 0,1%».
У 2004 р. Держлікінспекція з метою одержання об’єктивної інформації про кількість фальсифікованих лікарських засобів на фармацевтичному ринку провела перше в Україні (і на сьогодні поки що єдине) дослідження зразків таблеток сульфаметоксазолу та триметоприму, які в цей період були серед найчастіше фальсифікованих препаратів. Із 263 аптек у 26 різних регіонах України було відібрано 337 зразків 128 серій препаратів 8 виробників, які були досліджені в лабораторіях територіальних держлікінспекцій за візуальними і фармако-технологічними показниками та двома методиками тонкошарової хроматографії (Фармакопеї США та Індійської Фармакопеї).
За результатами цього масштабного дослідження було виявлено 4 зразки фальсифікованих лікарських засобів: 1 — у Донецькій і 3 — в Луганській областях, тобто 1,2% від кількості перевірених зразків. Усі вони мали маркування препарату Бісептол 480, таблетки, № 20 виробництва Паб’яницького фармацевтичного заводу «Польфа», Польща.
Таким чином, до 2005 р. в Україні була створена система для запобігання, виявлення та реагування на субстандартні та фальсифіковані лікарські засоби відповідно до рекомендацій ВООЗ 1999 р.
Серед недоліків цієї системи слід відмітити два основних моменти.
Стратегічним прорахунком керівників НРО України цього періоду була відсутність адекватної реакції на проблему врегулювання зростаючого продажу лікарських засобів через інтернет, необхідність якої у 2000–2005 рр. вже обговорювали на всіх міжнародних фармацевтичних конференціях, а регулятори в розвинених країнах почали ліцензувати такий вид торгівлі ліками. В Україні ж супротив керівників аптечних асоціацій та аптек, недооцінка швидкості розвитку інформаційних технологій та е-комерції призвели до переходу цього бізнесу на довгий час у «сіру», неврегульовану зону. Лише в травні 2020 р., в розпал епідемії COVID-19 і під час «жорсткого» карантину, Парламент і Уряд України прийняли вольове політичне рішення про запровадження е-торгівлі лікарськими засобами, поставивши перед цим фактом фармацевтичних фахівців, які не змогли своєчасно ініціювати такі зміни.
Ще однією тактичною помилкою керівництва НРО була недостатня пріоритезація завдання введення в Україні кримінальної відповідальності за фальсифікацію лікарських засобів, яка була поставлена в Програмі боротьби з виробництвом та розповсюдженням фальсифікованих лікарських засобів на 2003–2008 рр., але реалізована лише у 2011 р. Практика показала, що можливість лише адміністративного покарання за порушення законодавства щодо забезпечення якості лікарських засобів була слабким інструментом боротьби з фальсифікованими лікарськими засобами і у правоохоронних органів у цей період часто не було юридичних підстав для відкриття кримінальних справ, а у судів — винесення рішень за фактом виявлення фальсифікованих препаратів.
3. Епоха пошуку простих рішень для складних проблем. Розвиток і занепад системи боротьби з фальсифікованими ліками в Україні 2005–2020 рр.
Доборолась Україна
до самого краю…
Тарас Шевченко
Несподіваним і потужним ударом по системі боротьби з фальсифікованими лікарськими засобами в Україні, яку протягом багатьох років створювали сотні професіоналів, стала заява щойно призначеного Прем’єр-міністра Ю.В. Тимошенко на засіданні КМУ 02.03.2005 р. про наявність в аптеках 25% фальсифікованих ліків, які містять крейду замість активних речовин. Така заява політика, яка в той час мала високий рейтинг у населення, спричинила паніку і зниження довіри пацієнтів до ліків і системи охорони здоров’я взагалі; з різних регіонів повідомляли про випадки агресії, коли люди розбивали шибки в аптеках і кидали ліки в обличчя фармацевтів зі звинуваченнями їх у продажі фальсифікованих лікарських засобів. Співробітники НРО в стані глибокого когнітивного дисонансу губилися у здогадках щодо джерел інформації для заяви Прем’єр-міністра про такий високий відсоток фальсифікованих ліків в Україні; згідно з однією з версій, ними могли стати публікації російських масмедіа цього періоду щодо нібито 12,5% фальсифікованих лікарських засобів на ринку РФ, для переконливості помножені її «радниками» удвічі.
Ще однією гучною заявою на цьому засіданні КМУ стало проголошення мети знизити ціни на лікарські засоби на 15–40% до кінця 2005 р., що спричинило цілу низку негативних наслідків для фармацевтичного ринку протягом двох каденцій очільника Уряду (лютий–вересень 2005 р. та грудень 2007 р. – березень 2010 р.),
Розуміючи масштаб негативних наслідків цих заяв, тодішній заступник міністра охорони здоров’я В.О. Рибчук вже 25.03.2005 р. спробував пом’якшити їх своїми коментарями і уточненнями в .
Але ще кілька років керівникам НРО і фармацевтичній спільності довелося нервово реагувати і пояснювати політикам та чиновникам згубність численних радикальних ідей Ю.В. Тимошенко, які, на її думку, мали призвести до здешевлення ліків, таких як: проєкт створення мережі аптек на базі «Укрпошти»; перетворення державної компанії «Ліки України» на мега-гравця фармацевтичного ринку, створення «резерву» основних ліків для інтервенцій на ринку для зниження їх цін (за аналогією з валютними резервами Національного банку України); спроби жорсткого адміністративного обмеження розміру торгових націнок; використання прокуратури, Антимонопольного комітету України, контролюючих і податкових органів для тиску на фармацевтичні компанії для зниження цін на лікарські засоби тощо.
Апогеєм публічної демонстрації недовіри і негативного ставлення Прем’єр-міністра до фармацевтів та розхитування системи постачання ліків став епізод під час епідемії грипу Н1N1 у 2009 р. з надтерміновою закупкою Урядом партії капсул озельтамівіру. Під час персональної участі у розвантаженні літака в ніч на 2 листопада 2009 р. вона , в якому назвала фармацевтів «фармацевтичною мафією», злочинцями і «мародерами», які завищують ціни на ліки, і пообіцяла поставляти цей препарат не в аптеки, а безпосередньо в лікарні, де пацієнти будуть отримувати його безкоштовно. Можна тільки здогадуватися про реальну вартість цього лікарського засобу для країни, враховуючи ціни, за якими його було закуплено у Швейцарії протягом 2 вихідних днів (!), вартість його доставки в Україну спеціальним авіарейсом і розвантаження членами Уряду, Верховної Ради і Секретаріату Президента, трансферу в регіони не через стандартні дистриб’юторські канали, а потім — списання та утилізації невикористаних залишків препарату через 2 роки, після закінчення його терміну придатності.
У відповідь на шалений тиск Уряду, силових структур і Держлікінспекції (якій КМУ доручив проводити моніторинг цін на лікарські засоби) на фармацевтичну галузь з метою стримування підвищення цін у період економічної кризи 2008–2009 рр., вперше в історії України, в 2009 р. перед будинком КМУ відбулися акції протесту представників виробників, дистриб’юторів лікарських засобів і аптек. Однією з вимог протестувальників була вимога звільнити голову Держлікінспекції Г.В. Падалко.
У цей же час Комітет Верховної Ради України з питань охорони здоров’я ухвалив рішення від 08.07.2009 р. № 04-24/4-46 «Про виконання Державною інспекцією з контролю якості лікарських засобів і МОЗ України функцій щодо здійснення державного контролю якості лікарських засобів і запобігання потраплянню на фармацевтичний ринок фальсифікованих і неякісних лікарських засобів», у якому визнав стан державного контролю якості лікарських засобів незадовільним.
З 2005 до 2014 р. Держлікінспекцію та Держлікслужбу кілька разів перейменовували, реорганізували та об’єднували (див. рис. 2), що призводило до відволікання ресурсів і уваги НРО. Керівники, юристи і бухгалтери цих НРО скаженіли, раз за разом змінюючи і реєструючи нові статутні документи, печатки, банківські рахунки і численні нормативні документи, в яких були вказані права, обов’язки і повноваження їх організацій. Ця проблема також надовго загальмувала процес вступу Держлікслужби до PIC/S, який тривав рекордні 8 років, з 2003 до 2011 р.
Після об’єднання Держлікінспекції та Держлікслужби у 2008 р. новостворена установа, яка отримала статус центрального органу виконавчої влади, була переведена повністю на бюджетне фінансування (за винятком 2009 та 2001 р.). Озвучені у 2005 р. вражаючі цифри про 25% фальсифікованих лікарських засобів стали підставою для виділення і «освоєння» найбільш щедрого фінансування протягом 2009–2013 рр. (див. рис. 3) і збільшення граничної чисельності співробітників (з 600 до 1465 штатних одиниць у 2009 р.), коли керівники Держлікінспекції/Держлікслужби мали прямий доступ до прем’єр-міністрів України і підтримку від них.
Різке зменшення граничної чисельності у 2014 р. (до 560 працівників, в основному за рахунок співробітників територіальних держлікслужб) і фінансування Держлікслужби у 2014-2017 рр. відбулося на фоні агресії РФ, економічної кризи і 3-кратної девальвації гривні. Крім цього, 2013 р. став останнім роком, коли Державний бюджет України фінансував витрати розвитку Держлікслужби; надалі кошти виділялися лише на зарплатню і оплату комунальних послуг та енергоносіїв. У державних бюджетах України 2018–2021 рр. відбувалося відносне підвищення запланованих видатків на фінансування Держлікслужби, але, з урахуванням динаміки економічних показників з 2003 до 2019 рр. (темпів інфляції і зростання рівня цін; більше, ніж у 30 разів збільшення середньої зарплатні і вартості комунальних послуг в Україні тощо), можна говорити про 3-кратне погіршення реального рівня фінансування Держлікслужби у 2019 р. у порівнянні з фінансуванням Держлікінспекції у 2003 р.
На момент створення, у 2000-х роках Держлікслужба проголосила, що буде безкоштовно здійснювати функції ліцензування, інспектування, контролю якості лікарських засобів тощо, за виконання яких НРО інших країн отримують оплату від заявників. Потрапивши в пастку недостатнього і несвоєчасного бюджетного фінансування, Держлікслужба впродовж останніх років має низку проблем зі спроможністю виконувати статутні завдання, забезпечувати адекватний рівень оплати співробітників, оплачувати членські внески до PIC/S і навіть закуповувати нові випуски Державної Фармакопеї України. Спроби організувати виконання цих завдань через підпорядковані Держлікслужбі державні підприємства не вирішує старі і створює нові проблеми в юридичній та технічній площині.
У зв’язку з цим у 2020 р. комітет з охорони здоров’я Європейської Бізнес Асоціації заявив про серйозну проблему «».
Очевидно, щоб уникнути подальшого погіршення ситуації і колапсу роботи, політикам і чиновникам Уряду та МОЗ слід терміново зайнятися зміною організаційної форми і моделі фінансування Держлікслужби, взявши за приклад такі у НРО країн ЄС та США.
Найбільш дорогим, безглуздим і безрезультатним в історії українських НРО став проєкт Уряду і Держлікінспекції із закупівлі аналітичного обладнання для лабораторій контролю якості лікарських засобів на суму більше 60 млн грн (близько 7,5 млн дол.) у 2010–2011 рр. (див. рис. 3 а–в).
У розвинених країнах лабораторії з контролю лікарських засобів НРО є важливим і найбільш затратним, але допоміжним елементом системи забезпечення якості ліків. Головними, найбільш важливими і ефективними елементами такої системи вважаються процедури реєстрації лікарських засобів, ліцензування (виробників, дистриб’юторів, імпортерів і аптек) та їх регулярного інспектування (див. рис. 1).
Але в той період чиновники КМУ вважали, що знайшли просте та швидке рішення складної проблеми підвищення якості лікарських засобів; на нарадах в Уряді і МОЗ періодично звучала фраза «Ми закупимо обладнання для державних лабораторій, і ліки в Україні будуть якісними!». На жаль, це хибне уявлення до сьогодні продовжує існувати у керівників Держлікслужби, які періодично надсилають запити до КМУ з проханням виділити десятки мільйонів гривень на дооснащення лабораторій.
Лабораторії всіх територіальних органів Держлікінспекції, відповідно до їх завдання перевірки великої кількості зразків лікарських засобів простими і швидкими методами, у 2003–2004 рр. вже були оснащені новими сучасними аналітичними приладами для виконання цих завдань — електронними аналітичними вагами, УФ-спектрофотометрами, рН-метрами, наборами для тонкошарової хроматографії, приладами для дослідження розпадання таблеток і капсул тощо. Закуплене ж складне обладнання — для високоефективної рідинної хроматографії, ГРХ, інфрачервоної спектроскопії тощо маловідомих виробників, без гарантії та сервісного обслуговування були поставлені не в підпорядковані Держлікслужбі лабораторії другого рівня в Києві та Харкові, а в 10 лабораторій територіальних держлікінспекцій. Сім із них, за винятком київських міської та обласної і харківської, знаходилися далеко від більшості українських виробників та митниць із найбільшими потоками ввезення імпортних лікарських засобів, і всі вони не мали відповідних завдань і потреби в такому обладнанні, як і фахівців та фінансових ресурсів для його використання.
Найбільше такого обладнання отримали лабораторії державних інспекцій у Київській і Донецькій областях та АР Крим. З великими складнощами, лише через кілька років після закупівлі, це обладнання частково вдалося ввести в експлуатацію в лабораторії Київської обл. В лабораторіях у Донецькій обл. і АР Крим це обладнання так і не вдалося запустити в роботу. А згодом, у 2014 р., обладнання 4 із цих 10 лабораторій (в містах Сімферополь, Севастополь, Донецьк і Луганськ) залишилося на тимчасово окупованих територіях.
Дворівнева система лабораторного контролю в Україні була серйозно послаблена після 2007 р. у зв’язку з переведенням Держлікінспекції на фінансування лише з державного бюджету, відміною оплати за проведення контролю зразків і суттєвим зменшенням їх кількості в лабораторіях територіальних державних інспекцій після введення КМУ обмеження на перевірки аптек і дистриб’юторів. Після об’єднання Держлікінспекції і Держлікслужби у 2008 р. почався занепад роботи територіальних лабораторій, звільнення їх співробітників, консервація обладнання тощо.
Унікальна Програма професійного тестування лабораторій в Україні у 2008–2009 рр. та 2011 р. була взагалі призупинена, а з 2006 р. почалося зменшення участі в ній територіальних лабораторій першого рівня (див. табл. 2), для яких ця програма власне і була започаткована. Це свідчило про поступове руйнування в них системи забезпечення якості відповідно до рекомендацій GLP. З іншого боку, спостерігається збільшення участі в Програмі професійного тестування лабораторій відділів контролю якості та дослідних центрів українських фармацевтичних підприємств, що свідчить про підвищення рівня розробки, виробництва і якості ліків українських виробників.
У зв’язку із суттєвим зменшенням кількості зразків, що перевірялися територіальними лабораторіями, з 2010 р. Держлікінспекція змінила методику розрахунку кількості фальсифікованих лікарських засобів на ринку — замість відношення виявлених серій фальсифікованих ліків до кількості серій, зразки яких були проаналізовані в лабораторіях, вона стала використовувати відношення виявлених і вилучених фальсифікованих лікарських засобів (в упаковках або грошах) до загальної кількості лікарських засобів на ринку (упаковок або грошей).
Рішення не вказувати у приписах і розпорядженнях Держлікінспекції ознаки фальсифікації та показники, за якими перевірені зразки були визнані фальсифікованими або субстандартними, призвело до зниження ефективності виявлення інших серій цих небезпечних продуктів.
1 листопада 2011 р. набули чинності зміни до Кримінального Кодексу України (ККУ), які встановили за фальсифіковані лікарські засоби, що було передбачено ще у 2003 р. планом заходів Програми боротьби з виробництвом та розповсюдженням фальсифікованих лікарських засобів на 2003–2008 рр.
Одночасно в Закон України «Про лікарські засоби» вперше було введено визначення фальсифікованих лікарських засобів. «Фальсифікований лікарський засіб — лікарський засіб, який умисно промаркований неідентично (невідповідно) відомостям (одній або декільком з них) про лікарський засіб з відповідною назвою, що внесені до Державного реєстру лікарських засобів України, а так само лікарський засіб, умисно підроблений в інший спосіб, і не відповідає відомостям (одній або декільком з них), у тому числі складу, про лікарський засіб з відповідною назвою, що внесені до Державного реєстру лікарських засобів України».
На жаль, воно не було гармонізоване з рекомендованим ВООЗ в документі WHO/EDM/QSM/99.1, а «покращене» — переобтяжене зайвою інформацією і з додатковим обмеженням щодо наявності лікарського засобу, що фальсифікувався, в Державному реєстрі лікарських засобів.
Отже, згідно з цим визначенням фальсифікованого лікарського засобу, якщо фальсифікований був препарат, що не зареєстрований в Україні, то такий продукт не вважається фальсифікованим, що згодом стало призводити до проблем у судовій практиці. Як було згадано на початку цієї статті, в 2017 р. Всесвітня Асамблея Охорони Здоров’я в документі WHA70.23 рекомендувала використовувати нове, спрощене визначення фальсифікованих лікарських засобів, яке варто ввести в законодавство України, щоб виправити вищевказану колізію.
Майже одночасно, 28.10.2011 р. голова Держлікслужби О.А. Соловйов від імені України підписав «Конвенцію Ради Європи щодо протидії фальсифікації лікарських засобів та аналогічних злочинів, що несуть загрозу суспільному здоров’ю (Конвенцію Медікрім)». І вже 20 серпня 2012 р. Верховна Рада України ратифікувала цю Конвенцію першою серед парламентів країн-підписантів. На жаль, Україна до сьогодні не виконала ряд зобов’язань, взятих під час ратифікації Конвенції, зокрема і щодо введення кримінальної відповідальності за фальсифікацію медичних виробів і ветеринарних препаратів.
Безумовно, правильне рішення Верховної Ради про введення кримінальної відповідальності за діяльність, пов’язану з фальсифікованими лікарськими засобами, стало фетишем для багатьох політиків-популістів, які стверджували, що така норма сама по собі зможе вирішити проблему, і тричі (у 2012, 2016 та 2019 р.) в ст. 321-1 ККУ вносилися зміни щодо посилення відповідальності. У результаті наразі за виготовлення, збут, обіг тощо фальсифікованих лікарських засобів цією статтею передбачена кримінальна відповідальність у вигляді 5–8 років позбавлення волі; 8–10 років — за наявності обтяжуючих обставин та 10–15 років або довічне ув’язнення — у разі спричинення смерті або інших тяжких наслідків. Таке покарання є непропорційно жорстким та вищим, ніж за тяжкі злочини проти здоров’я та статевої недоторканості людини (тяжкі ушкодження, катування, зґвалтування та розбій), та таким самим, як за умисне вбивство.
Щодо ефективності такої норми, то після введення кримінальної відповідальності за діяльність, пов’язану з фальсифікованими лікарськими засобами, вона не призвела до помітного зменшення кількості виявлених і вилучених фальсифікованих препаратів (див. табл. 2). Згідно з даними Офісу Генерального прокурора, з кількох десятків кримінальних проваджень, зареєстрованих в Україні у 2016–2018 рр., менше 10% справ було передано до судів. У період 2013–2018 рр. українські суди щорічно виносили від 2 до 6 вироків за ст. 321-1 ККУ.
Така низька ефективність кримінальних розслідувань справ щодо фальсифікованих лікарських засобів може бути пояснена неповним впровадженням в Україні рекомендацій Конвенції Медікрім. Наприклад, згідно зі ст.16 Конвенції кримінальні розслідування мають проводити особи, підрозділи або служби, компетентні в питаннях фальсифікації медичних продуктів і що мають персонал, який пройшов відповідне навчання для виконання таких розслідувань, у тому числі у фінансовій сфері; такі підрозділи або служби повинні мати необхідні ресурси.
У Національній поліції України на сьогодні відсутні відповідні підрозділи, які б кваліфіковано проводили кримінальні розслідування за ст. 321-1 ККУ, що наразі проводять слідчі, а в судах рішення приймають судді, що не мають базових знань у фармацевтичній сфері. Також такі кримінальні розслідування ускладнюються неузгодженістю між ст. 321-1 ККУ та статтями Кримінально-процесуального кодексу, що унеможливлює в повній мірі застосовування деяких процесуальних інструментів для таких видів кримінальних розслідувань. Очевидно, розв’язання цих юридичних проблем має стати окремим напрямком у наступній редакції державної Програми боротьби з фальсифікованими лікарськими засобами.
13.12.2012 р. Держлікслужба повідомила про початок проєкту зі створення і впровадження «автоматизованої системи відстеження обігу лікарських засобів», а влітку 2013 р. — про його перші проміжні результати. Беручи до уваги досвід впровадження подібної системи в Туреччині і плани ЄС щодо виконання Директиви № 2011/62/EU, цей проєкт передбачав у 2013–2016 рр. впровадження в Україні системи відстеження за допомогою 2D-кодів обігу всіх упаковок лікарських засобів від виробників та імпортерів до дистриб’юторів, аптек та лікувальних закладів. Як запевнив голова Держлікслужби, «впровадження Системи в майбутньому повністю виключить можливість потрапляння фальсифікованих лікарських засобів у легальну мережу».
У 2013 р., на першому етапі, в пілотному проєкті з відстеження та ідентифікації упаковок лікарських засобів взяли участь по одному українському і зарубіжному виробнику, один дистриб’ютор і одна аптечна мережа. Держлікслужба для реалізації цього проєкту одержала з державного бюджету 15 млн грн і успішно освоїла їх. А вже у 2014 р. проєкт тихо зупинився після зміни Уряду і керівництва Держлікслужби внаслідок революційних подій того року.
У 2014 р. Уряд прийняв рішення про встановлення мораторію на перевірки бізнесу, який діє до сьогодні. У результаті Держлікслужба практично втратила можливість проводити позапланові перевірки аптек і дистриб’юторів, що суттєво знизило її можливості виявляти фальсифіковані лікарські засоби і карати порушників. Введення в березні 2020 р. в Україні карантину у зв’язку з епідемією COVID-19 ще більше обмежило можливість контролю і перевірок дистриб’юторів та аптек. І цим може бути пояснена найменша за всю історію спостереження кількість виявлених серій фальсифікованих лікарських засобів у 2020 р. (4 серії).
Теза про можливість остаточного вирішення проблеми фальсифікованих ліків шляхом впровадження 2D-кодування упаковок продовжувала активно озвучуватися політиками, чиновниками та фахівцями. При цьому ніхто не звертав уваги на той факт, що система 2D-кодування в ЄС призначалася лише для рецептурних лікарських засобів (за винятком більшості інфузійних розчинів) і одного безрецептурного (омепразолу з модифікованим вивільненням). При використанні такого ж підходу в Україні, з урахуванням часток рецептурних та безрецептурних препаратів на ринку (близько 40:60 в упаковках) тільки приблизно третина лікарських засобів в Україні буде мати обов’язкове маркування 2D-кодами. Відповідно, ця система не буде поширюватися на 2/3 упаковок ліків на ринку України і не зможе запобігти їх фальсифікації.
Крім цього, майже не обговорювалася доцільність і своєчасність впровадження системи 2D-кодування упаковок лікарських засобів в Україні, яка на сьогодні має обмежені ресурси, переживає глибоку економічну кризу у зв’язку з епідемією COVID-19 та має низку більш важливих і актуальних невирішених проблем у системі охорони здоров’я.
У результаті такої інформаційної кампанії, КМУ розпорядженням від 03.04.2019 р. № 301-р. схвалив «Концепцію реалізації державної політики щодо запобігання фальсифікації лікарських засобів та затвердження плану заходів з її реалізації». Замість комплексу заходів, рекомендованих , вказана Концепція передбачала лише один захід — створення системи маркування 2D-кодами упаковок лікарських засобів, без згадування будь-яких інших механізмів боротьби з обігом фальсифікованих препаратів.
Наступне рішення КМУ «Про запровадження пілотного проєкту щодо маркування контрольними (ідентифікаційними) знаками та проведення моніторингу обігу лікарських засобів», затверджене постановою від 24.07.2019 р. № 653, викликало ще більше питань і гарячі дискусії між регуляторами та операторами ринку. Відсутність зрозумілої концепції проєкту з коректними вихідними даними щодо кількості фальсифікованих лікарських засобів в Україні, розуміння обсягу необхідних інвестицій і витрат для створення, впровадження і підтримання цієї системи та джерел їх фінансування; KPIs — очікуваних кількісних результатів від впровадження; надійної інформації про досвід, результати та наслідки впровадження системи в ЄС та в інших країнах; плутанина з керівництвом і виконавцями проєкту; кризова ситуація в країні у зв’язку з епідемією COVID-19 — усе це змусило провідних українських виробників лікарських засобів 20.07.2020 р. звернутися з відкритим листом до керівників Уряду і профільного комітету Верховної Ради з пропозицією відтермінувати впровадження системи 2D-кодування лікарських засобів в Україні.
24 липня 2020 р. за результатами обговорення цього листа в МОЗ міністр охорони здоров’я, обґрунтовуючи свою позицію щодо необхідності цього проєкту, несподівано заявив, що «За даними ВООЗ, до 10% лікарських засобів на ринку України є фальсифікованими». Ці помилкові й значно завищені дані були широко розповсюджені електронними ЗМІ. Тому 31.08.2020 р. українські виробники лікарських засобів повторно звернулися з відкритим листом до міністра охорони здоров’я з пропозицією використовувати коректні вихідні дані і відкласти проєкт щодо впровадження системи 2D-кодування препаратів.
Уряд не прийняв ніякого рішення щодо внесення змін до вказаного пілотного проєкту, і формально він закінчився 31.12.2020 р. До 10.01.2021 р. МОЗ мав подати Уряду інформацію про його результати, у тому числі обсяги промаркованих ліків; сканування і перевірки відповідного кодування на всіх етапах обігу лікарського засобу та ін.
Однією з проблем системи боротьби з обігом фальсифікованих лікарських засобів стала втрата керівниками центрального апарату і більшості територіальних підрозділів Держлікслужби лідерства і репутації загальновизнаної експертної установи в цій сфері. Упродовж останніх років про проблему фальсифікації лікарських засобів населенню, фармацевтичній і медичній спільноті розказують викладачі університетів, керівники професійних асоціацій тощо, а виступів, заяв та інтерв’ю на цю тему керівники та співробітники Держлікслужби (за винятком окремих регіонів) майже не чути. Не відбувається ніякої реакції Держлікслужби і на оприлюднення некоректної інформації щодо масштабу фальсифікації ліків (на кшталт заяви міністра 24.07.2020 р. про нібито дані ВООЗ щодо 10% фальсифікованих лікарських засобів в Україні).
Керівники Уряду і МОЗ не використовують статистичні дані і висновки Держлікслужби щодо небезпеки і масштабу проблеми субстандартних і фальсифікованих лікарських засобів, що призводить до помилкових політичних та управлінських рішень. На сайті Держлікслужби на сьогодні немає регулярної узагальненої аналітичної інформації щодо кількості фальсифікованих лікарських засобів, яка б дозволила оцінити динаміку показників за періодами і оцінити їх небезпеку для суспільства та ефективність протидії з боку НРО.
Таким чином, у 2005–2020 рр. система боротьби з фальсифікацією лікарських засобів, створена у 1991–2005 рр., в цілому продовжувала виконувати свої функції, але відбувалося її послаблення у кількох важливих напрямках — інформування населення і медичних професіоналів про небезпеку фальсифікованих лікарських засобів; контролю зразків ліків у територіальних лабораторіях першого рівня; спроможність проводити позапланові перевірки операторів ринку; недостатність і нестабільність фінансового забезпечення НРО тощо.
Проєкт інвестування 60 млн грн в лабораторне обладнання для 10 територіальних лабораторій у 2010–2011 рр. закінчився безрезультатно і не вплинув на ефективність системи боротьби із субстандартними і фальсифікованими лікарськими засобами.
Позитивним здобутком цього періоду стало введення кримінальної відповідальності за фальсифікацію лікарських засобів в Україні, яке, однак, виявилося нерезультативним щодо кількості судових вироків у порівнянні з порушеними кримінальними справами і не призвело до помітного зменшення кількості фальсифікованих препаратів.
Кілька спроб розробки і впровадження системи 2D-кодування упаковок лікарських засобів у 2013–2020 рр. з різних причин не призвели до будь-яких результатів. У зв’язку з існуванням в системі охорони здоров’я України більш важливих, болючих і термінових проблем, вбачається, що цей проєкт слід відкласти на кілька років, до кращих часів.
Найбільш важливим позитивним здобутком цього періоду можна вважати загальне підвищення якості лікарських засобів в Україні. Цього результату вдалося досягти завдяки спільній довголітній і системній роботі фахівців фармацевтичних компаній і регуляторів, після впровадження в Україні в 2011–2013 рр. обов’язкового дотримання вимог GхP на всіх етапах життєвого циклу препарату; удосконалення системи реєстрації лікарських засобів; гармонізації Державної Фармакопеї України з Європейською Фармакопеєю; розвитку системи фармаконагляду тощо. Згідно з різними джерелами, на сьогодні кількість субстандартних лікарських засобів в Україні становить менше 1,0%. Наприклад, згідно зі звітом за 2019 р. Центральна лабораторія за результатами контролю більше 4 тис. зразків лікарських засобів визнала субстандартними 0,9% — у 2018 р. і 0,7% — у 2019 р.
Висновки
1. У 2017 р. ВООЗ оновила концепцію рекомендацій щодо боротьби з фальсифікацією лікарських засобів, об’єднавши в одну категорію субстандартні та фальсифіковані лікарські засоби два типи небезпечних продуктів — «субстандартні та фальсифіковані препарати».
2. У 2000–2005 рр. в Україні була створена і почала працювати система боротьби з фальсифікованими лікарськими засобами відповідно до рекомендацій ВООЗ WHO/EDM/QSM/99.1.
3. У 2005–2020 рр. відбувалося її послаблення у кількох важливих напрямках, натомість спостерігалися спроби керівників Уряду, МОЗ і Держлікслужби вирішити проблему фальсифікованих лікарських засобів за допомогою окремих проєктів — оснащенням лабораторій аналітичним обладнанням, введенням і посиленням кримінальної відповідальності за фальсифікацію лікарських засобів та введенням системи 2D-кодування упаковок лікарських засобів), які не призвели до очікуваних результатів.
4. Україні слід на рівні КМУ затвердити нову редакцію Програми боротьби із субстандартними і фальсифікованими лікарськими засобами з урахуванням Рекомендацій ВООЗ 2017 р. та Директиви ЄС № 2011/62/EU.
5. Важливою метою такої програми має стати зміна організаційної форми і моделі фінансування Держлікслужби для відновлення її інституційної спроможності, взявши за приклад концепції НРО країн зі строгою регуляторною системою.
доктор фармацевтичних наук, «АРТЕРІУМ ЛТД.»
(інформація у цій статті відображає персональну точку зору автора
і не є офіційною позицією компанії)
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим